Markov Chain-Based Sampling for Exploring RNA Secondary Structure under the Nearest Neighbor Thermodynamic Model and Extended Applications

Abstract
:1. Introduction
2. Methods
2.1. Derivation of Energy Functions
2.2. Mathematical Preliminaries
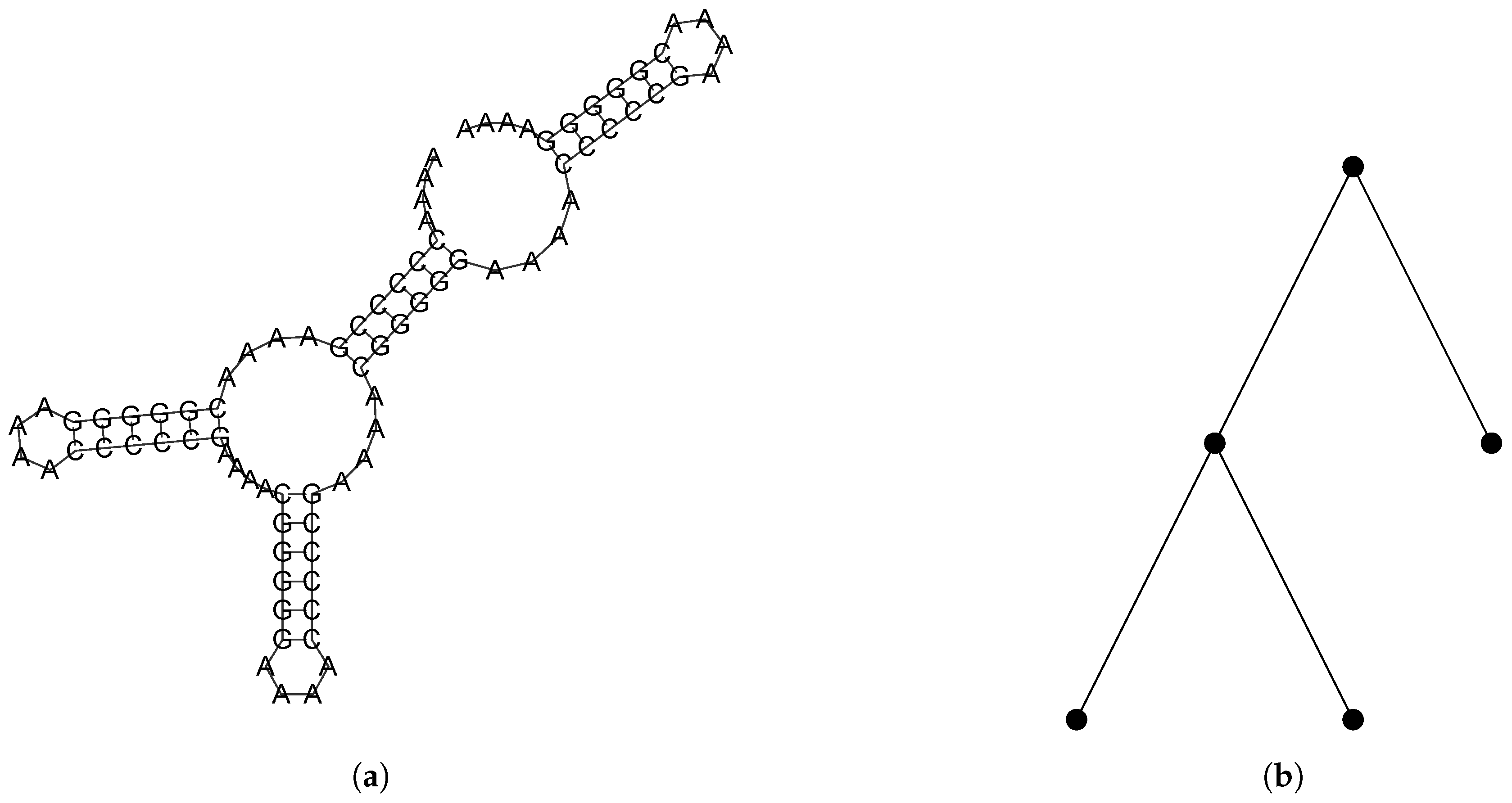
2.2.1. Combinatorial Objects
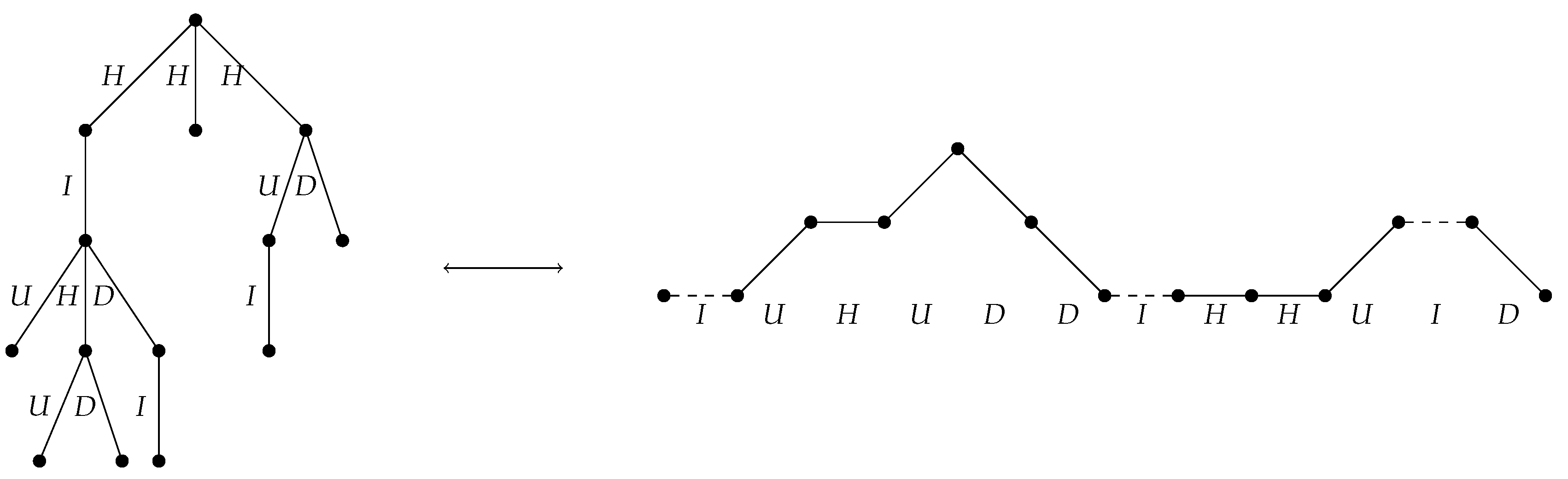
2.2.2. A Bijection Between and
- If e is the leftmost edge off a non-root node of down degree at least 2, assign the label U.
- If e is the rightmost edge off a non-root node of down degree at least 2, assign the label D.
- If e is the only edge off a non-root node of degree 1, assign the label I.
- If e is an edge off the root node, or if e is neither the leftmost nor the rightmost edge off its parent node, assign the label H.
2.2.3. Markov Chains
- Irreducibility: For any , there is some integer for which .
- Aperiodicity: For any state , we have .
2.2.4. Coupling
- Each chain and , when viewed in isolation, is a copy of , given initial states and .
- Whenever , we have .
