Artificial Intelligence Analysis of the Gene Expression of Follicular Lymphoma Predicted the Overall Survival and Correlated with the Immune Microenvironment Response Signatures

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Statistical Analysis
2.2. Follicular Lymphoma Gene Expression Dataset
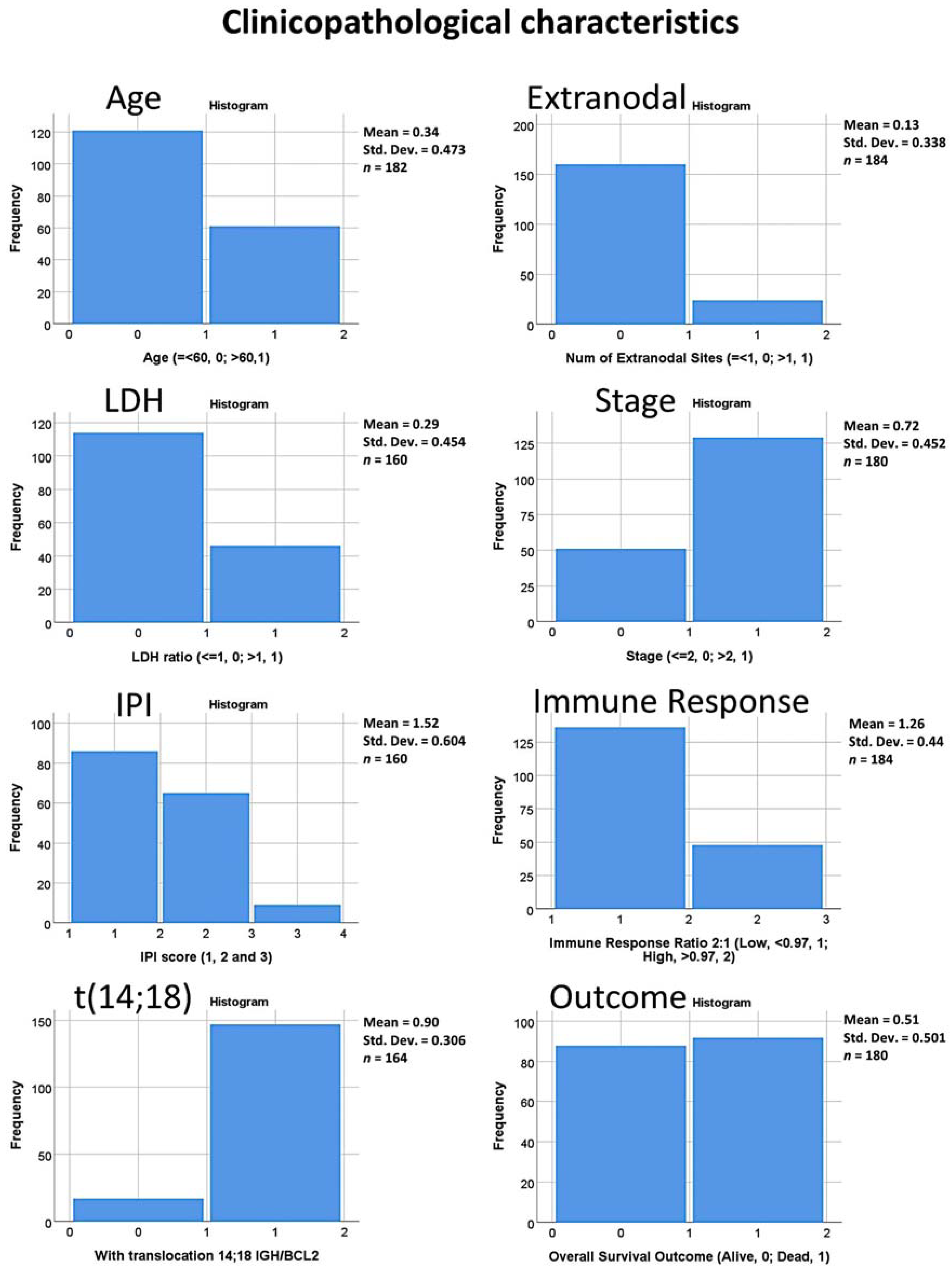
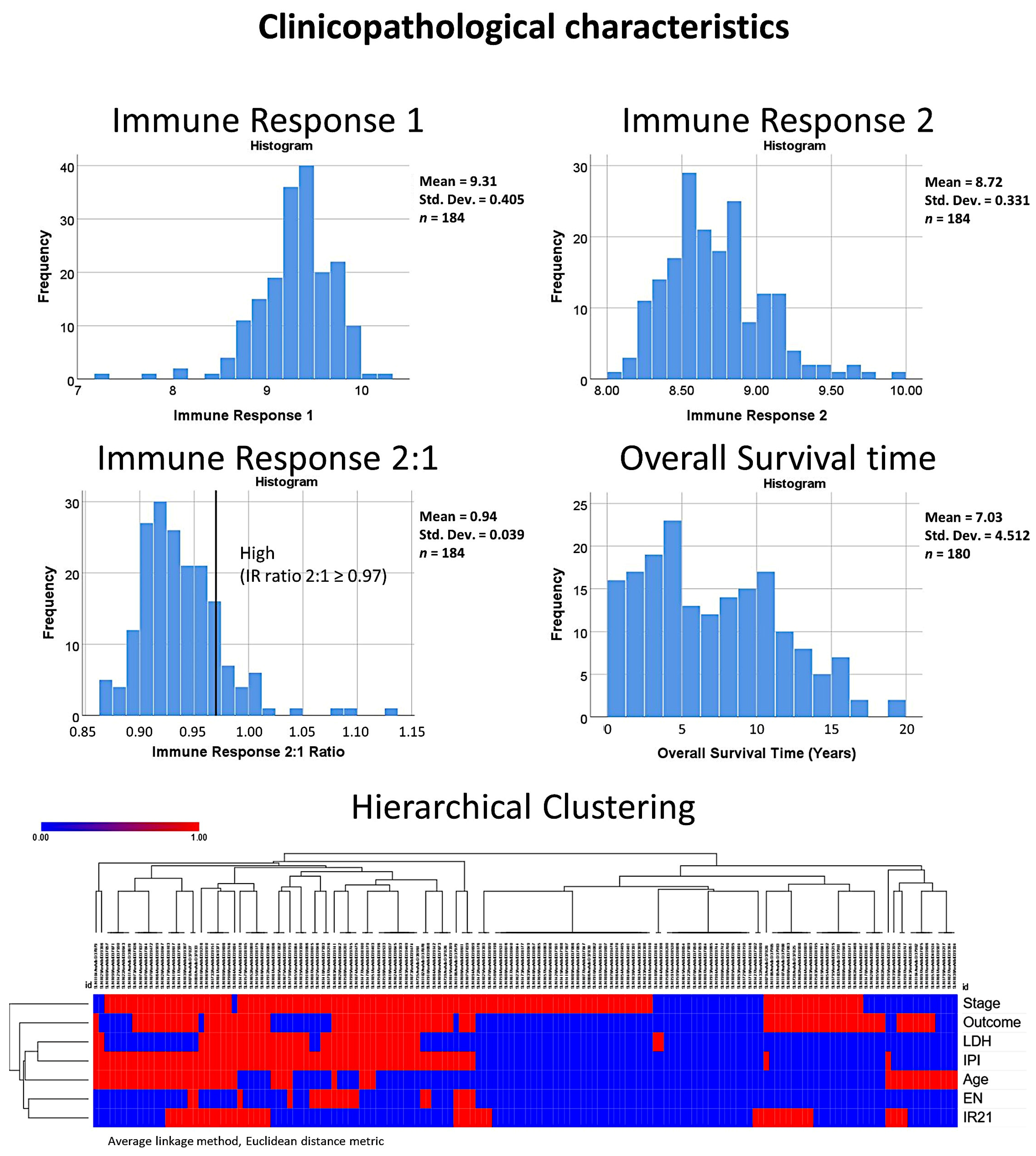
2.3. Patients and Clinicopathological Characteristic
2.4. Multilayer Perceptron and Radial Basis Function Neural Network Analysis
2.5. Gene Set Enrichment Analysis
3. Results
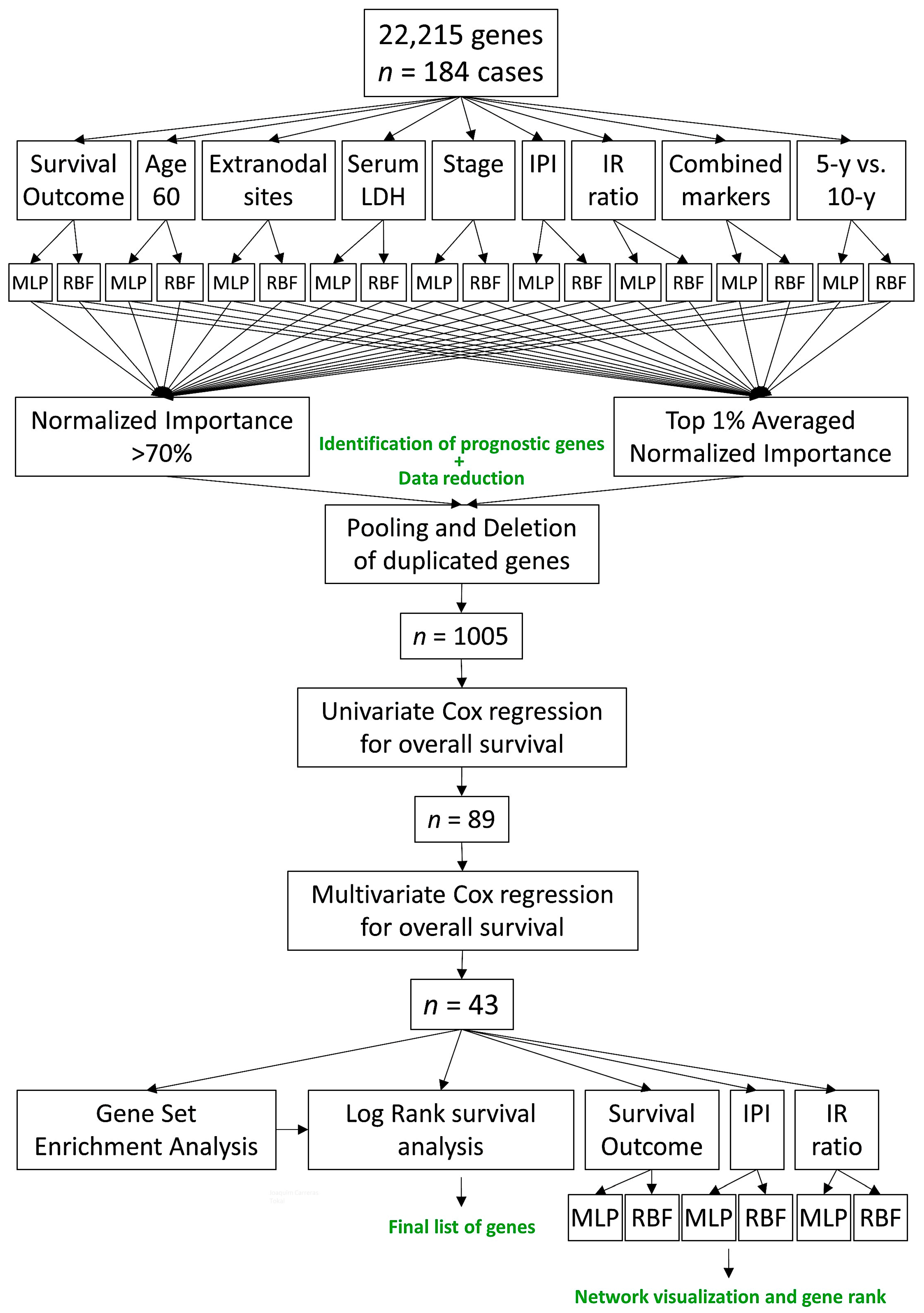
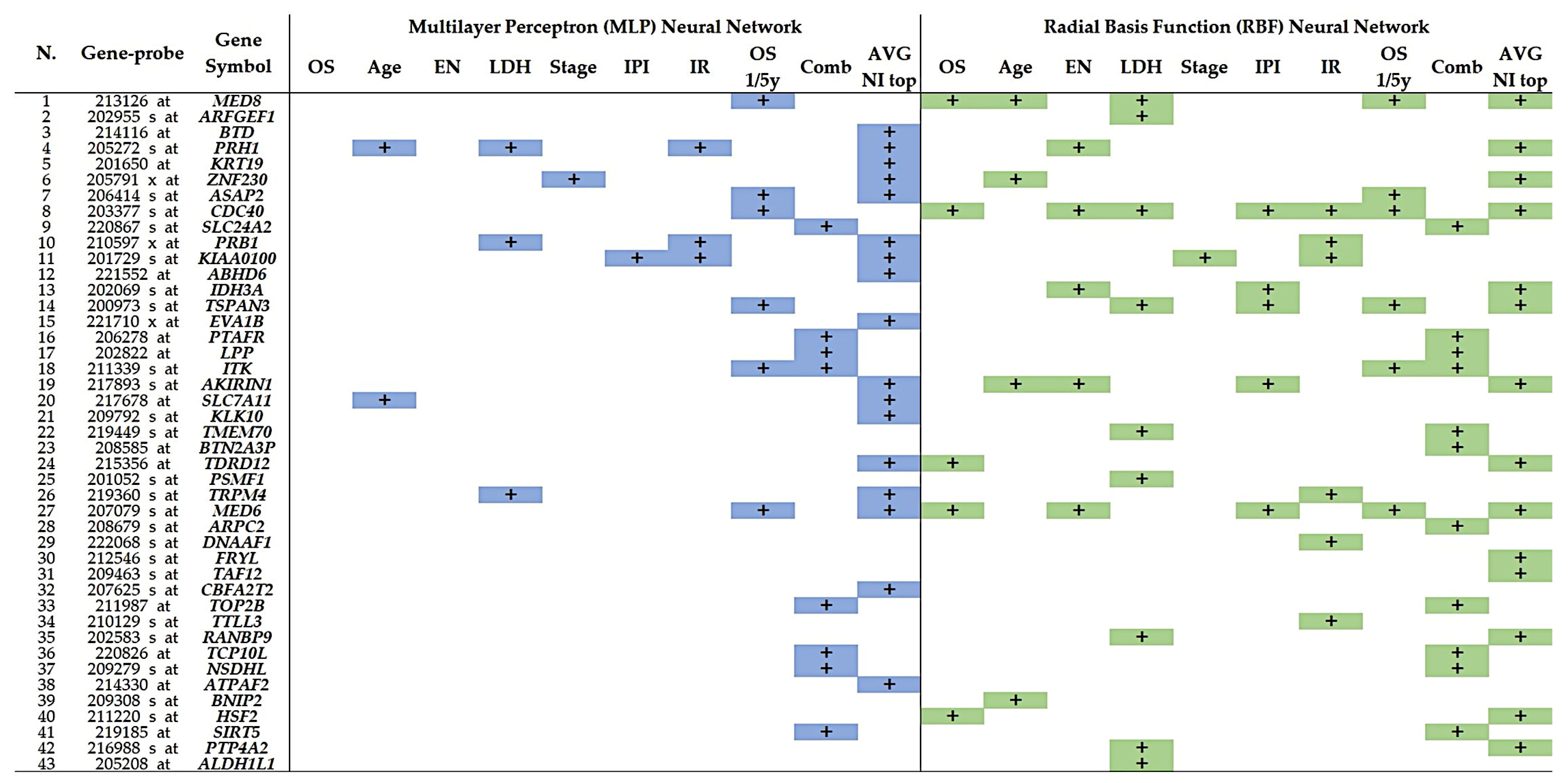
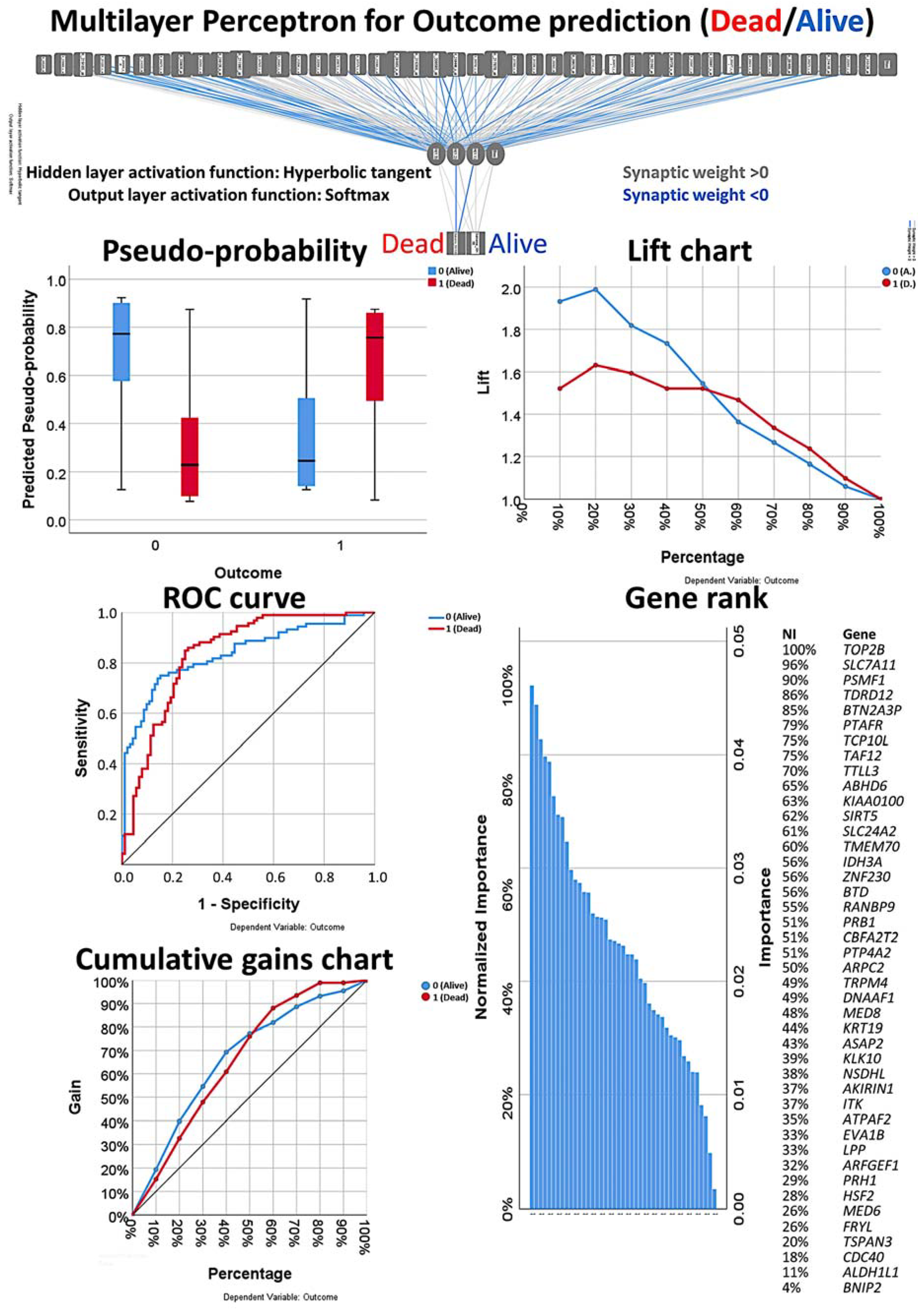
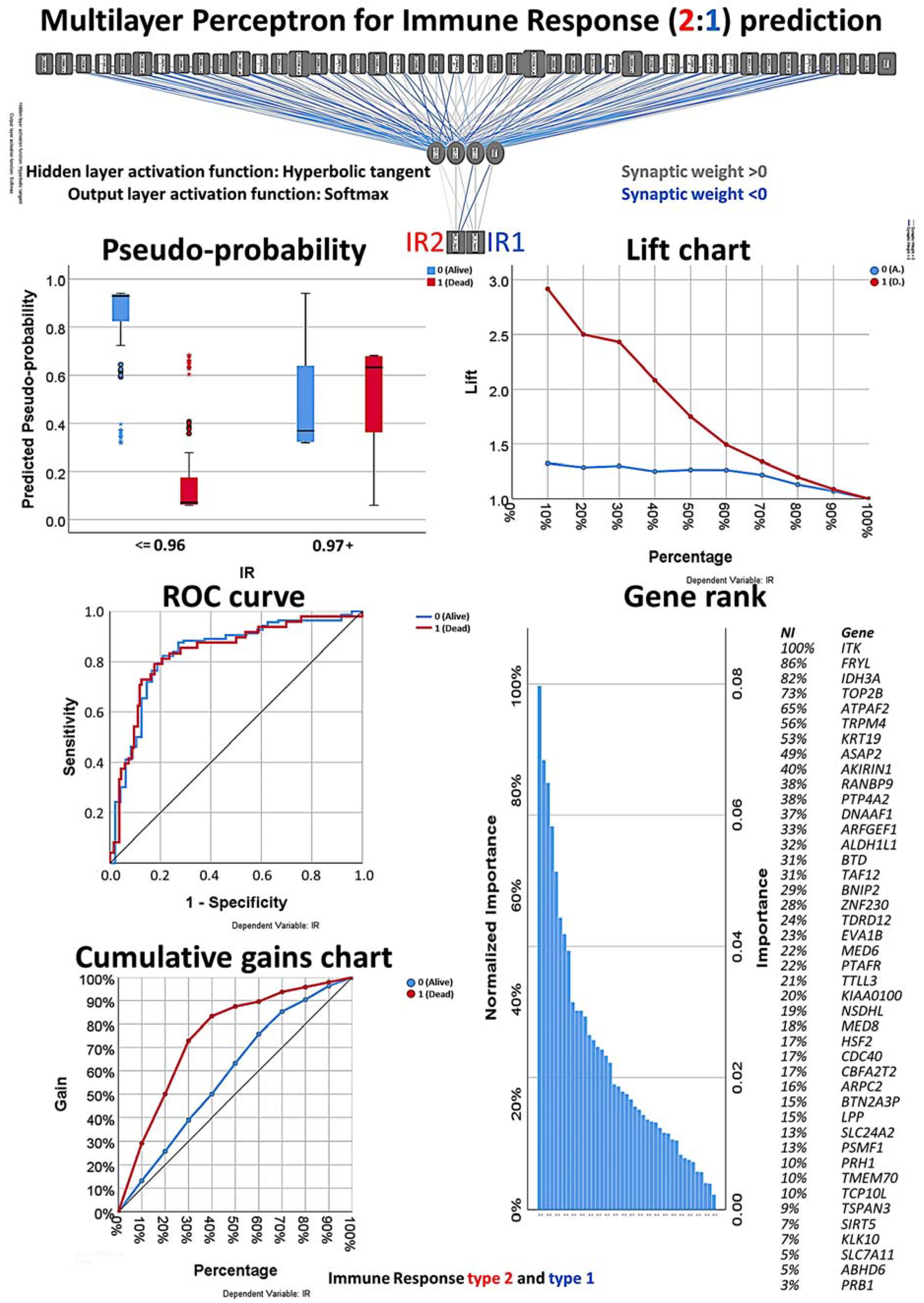
3.1. Multilayer Perceptron and Radial Basis Function Neural Network Analysis
3.2. Univariate and Multivariate Cox Survival Analysis
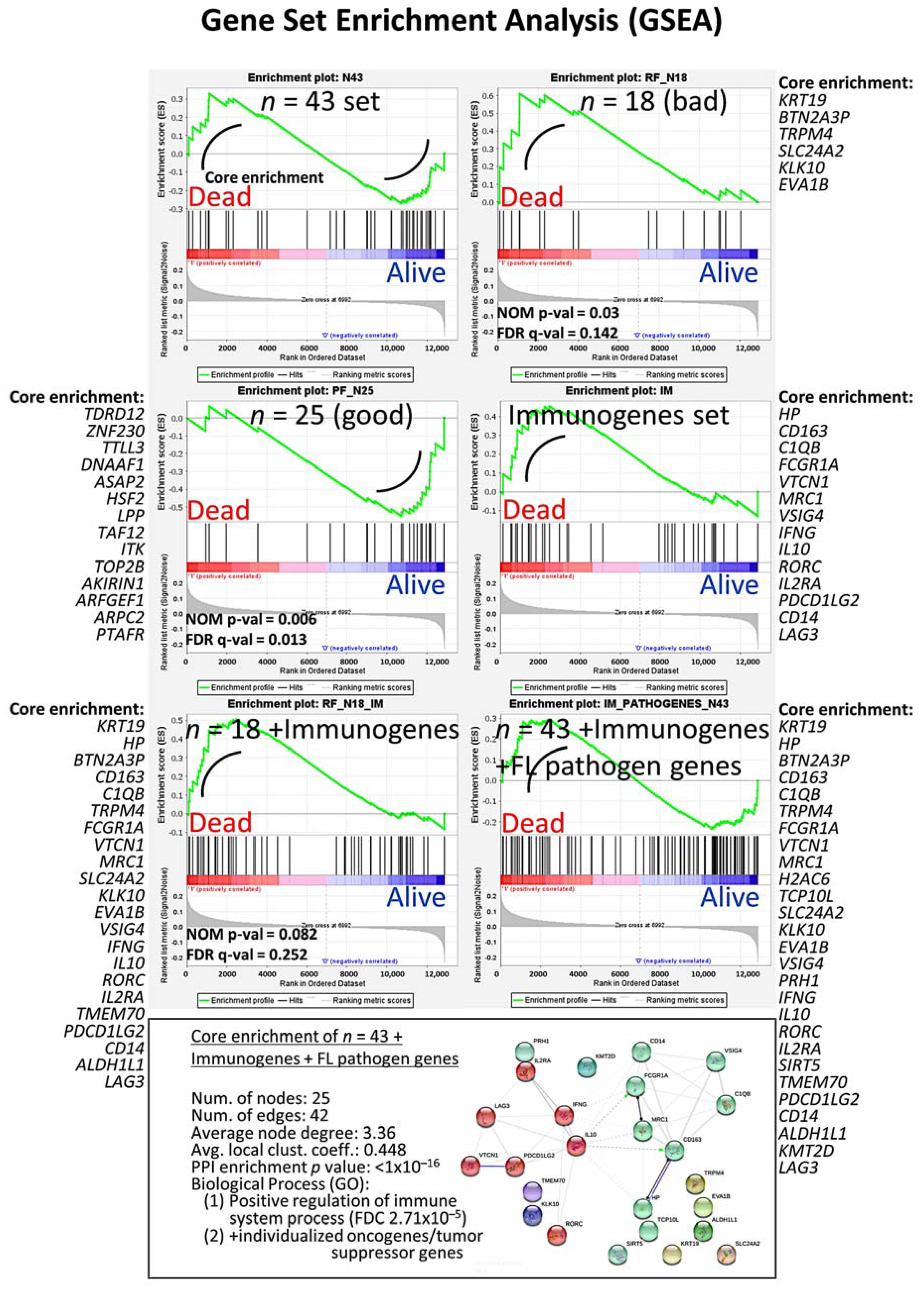
3.3. Gene Set Enrichment Analysis (GSEA)
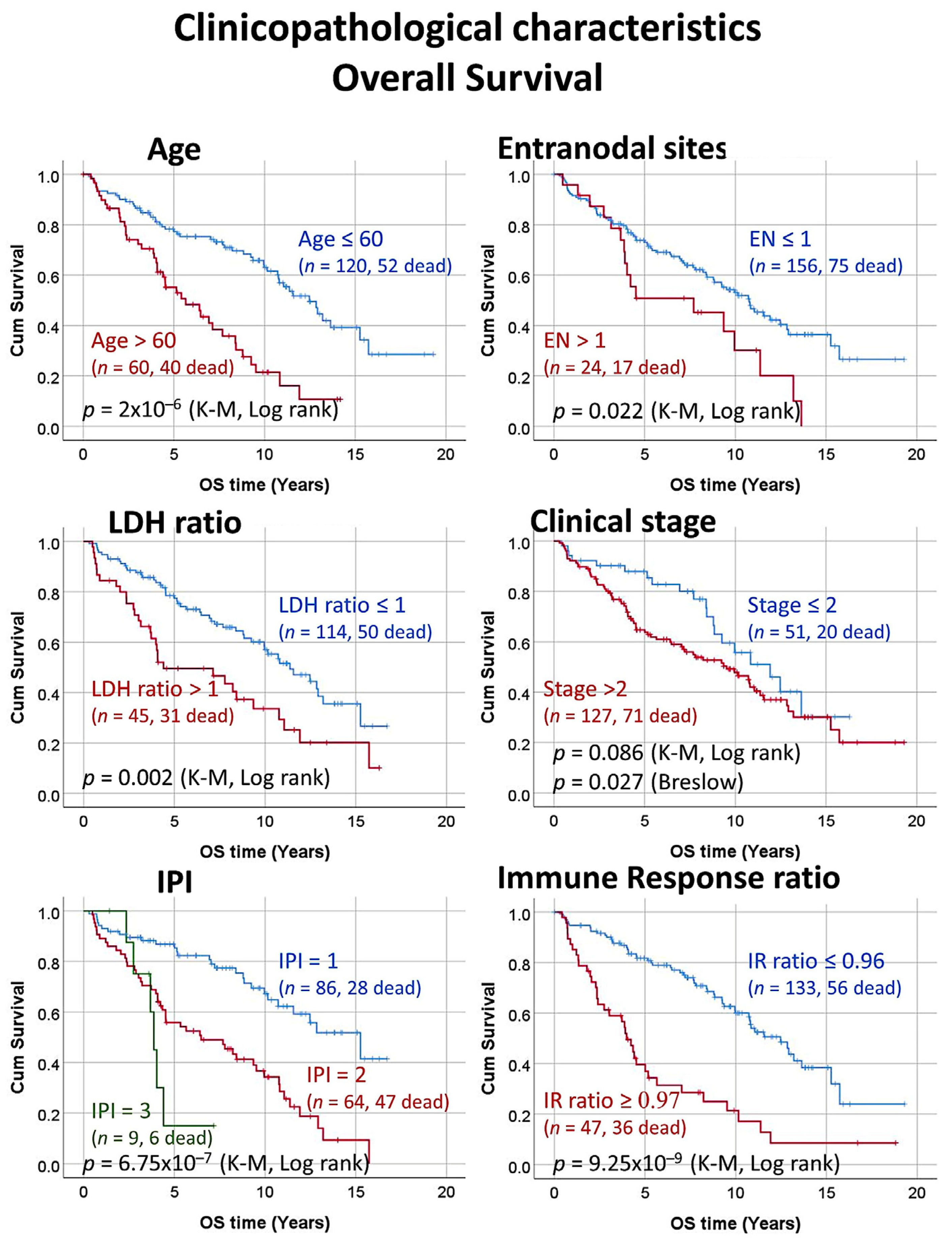
3.4. Univariate Survival Analysis with Kaplan–Meier and Log Rank Test
3.5. Final MLP and RBF Neural Network Analysis
3.6. Correlation between the 7 Highlighted Genes and the Clinicopathological Characteristics of the Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aunalytics. Artificial Intelligence, Machine Learning, and Deep Learning. Available online: https://www.aunalytics.com/artificial-intelligence-machine-learning-and-deep-learning%E2%81%A0/ (accessed on 8 November 2020).
- IBM SPSS Neural Netwoks 25; IBM Corporation: Armonk, NY, USA, 2018.
- IBM Corporation. IBM SPSS Neural Networks 20. New Tools for Building Predictive Models. IBM Business Analytics. Available online: https://www.ibm.com/support/pages/node/618179 (accessed on 28 October 2020).
- Carreras, J.; Hamoudi, R.; Nakamura, N. Artificial Intelligence Analysis of Gene Expression Data Predicted the Prognosis of Patients with Diffuse Large B-Cell Lymphoma. Tokai J. Exp. Clin. Med. 2020, 45, 37–48. [Google Scholar] [PubMed]
