Novel Point Mutations in the NKX2.5 Gene in Pediatric Patients with Non-Familial Congenital Heart Disease

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Mutation Screening
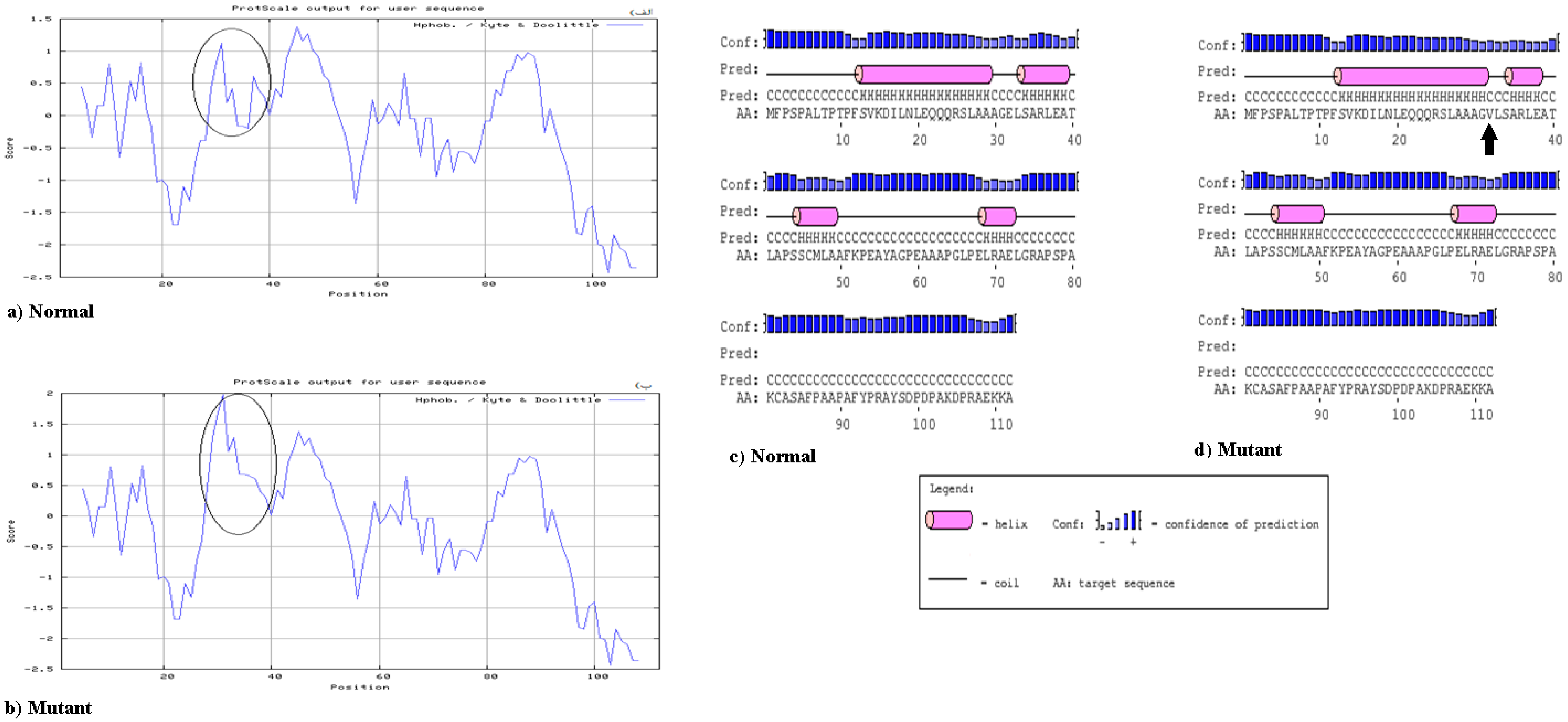
2.3. Bioinformatics Analysis
2.4. Statistical Analysis
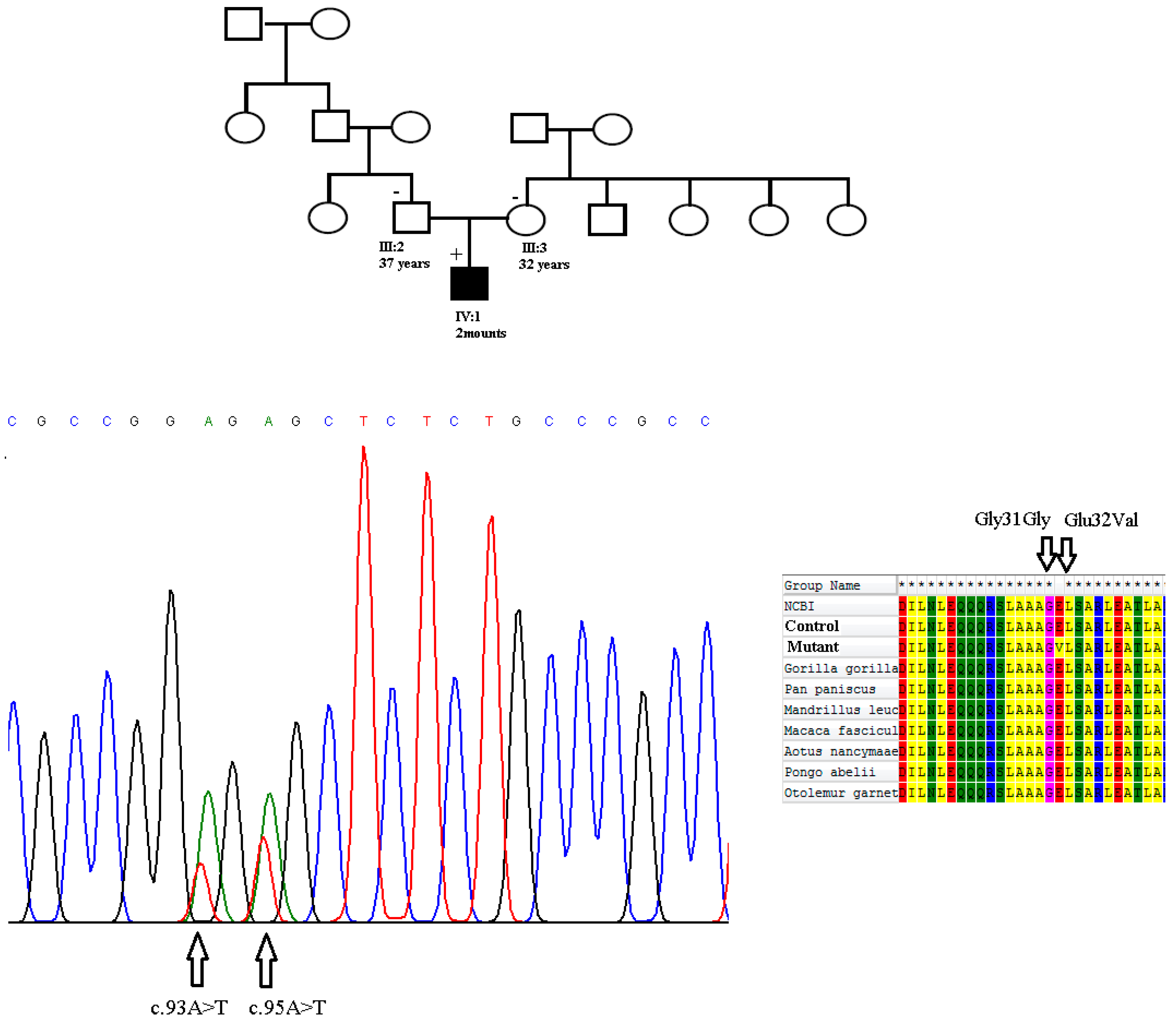
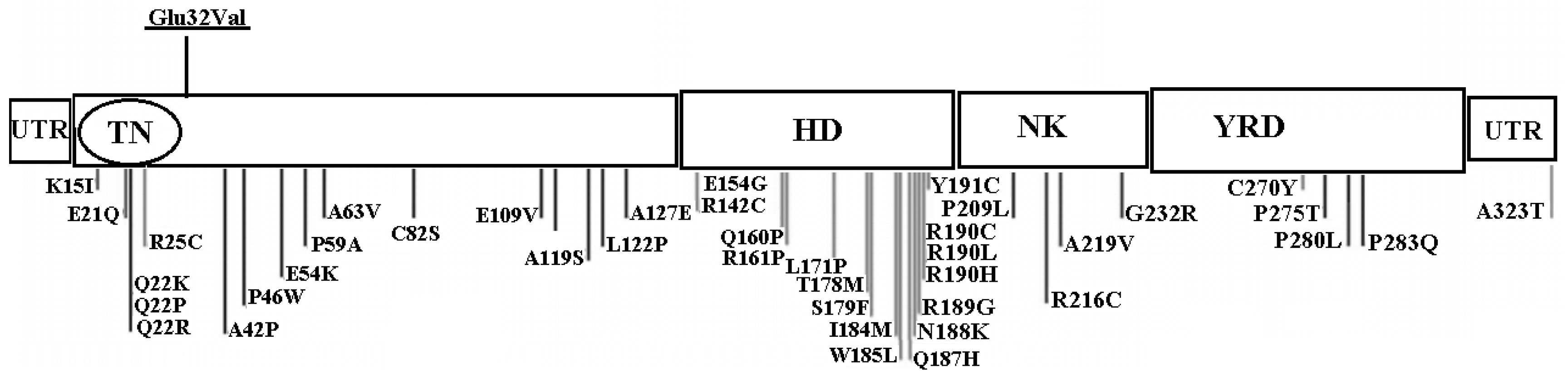
3. Results
4. Discussion
Limitations of the Study
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Ethical Standards
References
- Huang, J.B.; Liu, Y.L.; Sun, P.W.; Lv, X.D.; Du, M.; Fan, X.M. Molecular mechanisms of congenital heart disease. Cardiovasc. Pathol. 2010, 19, e183–e193. [Google Scholar] [PubMed] [Green Version]
- Garg, V. Insights into the genetic basis of congenital heart disease. Cell. Mol. Life Sci. 2006, 63, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Muntean, I.; Toganel, R.; Benedek, T. Genetics of Congenital Heart Disease: Past and Present. Biochem. Genet. 2017, 55, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.E.; Sander, T.L.; Klinkner, D.B.; Tomita-Mitchell, A. The molecular basis of congenital heart disease. Semin. Thorac. Cardiovasc. Surg. 2007, 19, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.M.; Rajakumar, G. Genetics of Congenital Heart Defects: The NKX2-5 Gene, a Key Player. Genes 2016, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Gioli-Pereira, L.; Pereira, A.C.; Mesquita, S.M.; Xavier-Neto, J.; Lopes, A.A.; Krieger, J.E. NKX2.5 mutations in patients with non-syndromic congenital heart disease. Int. J. Cardiol. 2010, 138, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Khatami, M.; Heidari, M.M.; Tabesh, F.; Ordooei, M.; Salehifar, Z. Mutation analysis of the NKX2.5 gene in Iranian pediatric patients with congenital hypothyroidism. J. Pediatr. Endocrinol. Metab. 