QSAR Prediction Model to Search for Compounds with Selective Cytotoxicity Against Oral Cell Cancer



1
Department of Medical Molecular Informatics, Meiji Pharmaceutical University, 2-522-1 Noshio, Kiyose, Tokyo 204-8588, Japan
2
Meikai University Research Institute of Odontology (M-RIO), 1-1 Keyakidai, Sakado, Saitama 350-0283, Japan
*
Author to whom correspondence should be addressed.
Medicines 2019, 6(2), 45; https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6020045
Submission received: 28 February 2019
/
Revised: 25 March 2019
/
Accepted: 26 March 2019
/
Published: 1 April 2019
(This article belongs to the Special Issue Biological Efficacy of Natural and Chemically Modified Products against Oral Inflammatory Lesions)
Abstract
:Background: Anticancer drugs often have strong toxicity against tumours and normal cells. Some natural products demonstrate high tumour specificity. We have previously reported the cytotoxic activity and tumour specificity of various chemical compounds. In this study, we constructed a database of previously reported compound data and predictive models to screen a new anticancer drug. Methods: We collected compound data from our previous studies and built a database for analysis. Using this database, we constructed models that could predict cytotoxicity and tumour specificity using random forest method. The prediction performance was evaluated using an external validation set. Results: A total of 494 compounds were collected, and these activities and chemical structure data were merged as database for analysis. The structure-toxicity relationship prediction model showed higher prediction accuracy than the tumour selectivity prediction model. Descriptors with high contribution differed for tumour and normal cells. Conclusions: Further study is required to construct a tumour selective toxicity prediction model with higher predictive accuracy. Such a model is expected to contribute to the screening of candidate compounds for new anticancer drugs.
1. Introduction
Various anticancer drugs are used to treat oral cancer; however, most of these drugs also affect normal cells. Damage to normal cell induces several adverse effects, one of these is oral mucositis (OM). OM of patients who receiving cancer therapy makes difficult to eat and to deprive volition of treatment. OM is an inflammation induced by various factors such as trauma, viruses and bacterial infections, genetic factors, stress, vitamin deficiency, and chemotherapy [1,2]. The mechanism of detail is still not well known; however, toxicity to normal cells is one of the causes. In addition, many anticancer drugs are toxic to normal cells and have low selectivity for tumour cells. For these reasons, anticancer drugs which have low toxicity on normal cells are urgently needed.
Compounds which are highly tumour-specific exist in natural products. Previously, we reported cytotoxic activity against human oral squamous cell carcinoma (OSCC) cell lines and human oral normal cells using a variety of natural and synthesized organic compounds with chromone and azulene, which are present in various natural products, as the mother nucleus [3]. We have recently reported that many anticancer drugs induce keratinocyte toxicity by inducing apoptosis [4]. However, very few reports have been published [5] about the exploration of new synthetic substances that show low keratinocyte toxicity except of our studies (Table 1).
Based on the notion that similar structures have similar activity, the relationship between chemical structure and activity is referred to as the structure-activity relationship (SAR). Currently, using information about chemical structure which called “descriptor” that is structural, physicochemical and quantum chemical variety of characteristics, data were calculated and used for relation analysis. Conventionally, multiple regression analysis, which is a standard statistical approach, has been employed to analyse the relationship between the characteristic amount and activity of such drugs. Recently, machine-learning methods have been applied to such analyses due to their high prediction performance, and the quantitative structure activity relationship (QSAR) model is used to screen lead compounds in drug discovery research [6,7,8].
We have also studied the properties of compounds relative to cytotoxicity activity using QSAR analysis of compound and cytotoxic activity reported in the literature [3]. However, we could not employ high performance analysis methods due to the limited number of compounds evaluated in each study.
Thus, in this study, we gathered compound data from our previous reports and developed a database with a sufficient number of compounds to facilitate the use of a more advanced prediction method than single regression analysis. We attempted to construct a prediction model to search for compounds with high cytotoxic activity and tumour specificity score, using the collected data of cytotoxic activity of various compounds against tumour and normal cells.
2. Materials and Methods
2.1. Data Collection and Preparation
We collected our original articles published up to May 2018 (with the exception of literature reviews), and compound and cytotoxicity data were extracted from the collected articles. All OSCC and normal human oral cells were incubated at 2 × 103/96-microwell and incubated for 48 h to produce near confluent cells (approximately half of the plate covered by cells) so that cells can further grow. Cells were then treated with various conditions of samples for 48 h. Controls contains the same concentration of DMSO, and subtracted from the experimental values to correct for DMSO cytotoxicity. Relative viable cell numbers were determined by MTT method. The conditions of cytotoxic assays were the same for all experiments we have done in our previous publications [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49].
Cytotoxicity data were used as a ratio of mean 50% cytotoxic concentration (CC50) against OSCC cell lines (HSC-2, HSC-3, and HSC-4) and human oral normal cells (human gingival fibroblast, HGF; human pulp cells, HPC; human periodontal ligament fibroblast, and HPLF), and these CC50 were converted to −logCC50 (pCC50), which is a negative common logarithm.
The tumour cell selective toxic index (selectivity index; SI) was defined as the ratio of the mean CC50 of OSCC cell lines to the mean CC50 of the human oral normal cells, and the SI was calculated for all individual compounds.
2.2. Chemical Structure Data Acquisition and Descriptor Calculation
The collected compounds were drawn using MarvinSketch 18.10.0 (ChemAxon, Budapest, Hungary) [50] and then converted to SMILES that is a form of a line notation based on graph theory, to obtain numerical data from the chemical structure.
The compound data were dealt with using the integrated computational chemistry system Molecular Operating Environment (MOE) version 2018.0101 (Chemical Computing Group Inc., Quebec, Canada) [51] as follows; salts were removed, structure optimization was calculated, and load partial charges were obtained. The structural data were converted to a 3D format by MOE using “Rebuild 3D” and structural optimization was realized by force field calculation (amber-10: EHT).
From this compound data, we calculated 2D and 3D descriptors using MOE and Dragon (version 7.0.2, Kode srl., Pisa, Italy) [52], respectively. Descriptors were treated independently by the software. Standard deviations were calculated with each descriptor; in cases where the value was zero, the descriptor was excluded. These descriptor data calculated by MOE and Dragon were merged for each compound.