- Carreras, J.K.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Shiraiwa, S.; Hamoudi, R.; et al. Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma. AI 2020, 1, 342–360. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, A.S.; Aster, J.C.; Lister, A.; Connor, R.F. Clinical Manifestations, Pathologic Features, Diagnosis, and Prognosis of Follicular Lymphoma. UptoDate. Available online: https://www.uptodate.com/contents/clinical-manifestations-pathologic-features-diagnosis-and-prognosis-of-follicular-lymphoma?search=lymphomas%20definition&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1 (accessed on 28 October 2020).
- Byers, R.J.; Sakhinia, E.; Joseph, P.; Glennie, C.; Hoyland, J.A.; Menasce, L.P.; Radford, J.A.; Illidge, T. Clinical quantitation of immune signature in follicular lymphoma by RT-PCR-based gene expression profiling. Blood 2008, 111, 4764–4770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras, J.; Lopez-Guillermo, A.; Fox, B.C.; Colomo, L.; Martinez, A.; Roncador, G.; Montserrat, E.; Campo, E.; Banham, A.H. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood 2006, 108, 2957–2964. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J.; Lopez-Guillermo, A.; Kikuti, Y.Y.; Itoh, J.; Masashi, M.; Ikoma, H.; Tomita, S.; Hiraiwa, S.; Hamoudi, R.; Rosenwald, A.; et al. High TNFRSF14 and low BTLA are associated with poor prognosis in Follicular Lymphoma and in Diffuse Large B-cell Lymphoma transformation. J. Clin. Exp. Hematop. 2019, 59, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras, J.; Lopez-Guillermo, A.; Roncador, G.; Villamor, N.; Colomo, L.; Martinez, A.; Hamoudi, R.; Howat, W.J.; Montserrat, E.; Campo, E. High numbers of tumor-infiltrating programmed cell death 1-positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J. Clin. Oncol. 2009, 27, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.S.; Wright, G.; Tan, B.; Rosenwald, A.; Gascoyne, R.D.; Chan, W.C.; Fisher, R.I.; Braziel, R.M.; Rimsza, L.M.; Grogan, T.M.; et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med. 2004, 351, 2159–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glas, A.M.; Kersten, M.J.; Delahaye, L.J.; Witteveen, A.T.; Kibbelaar, R.E.; Velds, A.; Wessels, L.F.; Joosten, P.; Kerkhoven, R.M.; Bernards, R.; et al. Gene expression profiling in follicular lymphoma to assess clinical aggressiveness and to guide the choice of treatment. Blood 2005, 105, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, W.J.; Kim, K.H.; Kim, J.Y.; Jeong, S.; Kim, N. Identification of DNA-Methylated CpG Islands Associated With Gene Silencing in the Adult Body Tissues of the Ogye Chicken Using RNA-Seq and Reduced Representation Bisulfite Sequencing. Front. Genet. 