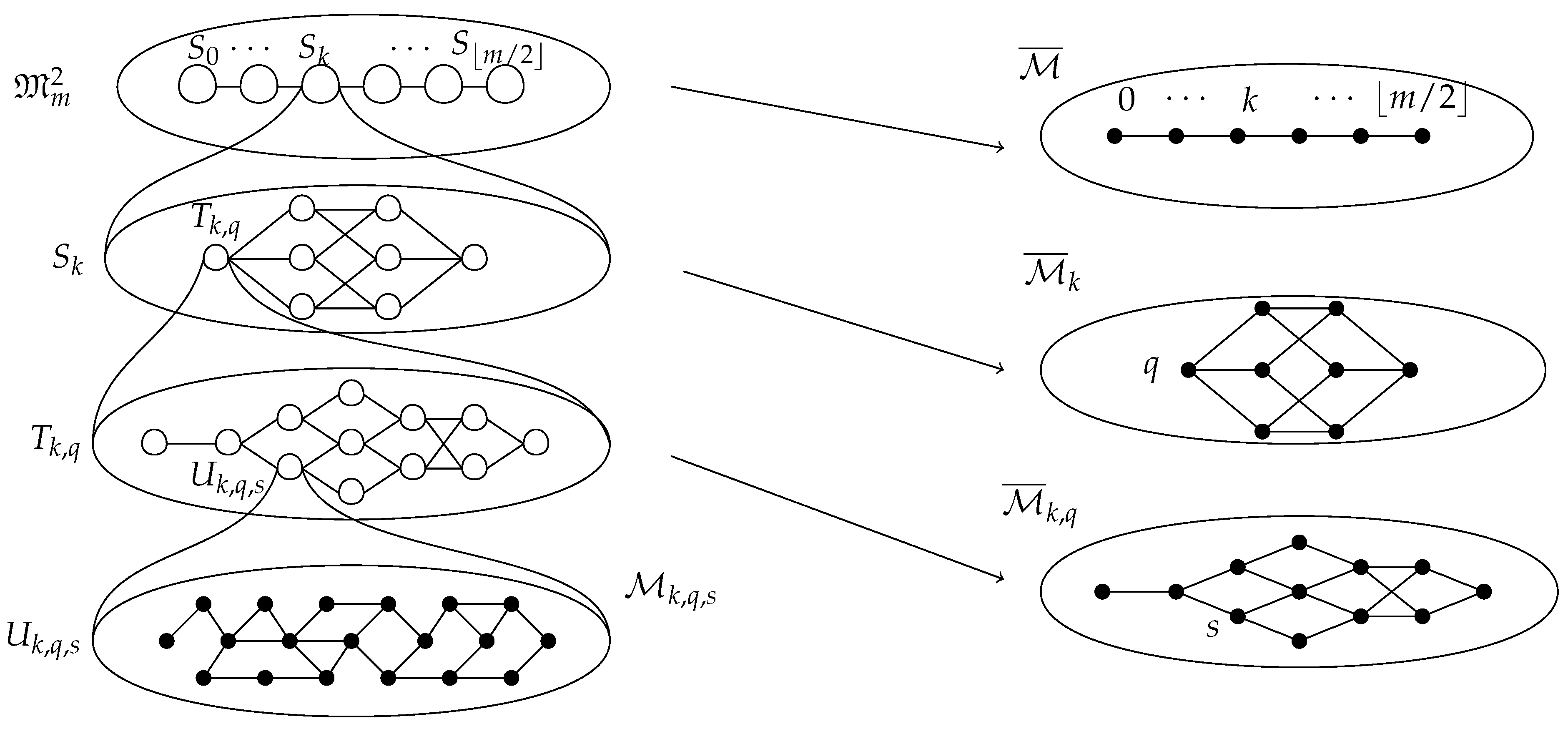
2.2.5. Decomposition
- 1.
- for all such that .
- 2.
- for all with .
- If heads, set . Let l be either 1 or , each with probability . If possible, let with probability . Otherwise, let .
- If tails, set , and update the same way as we did for in the previous case.
3. Results
3.1. Our Markov Chain on
- If , pick a random pair of consecutive symbols in , and call this pair s. If s is or , let be either or with probabilities and respectively. Let y be the string with s replaced by . Otherwise, let .
- If , pick i uniformly from . If is H or I, choose a symbol c to be either H or I with probabilities and respectively. Let y be the 2-Motzkin path given by changing the symbol in to c. Otherwise, we let .
- If , pick i and j each uniformly from . If each of and are either U or D, let y be the string with the symbols at indices i and j swapped. Otherwise, let .