2017, 30, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Schott, J.J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Ketharnathan, S.; Koshy, T.; Sethuratnam, R.; Paul, S.; Venkatesan, V. Investigation of NKX2.5 gene mutations in congenital heart defects in an Indian population. Genet. Test Mol. Biomark. 2015, 19, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Maury, P.; Gandjbakhch, E.; Baruteau, A.E.; Bessière, F.; Kyndt, F.; Bouvagnet, P.; Rollin, A.; Bonnet, D.; Probst, V.; Maltret, A. Cardiac Phenotype and Long-Term Follow-Up of Patients With Mutations in NKX2-5 Gene. J. Am. Coll. Cardiol. 2016, 68, 2389–2390. [Google Scholar] [CrossRef] [PubMed]
- Prall, O.W.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.J.; Olson, E.N. Building the heart piece by piece: Modularity of cis-elements regulating Nkx2-5 transcription. Development 1999, 126, 4187–4192. [Google Scholar] [PubMed]
- Shiojima, I.; Komuro, I.; Inazawa, J.; Nakahori, Y.; Matsushita, I.; Abe, T.; Nagai, R.; Yazaki, Y. Assignment of cardiac homeobox gene CSX to human chromosome 5q34. Genomics 1995, 27, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Stallmeyer, B.; Fenge, H.; Nowak-Gottl, U.; Schulze-Bahr, E. Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clin. Genet. 2010, 78, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Sathananthan, G.; Harris, L.; Nair, K. Ventricular Arrhythmias in Adult Congenital Heart Disease: Mechanisms, Diagnosis, and Clinical Aspects. Card. Electrophysiol. Clin. 2017, 9, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Reamon-Buettner, S.M.; Hecker, H.; Spanel-Borowski, K.; Craatz, S.; Kuenzel, E.; Borlak, J. Novel NKX2-5 mutations in diseased heart tissues of patients with cardiac malformations. Am. J. Pathol. 2004, 164, 2117–2125. [Google Scholar] [CrossRef]