2019, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Choi, H.Y.; Kim, B.W.; Dayem, A.A.; Yang, G.M.; Kim, K.S.; Yin, Y.F.; Cho, S.G. KRT19 directly interacts with beta-catenin/RAC1 complex to regulate NUMB-dependent NOTCH signaling pathway and breast cancer properties. Oncogene 2017, 36, 332–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, M.A.; Gamazon, E.R.; Al-Ejeh, F.; Aittomaki, K.; Andrulis, I.L.; Anton-Culver, H.; Arason, A.; Arndt, V.; Aronson, K.J.; Arun, B.K.; et al. Genome-wide association and transcriptome studies identify target genes and risk loci for breast cancer. Nat. Commun. 2019, 10, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappel, S.; Stoklosa, P.; Hauert, B.; Ross-Kaschitza, D.; Borgstrom, A.; Baur, R.; Galvan, J.A.; Zlobec, I.; Peinelt, C. TRPM4 is highly expressed in human colorectal tumor buds and contributes to proliferation, cell cycle, and invasion of colorectal cancer cells. Mol. Oncol. 2019, 13, 2393–2405. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Sun, W.; Tan, M.; Glazer, C.A.; Bhan, S.; Zhong, X.; Fakhry, C.; Sharma, R.; Westra, W.H.; Hoque, M.O.; et al. Integrated, genome-wide screening for hypomethylated oncogenes in salivary gland adenoid cystic carcinoma. Clin. Cancer Res. 2011, 17, 4320–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
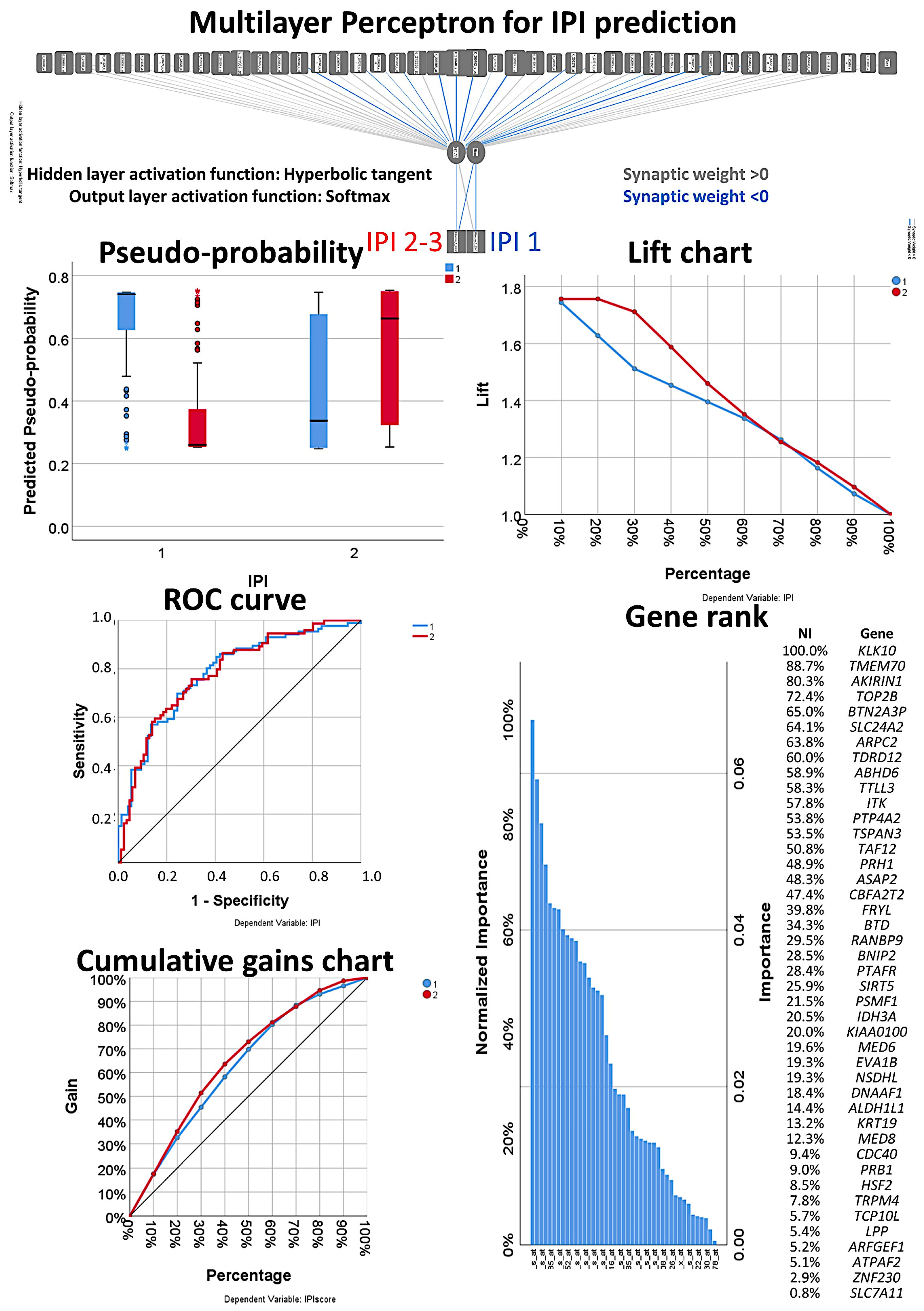{kind=link}
| Variable | Frequency | Percentage |
|---|---|---|
| Age > 60 years | 61 (182) | 22.5 |
| Number of extranodal sites > 1 | 24 (184) | 13 |
| LDH levels ratio > 1 | 46 (160) | 28.7 |
| Stage > 2 | 129 (180) | 71.7 |
| IPI score 2–3 | 74 (160) | 46.3 |
| Immune Response ratio 2:1 high (≥0.97) | 48 (184) | 26.1 |
| With translocation (14;18) positive | 147 (164) | 89.6 |
| Survival outcome dead | 92 (180) | 51.1 |
| Survival dead before 5 years | 35 (84) | 41.7 |
| Survival alive from 10 years | 49 (84) | 58.3 |