2.3. Preparation of Data Table
The cytotoxic activity and descriptor data were merged to a data table for analysis. The compounds in this data table were checked for duplication by using SMILES. Compounds that had one SMILES to several cytotoxic activity data from different articles adopted the mean pCC50.
2.4. Construction of Prediction Models by RF
The data table was randomly split (2:1 ratio) into a training set and an external validation set [53].
Eight structure-toxicity relationship prediction models were constructed by RF using the training set. The response variables of eight prediction models were three pCC50 against each OSCC cell line, three pCC50 against each human oral normal cell, the mean pCC50 against OSCC cell lines (mean tumour cell), and the mean pCC50 against human oral normal cells (i.e., the mean normal cell).
In the same manner, a tumour cell selective toxicity prediction model in which the response variable was the SI was constructed by using RF. Construction of prediction models by RF was performed “Bootstrap Forest” [54] in statistical software JMP® Pro. 13.1.0 (SAS Institute Inc., Cary, NC, USA) [55].
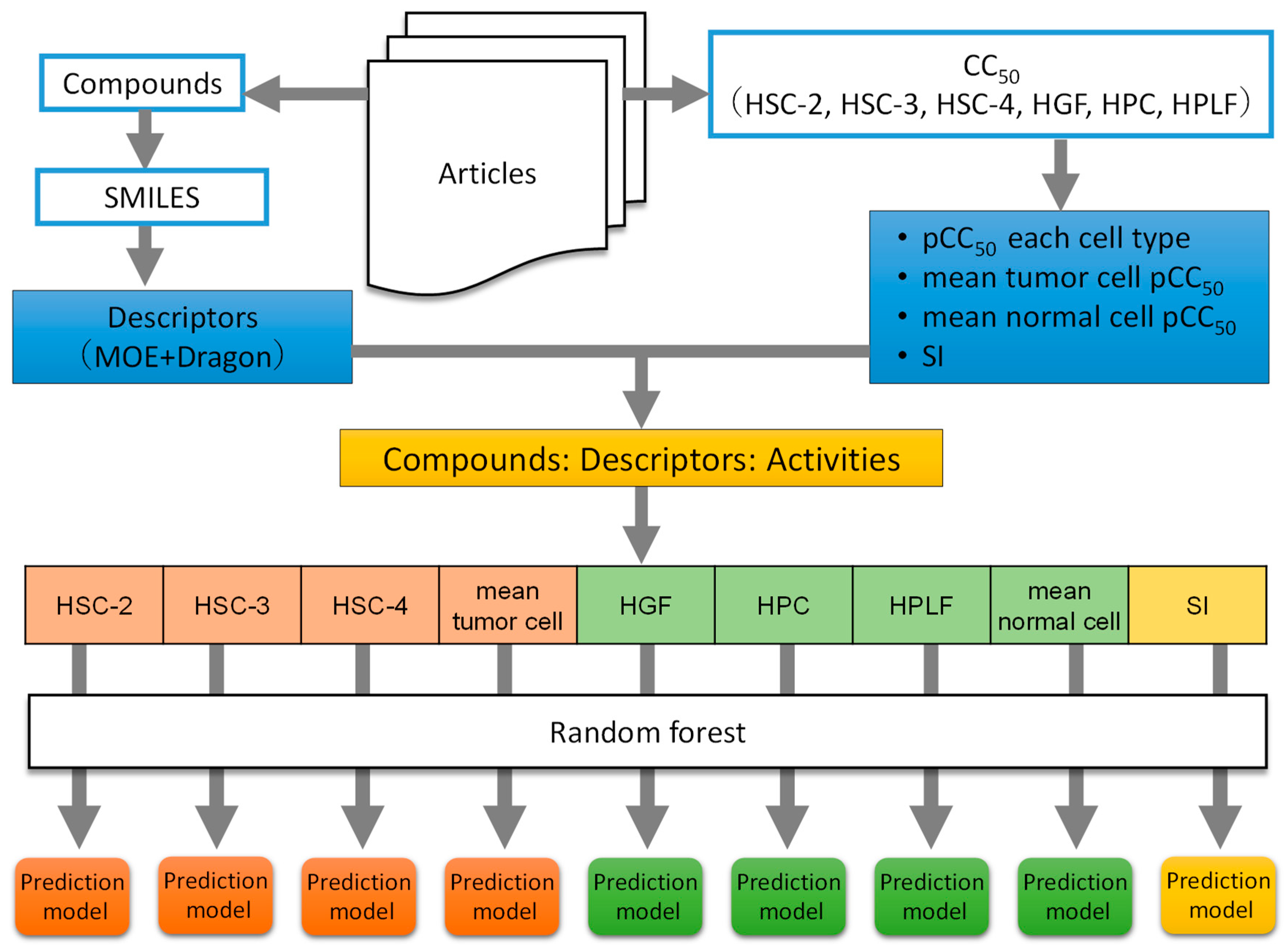
To construct the prediction model, changing parameter settings and largest coefficient of determination prediction model that was selected. Figure 1 shows the procedures from Section 2.1, Section 2.2, Section 2.3 and Section 2.4.
3. Results
3.1. Data Collection
We obtained 498 compounds from 39 articles [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49]. After eliminating duplicate compounds by SMILES, 494 compounds were analysed. Table 2 shows the articles and number of extracted compounds. These 494 compounds belong to the compound groups developed from various natural products, having skeletons shown in Table 2. SMILES data of these compounds are provided in Supplementary Materials (Table S1).
Descriptors were calculated from each software MOE and Dragon, subsequently excluded in case of the value is constant. After cleaning, 3750 descriptors were remained and used for analyses (319 descriptors calculated by MOE and 3431 descriptors calculated by Dragon).