- Draus, J.M.; Hauck, M.A., Jr.; Goetsch, M.; Austin, E.H., 3rd; Tomita-Mitchell, A.; Mitchell, M.E. Investigation of somatic NKX2-5 mutations in congenital heart disease. J. Med. Genet. 2009, 46, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Hirayama-Yamada, K.; Kamisago, M.; Akimoto, K.; Aotsuka, H.; Nakamura, Y.; Tomita, H.; Furutani, M.; Imamura, S.I.; Takao, A.; Nakazawa, M.; et al. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am. J. Med. Genet. Part A 2005, 135, 47–52. [Google Scholar] [CrossRef] [PubMed]
- McElhinney, D.B.; Geiger, E.; Blinder, J.; Benson, D.W.; Goldmuntz, E. NKX2.5 mutations in patients with congenital heart disease. J. Am. Coll. Cardiol. 2003, 42, 1650–1655. [Google Scholar] [CrossRef] [PubMed]
- Granados-Riveron, J.T.; Pope, M.; Bu’Lock, F.A.; Thornborough, C.; Eason, J.; Setchfield, K.; Ketley, A.; Kirk, E.P.; Fatkin, D.; Feneley, M.P.; et al. Combined mutation screening of NKX2-5, GATA4, and TBX5 in congenital heart disease: Multiple heterozygosity and novel mutations. Congenit. Heart Dis. 2012, 7, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Meng, H.; Qiao, Y.; Pang, S.; Chen, D.; Yan, B. Two novel and functional DNA sequence variants within an upstream enhancer of the human NKX2-5 gene in ventricular septal defects. Gene 2013, 524, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Soheili, F.; Darabi, P.; Dahmardeh, F.; Heidary, N.; Jalili, Z.; Fooladi, S.; Hakhamanesh, M.S.; Heidarizadeh, M. Nkx2-5 Mutations in Patients with Non-Syndrome Congenital Heart Disease. Zahedan J. Res. Med. Sci. 2015, 15, 29–32. [Google Scholar]
- Kheirollahi, M.; Khosravi, F.; Ashouri, S.; Ahmadi, A. Existence of mutations in the homeodomain-encoding region of NKX2.5 gene in Iranian patients with tetralogy of Fallot. J. Res. Med. Sci. 2016, 21, 24. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Zhu, J.F.; Yang, J.G.; Zhu, Q.H.; Du, R.; Li, J.; Li, W. Gene mutation in secundum atrial septal defect: Analysis of a Chinese family with 3 patients. Zhonghua Yi Xue Za Zhi 2008, 88, 250–253. [Google Scholar] [PubMed]