| Variable | Log Rank p Value | Cox p Value | Hazard Risk | 95% CI for HR | |
|---|---|---|---|---|---|
| Lower | Upper | ||||
| Age >60 years | 2 × 10−6 | 5 × 10−6 | 2.7 | 1.8 | 4.2 |
| Number of extranodal sites > 1 | 0.022 | 0.025 | 1.8 | 1.1 | 3.1 |
| LDH levels ratio > 1 | 0.002 | 0.002 | 2.0 | 1.3 | 3.2 |
| Stage > 2 1 | 0.086 | 0.088 | 1.5 | 0.9 | 2.5 |
| IPI score 2–3 | 4 × 10−7 | 2 × 10−6 | 3.1 | 2.0 | 5.0 |
| With translocation (14;18) positive | 0.249 | 0.253 | 1.6 | 0.7 | 3.7 |
| Immune Response ratio 2:1 high (≥0.97) | 9 × 10−9 | 5.3 × 10−8 | 3.3 | 2.1 | 5.0 |
| Survival: Dead up to 5-y vs. Alive from 10-years | 5 × 10−21 | 1.7 × 10−5 | 209.2 | 18.3 | 2393.0 |
| Multilayer Perceptron | Dependent Variable | Survival Outcome | Age 60 | Extranodal Sites | LDH | Stage | IPI | IR 2:1 Ratio | Translocation | Combined | 5 vs. 10-y | Mean | STD | Median |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case processing summary | Training | 129 | 116 | 137 | 108 | 130 | 109 | 132 | 109 | 44 | 59 | 107.1 | 33.3 | 116 |
| Training Percentage | 71.7 | 63.7 | 74.5 | 67.5 | 72.2 | 68.1 | 71.7 | 66.5 | 71 | 69.4 | 70.0 | 3.2 | 71 | |
| Testing | 51 | 66 | 47 | 52 | 50 | 51 | 52 | 55 | 18 | 26 | 45.9 | 14.7 | 51 | |
| Testing Percentage | 28.3 | 36.3 | 25.5 | 32.5 | 27.8 | 31.9 | 28.3 | 33.5 | 29 | 30.6 | 30.0 | 3.2 | 29 | |
| Valid | 180 | 182 | 184 | 160 | 180 | 160 | 184 | 164 | 62 | 85 | 153 | 46.4 | 180 | |
| Excluded | 4 | 2 | 0 | 24 | 4 | 24 | 0 | 20 | 122 | 99 | 31 | 46.4 | 4 | |
| Total | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 0 | 184 | |
| Network information | Num of Units | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | - | - | - |
| Rescaling Method for covariates | Standarized | - | - | - | ||||||||||
| Hidden layer | Num Hidden Layers | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 |
| Num Units in Hidden Layer | 9 | 8 | 5 | 15 | 10 | 8 | 11 | 6 | 8 | 8 | 9.1 | 2.8 | 8 | |
| Activation Function | Hyperbolic tangent | - | - | - | ||||||||||
| Output layer | Dep Variable | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 9 | 1 | - | - | - |
| Num of Units | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 18 | 2 | 3.8 | 5.3 | 2 | |