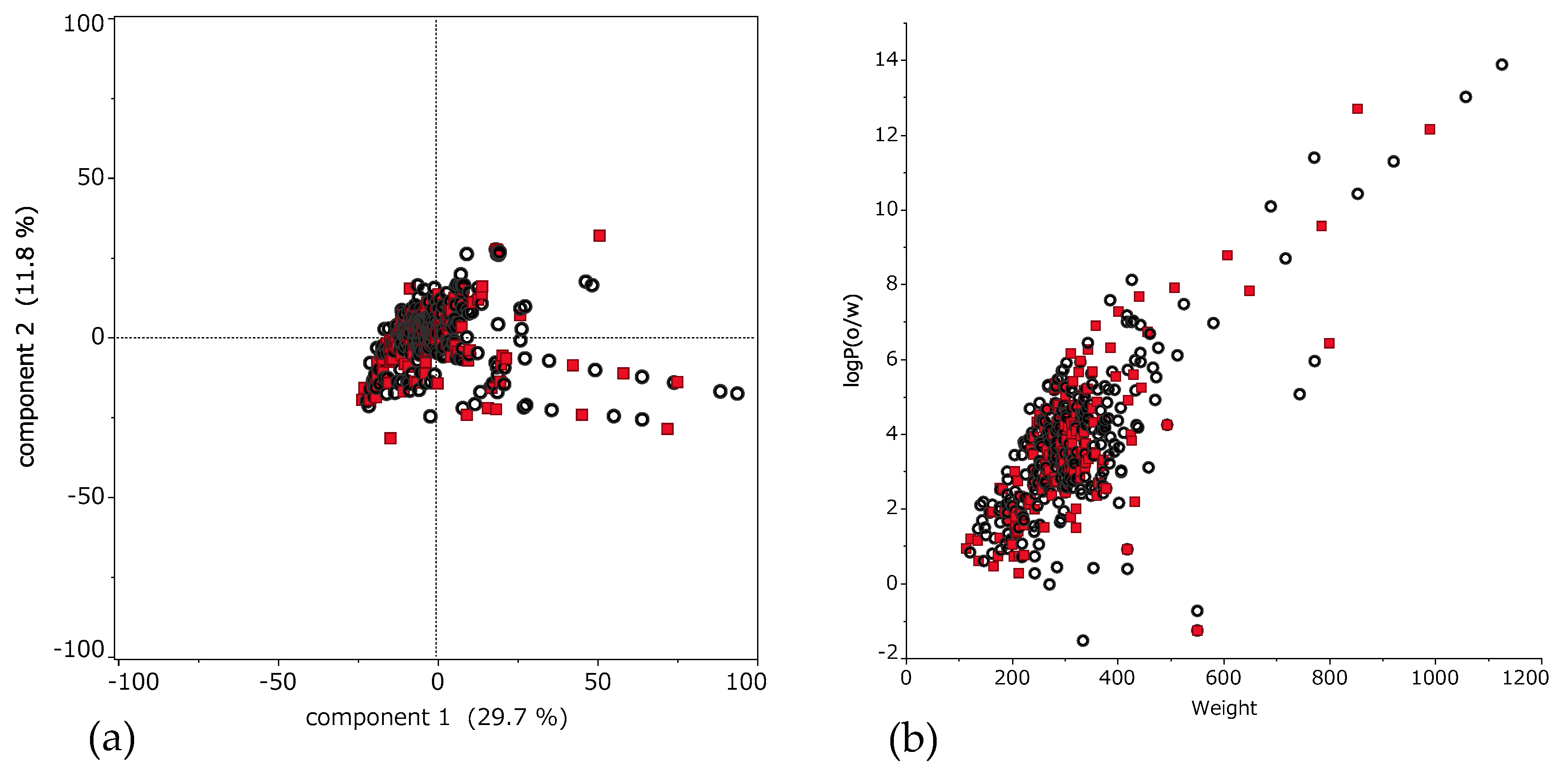
Figure 2a shows applicability domain (AD). AD is the range of molecular properties or structures for which the model is considered to be applicable [56]. This scatter plot shows the result of principal component analysis using descriptors. The horizontal axis is the first principal component, and the vertical axis is the second principal component. Training set and test set compounds distribute as well balanced.
Moreover, to indicate detailed properties of these compounds, scatter plot of molecular weight (MW) and octanol-water partitioning coefficient (logP) is shown in Figure 2b. These compounds showed characteristic distribution of MW from 114.2 to 1125.8 (median 297.9); and logP from −1.53 to 13.9 (median 3.46).
3.2. Construction of Prediction Models by RF
Several prediction models were built by parameter turning. Here, the model that demonstrated the largest value was selected. The prediction accuracy and parameters of each model are shown in Table 3. These models were evaluated by using parameters as follows; R2, root-mean-square error (RMSE), out of bag (OOB) RMSE, maximum absolute value of the residue, mean absolute error (MAE). The OOB RMSE is computed as the square root of the sum of squared errors divided by the number of OOB observations. OOB observations are training observations that are not used to construct a tree in RF. MAE is a mean of error at a model which the value is the closer to zero indicates the model is the higher accuracy.
Figure 3 shows scatter plots of each RF model obtained using the training and external validation sets, the measured pCC50, the predicted pCC50, the predicted SI, and the observed SI.
The model that demonstrated the greatest R2 value with the external validation set was the normal cell HPC model (R2 = 0.659, RMSE = 0.372), and the SI model (R2 = 0.404, RMSE = 0.340) demonstrated the smallest R2 value.
3.3. Large Contribution Descriptor for Prediction Model
Table 4 shows the top five contributing descriptors for the RF prediction model. Importance of descriptor were evaluated “LogWorth” in JMP® Pro software. “LogWorth” is calculated as negative common logarithm of p-value. This p-value is calculated in a complex manner that takes into account the number of different ways splits can occur. This calculation is very fair compared to the unadjusted p-value [57]. In the structure-toxicity relationship prediction model, most descriptors were classified into groups representing the topological shape. Note that descriptors meaning lipophilicity were observed in the tumour cell model, and electric charge descriptors were observed in the normal cell model. Topological or 3D shape descriptors were selected in the tumour cell selective toxicity prediction model.
4. Discussion
From our previous articles, 494 compounds and activity data were obtained. As reported in previous studies, compounds with higher tumour specificity than existing anticancer drugs are present among these 494 compounds. For example, in a study of 3-Styryl-2H-chromenes, several compounds showed higher tumour specificity than doxorubicin (at most approximately 4.8 times higher specificity) [39].