- If , pick a random pair of consecutive symbols in , and call this pair s. If s is of the form or for some and , let be the reverse of s, and let y be the string with s replaced by . Otherwise, let .
| Algorithm 1: The main Markov chain algorithm. This pseudocode calculates given . |
| Require: is a valid 2-Motzkin path of length m. |
| for do |
| if then |
| if and then |
| else if and then |
| else if then |
| if and then |
| else if and then |
| else if then |
| if ( and ) and then |
| if y is not a valid 2-Motzkin path then |
| else if then |
| if ( and ) or ( and ) and |
| then |
| return x |
| Algorithm 2: Algorithm to convert a sampled 2-Motzkin path to a plan tree. The pseudocode calculates . |
| Require: x is a valid 2-Motzkin path of length m. |
| root ← new Node() |
| // u will be where a new node will be added for an H or D symbol |
| root |
| // v will be always the last node added |
| new Node() |
| // the stack will keep track of previous values of u |
| stack = new Stack() |
| root.children.append(v) |
| for do |
| node ← new Node() |
| if then |
| v.children.append(node) |
| stack.push(u) |
| else if then |
| v.children.append(node) |
| else if then |
| u.children.append(node) |
| else if then |
| u.children.append(node) |
| stack.pop() |
| node |
| return root |
3.2. Mixing Time Results
- With probability , set .
- Otherwise, pick a random index . Let be a random symbol such that and . Now let and be and respectively, each with the jth symbol changed to a.
4. Discussion and Conclusions
4.1. Applications to RNA Modeling
4.2. Possibility of a Dynamic Programming Approach
4.3. Possibility of an SCFG Approach
4.4. Extended Applications
4.5. Independent Mathematical Research Interests
4.6. Limitations and Future Directions
- Can the mixing time bound in our main result be improved?
- Is there a rapidly mixing chain, with the same stationary distribution studied here, whose transitions correspond naturally to moves on the set plane trees? Mixing time bounds on the chain of matching exchange moves, as defined in [63], would be especially interesting, as such a chain may relate to RNA folding kinetics.
- Is there a rapidly mixing chain converging to the Gibbs distribution using the full energy function for the utilized NNTM model [16]? The chain presented here uses only the parameters and , setting .
- Is there a stochastic context-free grammar which generates secondary structures (in our simplified model or using the full NNTM) according to a Gibbs distribution with NNTM energy?
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RNA | ribonucleic acid |
| NNTM | Nearest Neighbor Thermodynamic Model |
| SCFG | stochastic context free grammar |
References
- Doudna, J.A. Structural genomics of RNA. Nat. Struct. Biol. 2000, 7, 954–956. [Google Scholar]
- Tinoco, I., Jr.; Bustamante, C. How RNA folds. J. Mol. Biol. 1999, 293, 271–281. [Google Scholar]
- Massire, C.; Westhof, E. MANIP: An interactive tool for modelling RNA. J. Mol. Gr. Model. 1998, 16, 197–205. [Google Scholar] [CrossRef]
- Seetin, M.G.; Mathews, D.H. Automated RNA tertiary structure prediction from secondary structure and low-resolution restraints. J. Comput. Chem. 2011, 32, 2232–2244. [Google Scholar] [CrossRef]
- Zhao, Y.; Gong, Z.; Xiao, Y. Improvements of the Hierarchical Approach for Predicting RNA Tertiary Structure. J. Biomol. Struct. Dyn. 2011, 28, 815–826. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, Y.; Gong, Z.; Wang, Y.; Man, J.; Xiao, Y. Automated and fast building of three-dimensional RNA structures. Sci. Rep. 2012, 2, 734. [Google Scholar]