- Kasahara, H.; Lee, B.; Schott, J.J.; Benson, D.W.; Seidman, J.G.; Seidman, C.E.; Izumo, S. Loss of function and inhibitory effects of human CSX/NKX2.5 homeoprotein mutations associated with congenital heart disease. J. Clin. Investig. 2000, 106, 299–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldmuntz, E.; Geiger, E.; Benson, D.W. NKX2.5 mutations in patients with tetralogy of fallot. Circulation 2001, 104, 2565–2568. [Google Scholar] [CrossRef] [PubMed]
- Reamon-Buettner, S.M.; Sattlegger, E.; Ciribilli, Y.; Inga, A.; Wessel, A.; Borlak, J. Transcriptional defect of an inherited NKX2-5 haplotype comprising a SNP, a nonsynonymous and a synonymous mutation, associated with human congenital heart disease. PLoS ONE 2013, 8, e83295. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.X.; Cartegni, L.; Zhang, M.Q.; Krainer, A.R. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat. Genet. 2001, 27, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Belvís, R.; Tizzano, E.F.; Martí-Fàbregas, J.; Leta, R.G.; Baena, M.; Carreras, F.; Pons-Lladó, G.; Baiget, M.; Martí-Vilalta, J.L. Mutations in the NKX2-5 gene in patients with stroke and patent foramen ovale. Clin. Neurol. Neurosurg. 2009, 111, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, J.; Wei, C.; Hou, Z.; Li, Y.; Zou, H.; Meng, M.; Wang, W.; Jiang, L. Genetic variations of NKX2-5 in sporadic atrial septal defect and ventricular septal defect in Chinese Yunnan population. Gene 2016, 575, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, X.; Shen, A.; Jiao, W.; Guan, X.; Li, Z. Screening NKX2.5 mutation in a sample of 230 Han Chinese children with congenital heart diseases. Genet. Test Mol. Biomark. 2009, 13, 159–162. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Exon | Primer Sequences | Product Size (bp) |
|---|---|---|
| Exon 1 | F: 5′-CCGCTTTCTGCCGCCCACC-3′ | 390 |
| R:5′-TCCTCCTCACCTTTCTTTTCG-3′ | ||
| Exon 2 (fragment 1) | F: 5′-CAAGCGTCTCTCTGCCTCTC-3′ | 242 |