In this study, we constructed a database of seed compounds for anticancer drugs, including a sufficient number of compounds for analysis.
Regarding cell type, the structure-toxicity relationship prediction models demonstrated the maximum R2 value for cytotoxic activity against normal cells. If toxicity against normal cells can be predicted accurately, it is hoped that such prediction models can be applied to the estimation of side effects caused by cytotoxicity against normal cells, such as OM, hematotoxicity alopecia and so on. In contrast, with the training set, the R2 values were greater than 0.9. In addition, the R2 values of all structure-toxicity relationship prediction models obtained with the external validation set were greater 0.5.
We consider that the structure responsible for lipophilicity or a combination of lipophilicity and another characteristic descriptors may contribute to cytotoxic activity prediction because lipophilicity was tend to observed in the tumour cell models and not in the normal cell models. Relationship of between lipophilicity and cytotoxicity against tumour cell might be considered that penetration mechanism of compounds into tumour cell is one of the reason, however, further study is needed.
We expect that these findings will be useful relative to examining prediction models in future. In construct to structure-toxicity relationship prediction model, the R2 results of the tumour cell selective toxicity prediction models were less than 0.5.
In light of these results, further study is required to construct a tumour cell selective toxicity prediction model. With the RF method, the meaning of the top five contributing descriptors tended to differ from the structure-toxicity relationship and tumour cell selective toxicity prediction models.
These results indicate that tumour cell selective toxicity prediction is difficult to realize using the methods employed in the cytotoxic activity prediction model.
Thus, further study involving other methods, parameter tuning, and so on is required to construct a tumour cell selective toxicity prediction model with high prediction accuracy. Superior anticancer drugs require both strong cytotoxicity against tumour cells and selectivity to tumour cells, therefore cytotoxic activity prediction model is needed.
In this study, we constructed prediction models that can estimate cytotoxicity and tumour selective toxicity based on cytotoxic activity data derived from various compounds. The RF machine learning method constructed models with higher prediction accuracy.
In future, using our findings as reference, we would like to construct a high-performance prediction model that can be used to search for candidate compounds for a new anticancer drug.
5. Conclusions
In this study, we constructed a database of different compounds with structure and cytotoxic activity data derived from various compounds reported in previous studies. With this database, cytotoxicity and tumour cell selective toxicity prediction models were constructed by RF method. It was found that the structure-toxicity relationship prediction model tended to demonstrate greater R2 values.
In future, we expect that collecting addition compound data and investigating various model construction methods will help realize a prediction model with good prediction accuracy, which would facilitate the search for candidate compounds for anticancer drugs.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2305-6320/6/2/45/s1, Table S1: SMILES of 494 compounds.
Author Contributions
J.N., M.I. performed QSAR analysis; H.S. performed cytotoxicity assay; Y.U. organized the review.
Funding
This work was partially supported by KAKENHI from the Japan Society for the Promotion of Science (JSPS): 15K08111(Y.U.) and 16K11519(S.H.).
Acknowledgments
The authors acknowledge Yoshiaki Sugita and Koichi Takao for the supply of chromones, and Meikai University School of Dentistry for the support for the research.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| HGF | Human gingival fibroblast |
| HPC | Human pulp cell |
| HPLF | Human periodontal fibroblast |
| Log P | Octanol-water partitioning coefficient |
| MOE | Molecular Operating Environment |
| OM | Oral mucositis |
| OOB | Out-of-bag |
| OSCC | Oral squamous cell carcinoma |
| pCC50 | −logCC50, a negative common logarithm |
| QASR | Quantitative structure-activity relationship |
| R2 | Coefficient of determination |
| RF | Random forest |
| RMSE | Root-mean-square error |
| SAR | Structure-activity relationship |
References
- Sonis, S.T. Mucositis: The impact, biology and therapeutic opportunities of oral mucositis. Oral Oncol. 2009, 45, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, F.; Yoshida, A.; Nakajima, A.; Wada-Takahashi, S.; Takahashi, S.S.; Lee, M.C. Alteration of the redox state with reactive oxygen species for 5-fluorouracil-induced oral mucositis in hamsters. PLoS ONE 2013, 8, e82834. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Takao, K.; Uesawa, Y.; Sakagami, H. Search for New Type of Anticancer Drugs with High Tumor Specificity and Less Keratinocyte Toxicity. Anticancer Res. 2017, 37, 5919–5924. [Google Scholar] [PubMed]
- Sakagami, H.; Okudaira, N.; Masuda, Y.; Amano, O.; Yokose, S.; Kanda, Y.; Suguro, M.; Natori, T.; Oizumi, H.; Oizumi, T. Induction of apoptosis in human oral keratinocyte by doxorubicin. Anticancer Res. 2017, 37, 1023–1029. [Google Scholar]
- SciFinder®. Available online: https://www.cas.org/products/scifinder (accessed on 5 February 2019).