| Activation Function | Softmax | - | - | - | ||||||||||
| Error Function | Cross-entropy | - | - | - | ||||||||||
| Model summary training | Cross Entropy Error | 87.8 | 70.7 | 50.9 | 51.4 | 70.8 | 70.5 | 59.3 | 30.2 | 173.5 | 30.0 | 73.9 | 40.8 | 70.5 |
| Percent of Incorrect Predictions | 40.3 | 35.3 | 14.6 | 22.2 | 23.8 | 33.0 | 16.7 | 11.0 | 19.9 | 22.0 | 25.3 | 8.8 | 22.2 | |
| Stopping Rule Used * | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | - | - | - | |
| Time in Seconds | 84.8 | 83.5 | 101.2 | 71.7 | 99.2 | 79.1 | 97.9 | 76.4 | 41.9 | 44.3 | 78.2 | 22.2 | 83.5 | |
| Model sum. testing | Cross Entropy Error | 28.1 | 32.7 | 11.3 | 27.8 | 24.0 | 30.2 | 23.9 | 9.2 | 84.2 | 13.9 | 30.7 | 21.3 | 27.8 |
| Percent Incorrect Predictions | 23.5 | 22.7 | 8.5 | 23.1 | 18.0 | 33.0 | 23.1 | 7.3 | 25.9 | 26.9 | 22.7 | 6.7 | 23.1 | |
| Classification | Training Overall Percent | 59.7 | 64.7 | 85.4 | 77.8 | 76.2 | 67.0 | 83.3 | 89.0 | 80.1 | 78.0 | 74.7 | 8.8 | 77.8 |
| Testing Overall Percent | 76.5 | 77.3 | 91.5 | 76.9 | 82.0 | 66.7 | 76.9 | 92.7 | 74.1 | 73.1 | 77.2 | 6.8 | 76.9 | |
| Area under the curve | Alive | 0.7 | 0.7 | 0.8 | 0.8 | 0.7 | 0.7 | 0.8 | 0.9 | 0.8 | 0.8 | 0.8 | 0.0 | 0.8 |
| Dead | 0.7 | 0.7 | 0.8 | 0.8 | 0.7 | 0.7 | 0.8 | 0.9 | 0.8 | 0.8 | 0.8 | 0.0 | 0.8 | |
| Radial Basis Function | Dependent Variable | Survival Outcome | Age 60 | Extranodal Sites | LDH | Stage | IPI | IR 2:1 Ratio | Translocation | Combined | 5 vs. 10-y | Mean | STD | Median |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case processing summary | Training | 127 | 123 | 125 | 116 | 127 | 109 | 134 | 119 | 47 | 66 | 108.2 | 30.5 | 123 |
| Training Percentage | 70.6 | 67.6 | 67.9 | 72.5 | 70.6 | 68.1 | 72.8 | 72.6 | 75.8 | 77.6 | 71.5 | 3.5 | 70.6 | |
| Testing | 53 | 59 | 59 | 44 | 53 | 51 | 50 | 45 | 15 | 19 | 44.8 | 16.4 | 51 | |
| Testing Percentage | 29.4 | 32.4 | 32.1 | 27.5 | 29.4 | 31.9 | 27.2 | 27.4 | 24.2 | 22.4 | 28.5 | 3.5 | 29.4 | |
| Valid | 180 | 182 | 184 | 160 | 180 | 160 | 184 | 164 | 62 | 85 | 153 | 46.4 | 180 | |
| Excluded | 4 | 2 | 0 | 24 | 4 | 24 | 0 | 20 | 122 | 99 | 31 | 46.4 | 4 | |