- Borodavka, A.; Singaram, S.W.; Stockley, P.G.; Gelbart, W.M.; Ben-Shaul, A.; Tuma, R. Sizes of long RNA molecules are determined by the branching patterns of their secondary structures. Biophys. J. 2016, 111, 2077–2085. [Google Scholar]
- Jaeger, J.A.; Turner, D.H.; Zuker, M. Improved predictions of secondary structures for RNA. Proc. Natl. Acad. Sci. USA 1989, 86, 7706–7710. [Google Scholar]
- Mathews, D.H.; Sabina, J.; Zuker, M.; Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004, 32, W135–W141. [Google Scholar] [CrossRef]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatshefte für Chemie/Chemical Monthly 1994, 125, 167–188. [Google Scholar]
- Mathuriya, A.; Bader, D.A.; Heitsch, C.E.; Harvey, S.C. GTfold: A scalable multicore code for RNA secondary structure prediction. In Proceedings of the 2009 ACM symposium on Applied Computing, Honolulu, HI, USA, 8–12 March 2009; pp. 981–988. [Google Scholar]
- Turner, D.H.; Mathews, D.H. NNDB: The nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2009, 38, D280–D282. [Google Scholar] [CrossRef]
- Doshi, K.J.; Cannone, J.J.; Cobaugh, C.W.; Gutell, R.R. Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinf. 2004, 5, 105. [Google Scholar]
- Hower, V.; Heitsch, C.E. Parametric analysis of RNA branching configurations. Bull. Math. Biol. 2011, 73, 754–776. [Google Scholar]
- Bakhtin, Y.; Heitsch, C.E. Large deviations for random trees and the branching of RNA secondary structures. Bull. Math. Biol. 2009, 71, 84–106. [Google Scholar]
- Heitsch, C.; Poznanović, S. Combinatorial insights into RNA secondary structure. In Discrete and Topological Models in Molecular Biology; Springer: Berlin, Germany, 2014; pp. 145–166. [Google Scholar]
- Lorenz, R.; Bernhart, S.H.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar]
- Donaghey, R.; Shapiro, L.W. Motzkin numbers. J. Comb. Theory Ser. A 1977, 23, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Bernhart, F.R. Catalan, Motzkin, and Riordan numbers. Discret. Math. 1999, 204, 73–112. [Google Scholar] [CrossRef]
- Eu, S.P.; Fu, T.S.; Hou, J.T.; Hsu, T.W. Standard Young tableaux and colored Motzkin paths. J. Combin. Theory Ser. A 2013, 120, 1786–1803. [Google Scholar] [CrossRef]
- Baril, J.L.; Kirgizov, S.; Petrossian, A. Motzkin paths with a restricted first return decomposition. Integers 2019, 19, A46. [Google Scholar]
- Fang, W. A partial order on Motzkin paths. Discrete Math. 2020, 343, 111802, 9. [Google Scholar] [CrossRef] [Green Version]
- Stanley, R.P. Enumerative Combinatorics: Volume 1, 2nd ed.; Cambridge University Press: New York, NY, USA, 2011. [Google Scholar]
- Deutsch, E.; Shapiro, L.W. A bijection between ordered trees and 2-Motzkin paths and its many consequences. Discret. Math. 2002, 256, 655–670. [Google Scholar]
- Madras, N.; Randall, D. Markov chain decomposition for convergence rate analysis. Ann. Appl. Probab. 2002, 12, 581–606. [Google Scholar] [CrossRef]
- Randall, D. Rapidly Mixing Markov Chains with Applications in Computer Science and Physics. Comput. Sci. Eng. 2006, 8, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Luby, M.; Randall, D.; Sinclair, A. Markov Chain Algorithms for Planar Lattice Structures. SIAM J. Comput. 2001, 31, 167–192. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.; Randall, D. Sampling adsorbing staircase walks using a new Markov chain decomposition method. In Proceedings of the 41st Annual Symposium on Foundations of Computer Science, Redondo Beach, CA, USA, 12–14 November 2000; pp. 492–502. [Google Scholar]