- Wolfgang, S.; Dina, R. QSAR/QSPR. In Applied Chemoinformatics; Thomas, E., Johann, G., Eds.; Wiley-VCH: Weinheim, Germany; Berlin, Germany, 2018; pp. 9–13. [Google Scholar]
- Guohui, S.; Tengjiao, F.; Xiaodong, S.; Yuxing, H.; Xin, C.; Lijiao, Z.; Ting, R.; Yue, Z.; Rugang, Z.; Yongzhen, P. In Silico Prediction of O6-Methylguanine-DNA Methyltransferase Inhibitory Potency of Base Analogs with QSAR and Machine Learning Methods. Molecules 2018, 23, 2892. [Google Scholar]
- Tengjiao, F.; Guohui, S.; Lijiao, Z.; Xin, C.; Rugang, Z. QSAR and Classification Study on Prediction of Acute Oral Toxicity of N-Nitroso Compounds. Int. J. Mol. Sci. 2018, 19, 3015. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Shirataki, Y.; Wakae, M.; Yamamoto, Y.; Hashimoto, K.; Satoh, K.; Ishihara, M.; Kikuchi, H.; Nishikawa, H.; Minagawa, K.; Motohashi, N.; et al. Cytotoxicity and radical modulating activity of isoflavones and isoflavanones from sophora species. Anticancer Res. 2004, 24, 1481–1488. [Google Scholar]
- Ishihara, M.; Sakagami, H. Re-evaluation of cytotoxicity and iron chelation activity of three β-diketones by semiempirical molecular orbital method. In Vivo 2005, 19, 119–124. [Google Scholar]
- Momoi, K.; Sugita, Y.; Ishihara, M.; Satoh, K.; Kikuchi, H.; Hashimoto, K.; Yokoe, I.; Nishikawa, H.; Fujisawa, S.; Sakagami, H. Cytotoxic activity of styrylchromones against human tumor cell lines. In Vivo 2005, 19, 157–164. [Google Scholar] [PubMed]
- Sakagami, H.; Ishihara, M.; Hoshino, Y.; Ishikawa, J.; Mikami, Y.; Fukai, T. Cytotoxicity of nocobactins NA-a, NA-b and their ferric complexes assessed by semiempirical molecular orbital method. In Vivo 2005, 19, 277–282. [Google Scholar] [PubMed]
- Ishihara, M.; Sakagami, H.; Liu, W.K. Quantitative structure-cytotoxicity relationship analysis of betulinic acid and its derivatives by semi-empirical molecular-orbital method. Anticancer Res. 2005, 25, 3951–3956. [Google Scholar] [PubMed]
- Inoue, K.; Kulsum, U.; Chowdhury, SA.; Fujisawa, S.; Ishihara, M.; Yokoe, I.; Sakagami, H. Tumor-specific cytotoxicity and apoptosis-inducing activity of berberines. Anticancer Res. 2005, 25, 4053–4060. [Google Scholar] [PubMed]
- Ishihara, M.; Yokote, Y.; Sakagami, H. Quantitative structure-cytotoxicity relationship analysis of coumarin and its derivatives by semiempirical molecular orbital method. Anticancer Res. 2006, 26, 2883–2886. [Google Scholar] [PubMed]
- Sasaki, M.; Okamura, M.; Ideo, A.; Shimada, J.; Suzuki, F.; Ishihara, M.; Kikuchi, H.; Kanda, Y.; Kunii, S.; Sakagami, H. Re-evaluation of tumor-specific cytotoxicity of mitomycin c, bleomycin and peplomycin. Anticancer Res. 2006, 26, 3373–3380. [Google Scholar]
- Ishihara, M.; Kawase, M.; Sakagami, H. Quantitative structure-activity relationship analysis of 4-trifluoromethylimidazole derivatives with the concept of absolute hardness. Anticancer Res. 2007, 27, 4047–4052. [Google Scholar]
- Ishihara, M.; Kawase, M.; Westman, G.; Samuelsson, K.; Motohashi, N.; Sakagami, H. Quantitative structure-cytotoxicity relationship analysis of phenoxazine derivatives by semiempirical molecular-orbital method. Anticancer Res. 2007, 27, 4053–4058. [Google Scholar] [PubMed]
- Ishihara, M.; Sakagami, H. QSAR of molecular structure and cytotoxic activity of vitamin K2 derivatives with concept of absolute hardness. Anticancer Res. 2007, 27, 4059–4064. [Google Scholar]
- Takekawa, F.; Nagumo, T.; Shintani, S.; Hashimoto, K.; Kikuchi, H.; Katayama, T.; Ishihara, M.; Amano, O.; Kawase, M.; Sakagami, H. Tumor-specific cytotoxic activity and type of cell death induced by 4-trifluoromethylimidazoles in human oral squamous cell carcinoma cell lines. Anticancer Res. 2007, 27, 4065–4070. [Google Scholar]
- Suzuki, F.; Hashimoto, K.; Ishihara, M.; Westman, G.; Samuelsson, K.; Kawase, M.; Motohashi, N.; Sakagami, H. Tumor-specificity and type of cell death induced by phenoxazines. Anticancer Res. 2007, 27, 4233–4238. [Google Scholar] [PubMed]
- Sakagami, H.; Hashimoto, K.; Suzuki, F.; Ishihara, M.; Kikuchi, H.; Katayama, T.; Satoh, K. Tumor-specificity and type of cell death induced by vitamin K2 derivatives and prenylalcohols. Anticancer Res. 2008, 28, 151–158. [Google Scholar] [PubMed]
- Ishihara, M.; Sakagami, H. Quantitative structure-cytotoxicity relationship analysis of 3-formylchromone derivatives by a semiempirical molecularorbital method with the concept of absolute hardness. Anticancer Res. 2008, 28, 277–282. [Google Scholar]
- Ishihara, M.; Kawase, M.; Sakagami, H. Quantitative structure-cytotoxicity relationship analysis of 5-trifluoromethyloxazole derivatives by a semiempirical molecular-orbital method with the concept of absolute hardness. Anticancer Res. 2008, 28, 997–1004. [Google Scholar]
- Ishihara, M.; Hatano, H.; Kawase, M.; Sakagami, H. Estimation of relationship between the structure of 1, 2, 3, 4-tetrahydroisoquinoline derivatives determined by a semiempirical molecular-orbital method and their cytotoxicity. Anticancer Res. 2009, 29, 2265–2272. [Google Scholar]
- Hatano, H.; Takekawa, F.; Hashimoto, K.; Ishihara, M.; Kawase, M.; Qing, C.; Qin-Tao, W.; Sakagami, H. Tumor-specific cytotoxic activity of 1, 2, 3, 4-tetrahydroisoquinoline derivatives against human oral squamous cell carcinoma cell lines. Anticancer Res. 2009, 29, 3079–3086. [Google Scholar]