| Total | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 184 | 0 | 184 | |
| Network information | Num of Units | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | 22,215 | - | - | - |
| Rescaling Method for Covariates | Standarized | - | - | - | ||||||||||
| Hidden layer | Num Hidden Layers | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 |
| Num Units in Hidden Layer | 2 | 2 | 3 | 7 | 6 | 2 | 10 | 2 | 3 | 2 | 4.1 | 2.9 | 3 | |
| Activation Function | Softmax | - | - | - | ||||||||||
| Output layer | Dep Variable | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 9 | 1 | - | - | - |
| Num of Units | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 18 | 2 | 3.8 | 5.3 | 2 | |
| Activation Function | Identity | - | - | - | ||||||||||
| Error Function | Sum of Squares | - | - | - | ||||||||||
| Model summary training | Sum of Squares Error | 31.7 | 26.7 | 12.1 | 25.5 | 27.6 | 27.2 | 20.1 | 9.7 | 72.2 | 14.7 | 28.7 | 17.5 | 26.7 |
| Percent of Incorrect Predictions | 47.2 | 32.5 | 11.2 | 36.2 | 33.1 | 47.7 | 21.6 | 9.2 | 26.5 | 36.4 | 32.5 | 11.6 | 33.1 | |
| Stopping Rule Used | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Time in Seconds | 487.5 | 481.9 | 451.5 | 436.2 | 477.2 | 336.7 | 587.6 | 406.0 | 84.4 | 94.4 | 381.9 | 178.0 | 451.5 | |
| Model sum. testing | Sum of Squares Error | 13.3 | 12.8 | 8.5 | 6.9 | 8.8 | 12.8 | 6.6 | 5.2 | 29.8 | 4.6 | 11.6 | 7.5 | 8.8 |
| Percent Incorrect Predictions | 56.6 | 35.6 | 16.9 | 11.4 | 17.0 | 56.9 | 18.0 | 13.3 | 35.6 | 36.8 | 31.6 | 17.1 | 35.6 | |
| Classification | Training Overall Percent | 52.8 | 67.5 | 88.8 | 63.8 | 66.9 | 52.3 | 78.4 | 90.8 | 73.5 | 63.6 | 67.5 | 11.6 | 66.9 |
| Testing Overall Percent | 43.4 | 64.4 | 83.1 | 88.6 | 83.0 | 43.1 | 82.0 | 86.7 | 64.4 | 63.2 | 68.4 | 17.1 | 64.4 | |
| Area under the curve | Alive | 0.5 | 0.6 | 0.6 | 0.6 | 0.6 | 0.5 | 0.8 | 0.6 | 0.7 | 0.7 | 0.6 | 0.1 | 0.6 |
| Dead | 0.5 | 0.6 | 0.6 | 0.6 | 0.6 | 0.5 | 0.8 | 0.6 | 0.7 | 0.7 | 0.6 | 0.1 | 0.6 | |
| Gene | B | p Value | HR | HR Lower | HR Higher | Cytoband |
|---|---|---|---|---|---|---|
| FRYL | 1.84 | 0.00963 | 6.27 | 1.56 | 25.16 | 4p11 |
| KIAA0100 | 1.82 | 0.00005 | 6.16 | 2.55 | 14.90 | 17q11.2 |