| R: 5′-CTGCGTGGACGTGAGTTTC-3′ | ||
| Exon 2 (Fragment 2) | F: 5′-TGCTGAAACTCACGTCCAC-3′ | 230 |
| R: 5′-ATAACCGTAGGGATTGAGGC-3′ | ||
| Exon 2 (Fragment 3) | F: 5′-AACGCCTACCCCGCCTATC-3′ | 224 |
| R: 5′-CCAGGCTCGGATACCATGC-3′ |
| Clinical Characteristics | Number | Percentage or Range |
|---|---|---|
| Median Age (years) | 2.8 | 0–4.5 |
| Male:female | 38:67 | 36.19:63.8 |
| Distribution of different types of CHD | ||
| Isolated ASD | 25 | 23.8 |
| Isolated VSD | 68 | 64.76 |
| Isolated TOF | 12 | 11.42 |
| ASD + VSD | 6 | 5.71 |
| ASD + TOF | 1 | 0.95 |
| VSD + TOF | 2 | 1.90 |
| ASD + VSD + TOF | 1 | 0.95 |
| Surgical treatment | 65 | 61.9 |
| Genetic Findings | Region | Function | Amino-Acid Change | Novel/Reported |
|---|---|---|---|---|
| c.95A > T | Exon 1 | Missense | Glu32Val | Novel |
| c.93A > T | Exon 1 | Synonymous | Gly31Gly | Novel |
| c.2357G > A | Exon 2 | Synonymous | Glu181Glu | Reported |
| rs72554028 | ||||
| c.606G > C | Exon 2 | Synonymous | Leu202Leu | Reported |
| rs3729753 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khatami, M.; Mazidi, M.; Taher, S.; Heidari, M.M.; Hadadzadeh, M. Novel Point Mutations in the NKX2.5 Gene in Pediatric Patients with Non-Familial Congenital Heart Disease. Medicina 2018, 54, 46. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina54030046
Khatami M, Mazidi M, Taher S, Heidari MM, Hadadzadeh M. Novel Point Mutations in the NKX2.5 Gene in Pediatric Patients with Non-Familial Congenital Heart Disease. Medicina. 2018; 54(3):46. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina54030046
Chicago/Turabian StyleKhatami, Mehri, Mansoureh Mazidi, Shabnam Taher, Mohammad Mehdi Heidari, and Mehdi Hadadzadeh. 2018. "Novel Point Mutations in the NKX2.5 Gene in Pediatric Patients with Non-Familial Congenital Heart Disease" Medicina 54, no. 3: 46. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina54030046