- Takekawa, F.; Sakagami, H.; Ishihara, M. Estimation of relationship between structure of newly synthesized dihydroimidazoles determined by a semiempirical molecular-orbital method and their cytotoxicity. Anticancer Res. 2009, 29, 5019–5022. [Google Scholar] [PubMed]
- Ishihara, M.; Wakabayashi, H.; Motohashi, N.; Sakagami, H. Quantitative structure-cytotoxicity relationship of newly synthesized tropolones determined by a semiempirical molecular-orbital method (PM5). Anticancer Res. 2010, 30, 129–134. [Google Scholar]
- Ishihara, M.; Wakabayashi, H.; Motohashi, N.; Sakagami, H. Quantitative structure–cytotoxicity relationship of newly synthesized trihaloacetylazulenes determined by a semi-empirical molecular-orbital method (PM5). Anticancer Res. 2011, 31, 515–520. [Google Scholar]
- Ishihara, M.; Wakabayashi, H.; Motohashi, N.; Sakagami, H. Estimation of relationship between the structure of trihaloacetylazulene derivatives determined by a semiempirical molecular–orbital method (PM5) and their cytotoxicity. Anticancer Res. 2010, 30, 837–842. [Google Scholar] [PubMed]
- Ohno, H.; Araho, D.; Uesawa, Y.; Kagaya, H.; Ishihara, M.; Sakagami, H.; Yamamoto, M. Evaluation of cytotoxicity and tumor-specificity of licorice flavonoids based on chemical structure. Anticancer Res. 2013, 33, 3061–3068. [Google Scholar]
- Uesawa, Y.; Mohri, K.; Kawase, M.; Ishihara, M.; Sakagami, H. Quantitative structure–activity relationship (QSAR) analysis of tumor-specificity of 1, 2, 3, 4-tetrahydroisoquinoline derivatives. Anticancer Res. 2011, 31, 4231–4238. [Google Scholar] [PubMed]
- Sekine, S.; Shimodaira, C.; Uesawa, Y.; Kagaya, H.; Kanda, Y.; Ishihara, M.; Amano, O.; Sakagami, H.; Wakabayashi, H. Quantitative structure–activity relationship analysis of cytotoxicity and anti-uv activity of 2-aminotropones. Anticancer Res. 2014, 34, 1743–1750. [Google Scholar]
- Shimada, C.; Uesawa, Y.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Saito, T.; Sugita, Y.; et al. Quantitative structure–cytotoxicity relationship of phenylpropanoid amides. Anticancer Res. 2014, 34, 3543–3548. [Google Scholar]
- Shimada, C.; Uesawa, Y.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Miyashiro, T.; Sugita, Y.; et al. Quantitative structure–cytotoxicity relationship of piperic acid amides. Anticancer Res. 2014, 34, 4877–4884. [Google Scholar]
- Shimada, C.; Uesawa, Y.; Ishii-Nozawa, R.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Sugita, Y.; et al. Quantitative structure–cytotoxicity relationship of 3-styrylchromones. Anticancer Res. 2014, 34, 5405–5412. [Google Scholar]
- Uesawa, Y.; Sakagami, H.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Yahagi, H.; Takao, K.; Sugita, Y. Quantitative structure–cytotoxicity relationship of 3-styryl-2H-chromenes. Anticancer Res. 2015, 35, 5299–5308. [Google Scholar] [PubMed]
- Sakagami, H.; Uesawa, Y.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Sugita, Y. Quantitative structure–cytotoxicity relationship of oleoylamides. Anticancer Res. 2015, 35, 5341–5355. [Google Scholar] [PubMed]
- Uesawa, Y.; Sakagami, H.; Kagaya, H.; Yamashita, M.; Takao, K.; Sugita, Y. Quantitative structure-cytotoxicity relationship of 3-benzylidenechromanones. Anticancer Res. 2016, 36, 5803–5812. [Google Scholar] [CrossRef]
- Fukuchi, K.; Okudaira, N.; Adachi, K.; Odai-Ide, R.; Watanabe, S.; Ohno, H.; Yamamoto, M.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; et al. Antiviral and antitumor activity of licorice root extracts. In Vivo 2016, 30, 777–786. [Google Scholar] [CrossRef]
- Sakagami, H.; Masuda, Y.; Tomomura, M.; Yokose, S.; Uesawa, Y.; Ikezoe, N.; Asahara, D.; Takao, K.; Kanamoto, T.; Terakubo, S.; et al. Quantitative structure–cytotoxicity relationship of chalcones. Anticancer Res. 2017, 37, 1091–1098. [Google Scholar]
- Sakagami, H.; Uesawa, Y.; Masuda, Y.; Tomomura, M.; Yokose, S.; Miyashiro, T.; Murai, J.; Takao, K.; Kanamoto, T.; Terakubo, S.; et al. Quantitative structure–cytotoxicity relationship of newly synthesized piperic acid esters. Anticancer Res. 2017, 37, 6161–6168. [Google Scholar]
- Uesawa, Y.; Sakagami, H.; Ikezoe, N.; Takao, K.; Kagaya, H.; Sugita, Y. Quantitative structure–cytotoxicity relationship of aurones. Anticancer Res. 2017, 37, 6169–6176. [Google Scholar]
- Sakagami, H.; Okudaira, N.; Uesawa, Y.; Takao, K.; Kagaya, H.; Sugita, Y. Quantitative structure–cytotoxicity relationship of 2-azolylchromones. Anticancer Res. 2018, 38, 763–770. [Google Scholar]
- Uesawa, Y.; Sakagami, H.; Okudaira, N.; Toda, K.; Takao, K.; Kagaya, H.; Sugita, Y. Quantitative structure–cytotoxicity relationship of cinnamic acid phenetyl esters. Anticancer Res. 2018, 38, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Maruyama, R.; Irie, Y.; Hashimoto, M.; Wakabayashi, H.; Okudaira, N.; Uesawa, Y.; Kagaya, H.; Sakagami, H. In vitro anti-tumor activity of azulene amide derivatives. In Vivo 2018, 32, 479–486. [Google Scholar] [PubMed]
- Uehara, M.; Minemura, H.; Ohno, T.; Hashimoto, M.; Wakabayashi, H.; Okudaira, N.; Sakagami, H. In vitro antitumor activity of alkylaminoguaiazulenes. In Vivo 2018, 32, 541–547. [Google Scholar] [PubMed]
- Marvin. Available online: https://chemaxon.com/products/marvin (accessed on 15 November 2018).
- MOE. Available online: http://www.chemcomp.com/MOE-Molecular_Operating_Environment.htm (accessed on 15 November 2018).
- Dragon. Available online: https://chm.kode-solutions.net/products_dragon.php (accessed on 15 November 2018).