- Hermon, J.; Salez, J. Modified log-Sobolev inequalities for strong-Rayleigh measures. arXiv 2019, arXiv:1902.02775. [Google Scholar]
- Jerrum, M.; Son, J.B.; Tetali, P.; Vigoda, E. Elementary bounds on Poincaré and log-Sobolev constants for decomposable Markov chains. Ann. Appl. Probab. 2004, 14, 1741–1765. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E. Problems in catalan mixing and matchings in regular hypergraphs. Ph.D. Thesis, Georgia Institute of Technology, Atlanta, GA, USA, 2016. [Google Scholar]
- Wilson, D.B. Mixing times of lozenge tiling and card shuffling Markov chains. Ann. Appl. Probab. 2004, 14, 274–325. [Google Scholar] [CrossRef]
- Yoffe, A.M.; Prinsen, P.; Gopal, A.; Knobler, C.M.; Gelbart, W.M.; Ben-Shaul, A. Predicting the sizes of large RNA molecules. Proc. Natl. Acad. Sci. USA 2008, 105, 16153–16158. [Google Scholar]
- Andronescu, M.; Bereg, V.; Hoos, H.H.; Condon, A. RNA STRAND: The RNA secondary structure and statistical analysis database. BMC Bioinf. 2008, 9, 340. [Google Scholar]
- Alonso, L. Uniform generation of a Motzkin word. Theor. Comput. Sci. 1994, 134, 529–536. [Google Scholar]
- Nebel, M.E.; Scheid, A. Evaluation of a sophisticated SCFG design for RNA secondary structure prediction. Theory Biosci. 2011, 130, 313–336. [Google Scholar]
- Rivas, E.; Eddy, S.R. Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics 2000, 16, 583–605. [Google Scholar]
- Rivas, E.; Eddy, S.R. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. Mol. Biol. 1999, 285, 2053–2068. [Google Scholar]
- Knudsen, B.; Hein, J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 2003, 31, 3423–3428. [Google Scholar]
- Andrieu, C.; De Freitas, N.; Doucet, A.; Jordan, M.I. An introduction to MCMC for machine learning. Mach. Learn. 2003, 50, 5–43. [Google Scholar]
- Chib, S. Introduction to simulation and MCMC methods. In The Oxford Handbook of Bayesian Econometrics; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Jackman, S. Estimation and inference via Bayesian simulation: An introduction to Markov chain Monte Carlo. Am. J. Political Sci. 2000, 1, 375–404. [Google Scholar]
- Huelsenbeck, J.P.; Larget, B.; Alfaro, M.E. Bayesian Phylogenetic Model Selection Using Reversible Jump Markov Chain Monte Carlo. Mol. Biol. Evolut. 2004, 21, 1123–1133. [Google Scholar] [CrossRef]
- Kim, S.; Li, H.; Dougherty, E.R.; Cao, N.; Chen, Y.; Bittner, M.; Suh, E.B. Can Markov chain models mimic biological regulation? J. Biol. Syst. 2002, 10, 337–357. [Google Scholar]
- Lusseau, D. Effects of tour boats on the behavior of bottlenose dolphins: Using Markov chains to model anthropogenic impacts. Conserv. Biol. 2003, 17, 1785–1793. [Google Scholar]
- Said, M.R.; Oppenheim, A.V.; Lauffenburger, D.A. Modeling cellular signal processing using interacting Markov chains. In Proceedings of the 2003 IEEE International Conference on Acoustics, Speech, and Signal Processing, Hong Kong, China, 6–10 April 2003. [Google Scholar]
- Gelman, A.; Rubin, D.B. Markov chain Monte Carlo methods in biostatistics. Stat. Methods Med. Res. 1996, 5, 339–355. [Google Scholar]
- Hamra, G.; MacLehose, R.; Richardson, D. Markov Chain Monte Carlo: An Introduction for Epidemiologists. Int. J. Epidemiol. 2013, 42, 627–634. [Google Scholar]