| CDC40 | 1.64 | 0.00004 | 5.13 | 2.34 | 11.24 | 6q21 |
| MED8 | 1.57 | 1.6 × 10−8 | 4.79 | 2.78 | 8.25 | 1p34.2 |
| PTP4A2 | 1.51 | 0.05252 | 4.55 | 0.98 | 21.00 | 1p35 |
| BNIP2 | 1.48 | 0.04624 | 4.38 | 1.02 | 18.73 | 15q22.2 |
| TMEM70 | 1.46 | 0.00308 | 4.30 | 1.64 | 11.31 | 8q21.11 |
| MED6 | 1.41 | 0.00627 | 4.08 | 1.49 | 11.20 | 14q24.2 |
| SLC24A2 | 1.39 | 0.00005 | 4.03 | 2.06 | 7.89 | 9p22.1 |
| KLK10 | 1.34 | 0.00265 | 3.81 | 1.59 | 9.13 | 19q13 |
| RANBP9 | 1.29 | 0.01825 | 3.63 | 1.24 | 10.60 | 6p23 |
| PRB1 | 1.08 | 0.00005 | 2.94 | 1.74 | 4.95 | 12p13.2 |
| EVA1B | 1.00 | 0.00041 | 2.71 | 1.56 | 4.72 | 1p34.3 |
| CBFA2T2 | 0.99 | 0.01269 | 2.69 | 1.24 | 5.86 | 20q11 |
| ALDH1L1 | 0.74 | 0.08266 | 2.09 | 0.91 | 4.80 | 3q21.3 |
| KRT19 | 0.71 | 0.00002 | 2.04 | 1.47 | 2.81 | 17q21.2 |
| BTN2A3P | 0.71 | 0.00320 | 2.03 | 1.27 | 3.25 | 6p22.1 |
| TRPM4 | 0.56 | 0.00449 | 1.76 | 1.19 | 2.60 | 19q13.33 |
| Gene | B | p Value | HR | HR Lower | HR Higher | Cytoband |
|---|---|---|---|---|---|---|
| HSF2 | −0.48 | 0.04873 | 0.62 | 0.38 | 1.00 | 6q22.31 |
| ATPAF2 | −0.48 | 0.04152 | 0.62 | 0.39 | 0.98 | 17p11.2 |
| SLC7A11 | −0.51 | 0.00208 | 0.60 | 0.43 | 0.83 | 4q28.3 |
| PTAFR | −0.64 | 0.00060 | 0.53 | 0.37 | 0.76 | 1p35-p34.3 |
| TTLL3 | −0.74 | 0.01720 | 0.48 | 0.26 | 0.88 | 3p25.3 |
| TCP10L | −0.75 | 0.03536 | 0.47 | 0.24 | 0.95 | 21q22.11 |
| DNAAF1 | −0.8 | 0.00807 | 0.45 | 0.25 | 0.81 | 16q24.1 |
| PRH1 | −0.85 | 1 × 10−5 | 0.43 | 0.29 | 0.62 | 12p13.2 |
| NSDHL | −0.89 | 0.04102 | 0.41 | 0.17 | 0.96 | Xq28 |
| TAF12 | −0.99 | 0.01139 | 0.37 | 0.17 | 0.80 | 1p35.3 |
| TSPAN3 | −1 | 0.00040 | 0.37 | 0.21 | 0.64 | 15q24.3 |
| AKIRIN1 | −1.03 | 0.00195 | 0.36 | 0.19 | 0.69 | 1p34.3 |
| ITK | −1.04 | 0.00102 | 0.35 | 0.19 | 0.66 | 5q31-q32 |
| TDRD12 | −1.09 | 0.00392 | 0.34 | 0.16 | 0.70 | 19q13.11 |
| LPP | −1.12 | 0.00097 | 0.33 | 0.17 | 0.63 | 3q28 |
| BTD | −1.13 | 9.5 × 10−6 | 0.32 | 0.20 | 0.53 | 3p25 |
| SIRT5 | −1.22 | 0.04956 | 0.30 | 0.09 | 1.00 | 6p23 |
| ZNF230 | −1.29 | 0.00002 | 0.27 | 0.15 | 0.50 | 19q13.31 |
| ABHD6 | −1.38 | 7.2 × 10−5 | 0.25 | 0.13 | 0.50 | 3p14.3 |
| TOP2B | −1.49 | 0.01673 | 0.23 | 0.07 | 0.76 | 3p24 |