- Paola, G. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar]
- SAS Institute Inc. Chapter 6 Bootstrap Forest. In JMP® 13 Predictive and Specialized Modeling, 2nd ed.; SAS Institute Inc.: Cary, NC, USA, 2017; pp. 107–122. [Google Scholar]
- JMP®. Available online: https://www.jmp.com/en_us/home.html (accessed on 15 November 2018).
- Wolfgang, S.; Dina, R. Applicability domain and model acceptability critera. In Applied Chemoinformatics; Thomas, E., Johann, G., Eds.; Wiley-VCH: Weinheim, Germany; Berlin, Germany, 2018; pp. 41–43. [Google Scholar]
- SAS Institute Inc. Chapter 5 Partition Models. In JMP® 13 Predictive and Specialized Modeling, 2nd ed.; SAS Institute Inc.: Cary, NC, USA, 2017; pp. 104–106. [Google Scholar]
Figure 1.
Schematic diagram of data collection and analysis.

Figure 2.
Chemical space of 494 compounds. (a) Applicability domain (AD) of 494 compounds. Scatter plot of principal component analysis using descriptors. The horizontal axis is the first principal component, and the vertical axis is the second principal component. These percentage are eigenvalue that represent a partition of the total variation in the multivariate sample. Each dot represents a compound; black circle is the training set and red square is the external validation set. (b) The horizontal axis is molecular weight (MW) and the vertical axis is octanol-water partitioning coefficient (logP). Here, each dot represents a compound; black is the training set and red is the external validation set.
Figure 2.
Chemical space of 494 compounds. (a) Applicability domain (AD) of 494 compounds. Scatter plot of principal component analysis using descriptors. The horizontal axis is the first principal component, and the vertical axis is the second principal component. These percentage are eigenvalue that represent a partition of the total variation in the multivariate sample. Each dot represents a compound; black circle is the training set and red square is the external validation set. (b) The horizontal axis is molecular weight (MW) and the vertical axis is octanol-water partitioning coefficient (logP). Here, each dot represents a compound; black is the training set and red is the external validation set.

Figure 3.
Scatter plot of training set and external validation set. In scatter plots of tumour and normal cell, the horizontal axis is the predicted pCC50, and the vertical axis is the observed pCC50 of tumour and normal cell. In scatter plot of SI, the horizontal axis is the predicted SI, and the vertical axis is the observed SI. Each dot represents a compound; black circle is the training set and red square is the external validation set.
Figure 3.
Scatter plot of training set and external validation set. In scatter plots of tumour and normal cell, the horizontal axis is the predicted pCC50, and the vertical axis is the observed pCC50 of tumour and normal cell. In scatter plot of SI, the horizontal axis is the predicted SI, and the vertical axis is the observed SI. Each dot represents a compound; black circle is the training set and red square is the external validation set.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Urgency of manufacturing new anticancer drugs with low keratinocyte toxicity (data obtained from SciFinder® [5] on 5 February 2019)
Table 1.
Urgency of manufacturing new anticancer drugs with low keratinocyte toxicity (data obtained from SciFinder® [5] on 5 February 2019)
| Search Terms | Number of Total Reports (A) | Number of Our Reports (B) | % (B/A) × 100 |
|---|---|---|---|
| OSCC | 8951 (100) | 141 | 1.6 |
| OSCC + Anticancer Drug | 335 (3.70) | 60 | 17.9 |
| OSCC + Anticancer Drug + Tumour-Specificity | 50 (0.56) | 40 | 80.0 |
| OSCC + Anticancer Drug + Tumour-Specificity + Newly Synthesized | 2 (0.02) | 2 | 100.0 |
| OSCC + Anticancer Drug + Keratinocyte Toxicity | 5 (0.06) | 4 | 80.0 |
| OSCC + anticancer drug + QSAR | 27 (0.30) | 25 | 92.6 |
| OSCC + Anticancer Drug + QSAR+ Newly Synthesized | 3 (0.03) | 3 | 100.0 |
Table 2.
Number of compounds and basic skeleton extracted from articles.
| No. | Number of Compounds | Basic Skeleton | Ref. |
|---|---|---|---|
| 1 | 9 | Isoflavones and Isoflavanones | [9] |
| 2 | 3 | Three β-Diketones | [10] |
| 3 | 6 | Styrylchromones | [11] |
| 4 | 3 | Nocobactins NA-a, NA-b and Their Ferric Complexes | [12] |
| 5 | 5 | Betulinic Acid and Its Derivatives | [13] |
| 6 | 2 | Berberines | [14] |
| 7 | 20 | Coumarin and Its Derivatives | [15] |
| 8 | 1 | Mitomycin C, Bleomycin and Peplomycin | [16] |
| 9 | 13 | 4-Trifluoromethylimidazole Derivatives | [17] |
| 10 | 15 | Phenoxazine Derivatives | [18] |
| 11 | 7 | Vitamin K2 Derivatives | [19] |
| 12 | 2 | 4-Trifluoromethylimidazoles | [20] |
| 13 | 10 | Phenoxazines | [21] |
| 14 | 18 | Vitamin K2 Derivatives and Prenylalcohols | [22] |
| 15 | 10 | 3-Formylchromone Derivatives | [23] |
| 16 | 12 | 5-Trifluoromethyloxazole Derivatives | [24] |
| 17 | 19 | 1,2,3,4-Tetrahydroisoquinoline Derivatives | [25] |
| 18 | 19 | 1,2,3,4Tetrahydroisoquinoline Derivatives | [26] |
| 19 | 12 | Dihydroimidazoles | [27] |
| 20 | 24 | Tropolones | [28] |
| 21 | 24 | Trihaloacetylazulenes | [29] |
| 22 | 22 | Trihaloacetylazulene Derivatives | [30] |
| 23 | 10 | Licorice Flavonoids | [31] |
| 24 | 4 | 1,2,3,4-Tetrahydroisoquinoline Derivatives | [32] |
| 25 | 19 | 2-Aminotropones | [33] |
| 26 | 12 | Phenylpropanoid Amides | [34] |
| 27 | 12 | Piperic Acid Amides | [35] |
| 28 | 15 | 3-Styrylchromones | [36] |
| 29 | 16 | 3-Styryl-2H-chromenes | [37] |
| 30 | 18 | Oleoylamides | [38] |
| 31 | 17 | 3-Benzylidenechromanones | [39] |
| 32 | 18 | Licorice Root Extracts | [40] |
| 33 | 15 | Chalcones | [41] |
| 34 | 11 | Piperic Acid Esters | [42] |
| 35 | 17 | Aurones | [43] |
| 36 | 24 | 2-Azolylchromones | [44] |
| 37 | 10 | Cinnamic Acid Phenetyl Esters | [45] |
| 38 | 10 | Azulene Amide Derivatives | [46] |
| 39 | 10 | Alkylaminoguaiazulenes | [47] |
Table 3.