- Nascimento, F.F.; dos Reis, M.; Yang, Z. A biologist’s guide to Bayesian phylogenetic analysis. Nat. Ecol. Evolut. 2017, 1, 1446–1454. [Google Scholar]
- Van Ravenzwaaij, D.; Cassey, P.; Brown, S.D. A simple introduction to Markov Chain Monte–Carlo sampling. Psychon. Bull. Rev. 2018, 25, 143–154. [Google Scholar]
- Levin, D.A.; Peres, Y.; Wilmer, E.L. Markov Chains and Mixing Times; American Mathematical Society: Providence, RI, USA, 2009. [Google Scholar]
- Montenegro, R.R.; Tetali, P. Mathematical Aspects of Mixing Times in Markov Chains; Now Publishers Inc.: Boston, MA, USA, 2006. [Google Scholar]
- Jerrum, M. Counting, Sampling and Integrating: Algorithms and Complexity; Springer Science & Business Media: New York, NY, USA, 2003. [Google Scholar]
- Aldous, D.J. Mixing time for a Markov chain on cladograms. Comb. Probab. Comput. 2000, 9, 191–204. [Google Scholar]
- Schweinsberg, J. An O (n2) bound for the relaxation time of a Markov chain on cladograms. Random Struct. Algorithms 2002, 20, 59–70. [Google Scholar]
- Dershowitz, N.; Zaks, S. Ordered trees and non-crossing partitions. Discret. Math. 1986, 62, 215–218. [Google Scholar]
- Cohen, E.; Tetali, P.; Yeliussizov, D. Lattice path matroids: Negative correlation and fast mixing. arXiv 2015, arXiv:1505.06710. [Google Scholar]
- McShine, L.; Tetali, P. On the mixing time of the triangulation walk and other Catalan structures. In Proceedings of the Randomization Methods in Algorithm Design, Princeton, NJ, USA, 12–14 December 1997; pp. 147–160. [Google Scholar]
- Sohoni, M. Rapid mixing of some linear matroids and other combinatorial objects. Graphs Combin. 1999, 15, 93–107. [Google Scholar] [CrossRef]
- Robert, C.P.; Casella, G.; Casella, G. Introducing Monte Carlo Methods with R; Springer: Berlin, Germany, 2010; Volume 18. [Google Scholar]
- Heitsch, C.E.; Tetali, P. Meander graphs. In Proceedings of the 23rd International Conference on Formal Power Series and Algebraic Combinatorics, Reykjavik, Iceland, 13–17 June 2011; pp. 469–480. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Y | Z | Turner | a | b | c | h | f | i | g | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | G | 89 | 4.6 | 0.4 | 0.1 | 3.8 | 3.0 | |||||
| G | C | 89 | 4.6 | 0.4 | 0.1 | 3.5 | 3.0 | |||||
| C | G | 99 | 3.4 | 0 | 0.4 | 4.5 | 2.3 | 2.3 | 1.3 | |||
| G | C | 99 | 3.4 | 0 | 0.4 | 4.1 | 2.3 | 2.2 | 1.9 | |||
| C | G | 04 | 9.3 | 0 | 4.5 | 2.3 | 0.9 | |||||
| G | C | 04 | 9.3 | 0 | 4.1 | 2.3 | 0.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirkpatrick, A.; Patton, K.; Tetali, P.; Mitchell, C. Markov Chain-Based Sampling for Exploring RNA Secondary Structure under the Nearest Neighbor Thermodynamic Model and Extended Applications. Math. Comput. Appl. 2020, 25, 67. https://0-doi-org.brum.beds.ac.uk/10.3390/mca25040067
Kirkpatrick A, Patton K, Tetali P, Mitchell C. Markov Chain-Based Sampling for Exploring RNA Secondary Structure under the Nearest Neighbor Thermodynamic Model and Extended Applications. Mathematical and Computational Applications. 2020; 25(4):67. https://0-doi-org.brum.beds.ac.uk/10.3390/mca25040067
Chicago/Turabian StyleKirkpatrick, Anna, Kalen Patton, Prasad Tetali, and Cassie Mitchell. 2020. "Markov Chain-Based Sampling for Exploring RNA Secondary Structure under the Nearest Neighbor Thermodynamic Model and Extended Applications" Mathematical and Computational Applications 25, no. 4: 67. https://0-doi-org.brum.beds.ac.uk/10.3390/mca25040067