| ARPC2 | −1.7 | 0.00804 | 0.18 | 0.05 | 0.64 | 2q36.1 |
| ASAP2 | −1.96 | 0.00003 | 0.14 | 0.06 | 0.36 | 2p25|2p24 |
| IDH3A | −2.03 | 0.00009 | 0.13 | 0.05 | 0.36 | 15q25.1-q25.2 |
| PSMF1 | −2.44 | 0.00415 | 0.09 | 0.02 | 0.46 | 20p13 |
| ARFGEF1 | −2.69 | 0.00000 | 0.07 | 0.02 | 0.20 | 8q13 |
| Crosstabulations | OS Prognosis | Outcome Dead/Alive | Age | EN | LDH | Stage | IPI | IR |
|---|---|---|---|---|---|---|---|---|
| EVA1B | Bad | 0.7840 | 0.7045 | 0.1580 | 0.1046 | 0.0668 | 0.1657 | |
| KRT19 | Bad | 0.0148 | 0.0384 | 0.1236 | 0.1815 | 0.4020 | 0.9878 | |
| BTN2A3P | Bad | 0.0820 | 0.7045 | 0.5451 | 0.5724 | 0.0440 | 0.0751 | |
| KLK10 | Bad | 0.4673 | 0.0481 | 0.0184 | 0.6009 | 0.8798 | 0.0258 | 0.6764 |
| TRPM4 | Bad | 0.1646 | 0.3882 | 0.1212 | 0.1698 | 0.1710 | 0.9865 | 0.0974 |
| TDRD12 | Good | 0.5468 | 0.8321 | 0.1934 | 0.1616 | 0.4593 | 0.8869 | |
| ZNF230 | Good | 0.0698 | 0.0310 | 0.9695 | 0.6875 | 0.2887 | 0.5414 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Nakamura, N.; Hamoudi, R. Artificial Intelligence Analysis of the Gene Expression of Follicular Lymphoma Predicted the Overall Survival and Correlated with the Immune Microenvironment Response Signatures. Mach. Learn. Knowl. Extr. 2020, 2, 647-671. https://0-doi-org.brum.beds.ac.uk/10.3390/make2040035
Carreras J, Kikuti YY, Miyaoka M, Hiraiwa S, Tomita S, Ikoma H, Kondo Y, Ito A, Nakamura N, Hamoudi R. Artificial Intelligence Analysis of the Gene Expression of Follicular Lymphoma Predicted the Overall Survival and Correlated with the Immune Microenvironment Response Signatures. Machine Learning and Knowledge Extraction. 2020; 2(4):647-671. https://0-doi-org.brum.beds.ac.uk/10.3390/make2040035
Chicago/Turabian StyleCarreras, Joaquim, Yara Yukie Kikuti, Masashi Miyaoka, Shinichiro Hiraiwa, Sakura Tomita, Haruka Ikoma, Yusuke Kondo, Atsushi Ito, Naoya Nakamura, and Rifat Hamoudi. 2020. "Artificial Intelligence Analysis of the Gene Expression of Follicular Lymphoma Predicted the Overall Survival and Correlated with the Immune Microenvironment Response Signatures" Machine Learning and Knowledge Extraction 2, no. 4: 647-671. https://0-doi-org.brum.beds.ac.uk/10.3390/make2040035