Parameters of each model by random forest
| Parameters | Tumour Cells | Normal Cells | SI | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HSC-2 | HSC-3 | HSC-4 | Mean | HGF | HPC | HPLF | Mean | ||
| Number of Tree | 100 | 300 | 100 | 100 | 100 | 100 | 100 | 100 | 300 |
| Number of Term | 952 | 1000 | 952 | 952 | 952 | 952 | 952 | 952 | 1000 |
| Number of Maximum Split at Tree | 100 | 1000 | 2000 | 2000 | 2000 | 2000 | 2000 | 2000 | 2000 |
| Minimum Node Size | 3 | 5 | 5 | 5 | 5 | 5 | 3 | 5 | 5 |
| Seed Value | 29 | 36 | 44 | 77 | 93 | 91 | 730 | 9045 | 124 |
| Number of Tree | 23 | 8 | 21 | 20 | 9 | 4 | 34 | 12 | 8 |
| Number of Term at a Split | 1000 | 1000 | 952 | 952 | 952 | 952 | 952 | 952 | 1000 |
| R2(Training Set) | 0.904 | 0.847 | 0.868 | 0.876 | 0.862 | 0.815 | 0.908 | 0.858 | 0.817 |
| R2(External Validation Set) | 0.564 | 0.568 | 0.631 | 0.563 | 0.554 | 0.659 | 0.515 | 0.576 | 0.404 |
| RMSE (External Validation Set) | 0.480 | 0.496 | 0.496 | 0.473 | 0.435 | 0.372 | 0.442 | 0.397 | 0.340 |
| OOB RMSE | 0.808 | 0.778 | 0.742 | 0.760 | 0.593 | 0.587 | 0.618 | 0.573 | 0.579 |
| Maximum Absolute Value of the Residue | 2.052 | 1.875 | 1.424 | 1.408 | 1.347 | 1.758 | 1.331 | 1.582 | 1.188 |
| Mean Absolute Error | 0.236 | 0.255 | 0.232 | 0.216 | 0.216 | 0.199 | 0.240 | 0.198 | 0.191 |
Table 4.
Top five contributing descriptors for random forest prediction model.
| Cell Type | Descriptor | Meaning | |
|---|---|---|---|
| Tumour Cells | HSC-2 | vsurf_D7 | Lipophilicity |
| vsurf_D2 | Lipophilicity | ||
| GCUT_SMR_0 | Topological shape | ||
| CATS2D_07_LL | Lipophilicity | ||
| SpMin2_Bh(e) | Topological shape and electric state | ||
| HSC-3 | SssNH | Topological shape and electric state | |
| b_max1len | Topological shape | ||
| Mor13s | 3D shape and electric state | ||
| Mor15s | 3D shape and electric state | ||
| F01[C-C] | Topological shape | ||
| HSC-4 | SpMax_L | Topological shape | |
| SpAD_EA(dm) | Topological shape and dipole moment | ||
| ATSC2s | Topological shape and electric state | ||
| vsurf_D7 | Lipophilicity | ||
| ATSC5s | Topological shape and electric state | ||
| Mean | logP(o/w) | Lipophilicity | |
| vsurf_D2 | Lipophilicity | ||
| vsurf_D6 | Lipophilicity | ||
| P_VSA_ppp_L | Topological shape and lipophilicity | ||
| SssNH | Topological shape and electric state | ||
| Normal Cells | HGF | GCUT_SLOGP_0 | Topological shape |
| F10[C-N] | Topological shape | ||
| SssNH | Topological shape and electric state | ||
| SpMin2_Bh(s) | Topological shape | ||
| GCUT_SMR_0 | Topological shape | ||
| HPC | VE1_B(p) | Topological shape and polarizability | |
| b_max1len | Topological shape | ||
| CATS3D_10_PL | 3D shape and electric state | ||
| h_pKb | Topological shape and electric state | ||
| SpMin1_Bh(p) | Topological shape and polarizability | ||
| HPLF | P_VSA_e_3 | Topological shape and electric state | |
| GCUT_SLOGP_0 | Topological shape | ||
| F10[C-N] | Topological shape | ||
| SssNH | Topological shape and electric state | ||
| h_pavgQ | Topological shape and electric state | ||
| Mean | h_pstrain | Topological shape and electric state | |
| h_pavgQ | Topological shape and electric state | ||
| b_max1len | Topological shape | ||
| SssNH | Topological shape and electric state | ||
| F10[C-N] | Topological shape | ||
| SI | TDB08p | 3D shape and polarizability | |
| F06[C-N] | Topological shape | ||
| PEOE_VSA+1 | Topological shape and electric state | ||
| R5u+ | 3D shape and size | ||
| RDF035m | 3D shape and size | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nagai, J.; Imamura, M.; Sakagami, H.; Uesawa, Y. QSAR Prediction Model to Search for Compounds with Selective Cytotoxicity Against Oral Cell Cancer. Medicines 2019, 6, 45. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6020045
AMA Style
Nagai J, Imamura M, Sakagami H, Uesawa Y. QSAR Prediction Model to Search for Compounds with Selective Cytotoxicity Against Oral Cell Cancer. Medicines. 2019; 6(2):45. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6020045
Chicago/Turabian StyleNagai, Junko, Mai Imamura, Hiroshi Sakagami, and Yoshihiro Uesawa. 2019. "QSAR Prediction Model to Search for Compounds with Selective Cytotoxicity Against Oral Cell Cancer" Medicines 6, no. 2: 45. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6020045
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.