
Reduced Fatty Acid Use from CD36 Deficiency Deteriorates Streptozotocin-Induced Diabetic Cardiomyopathy in Mice

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
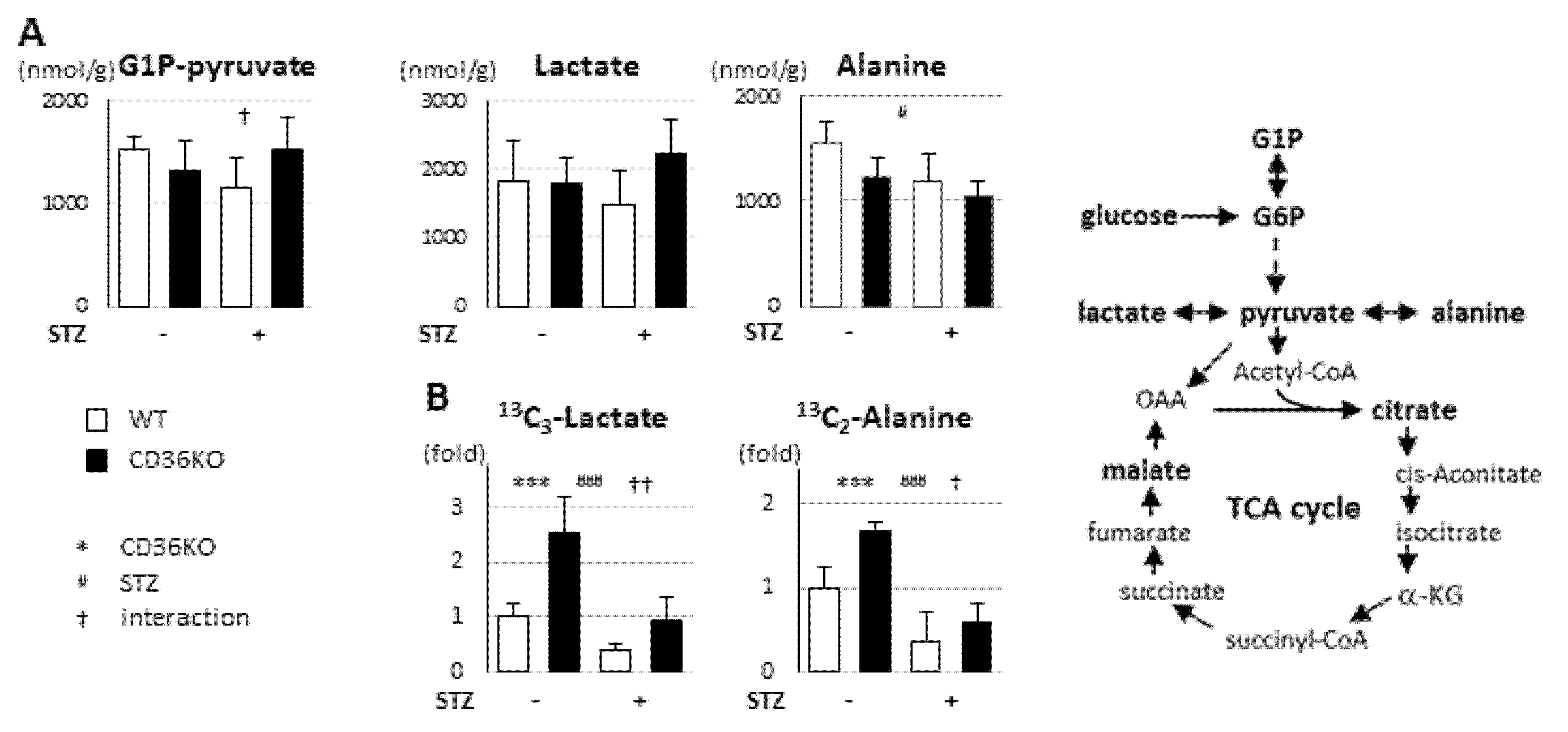{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
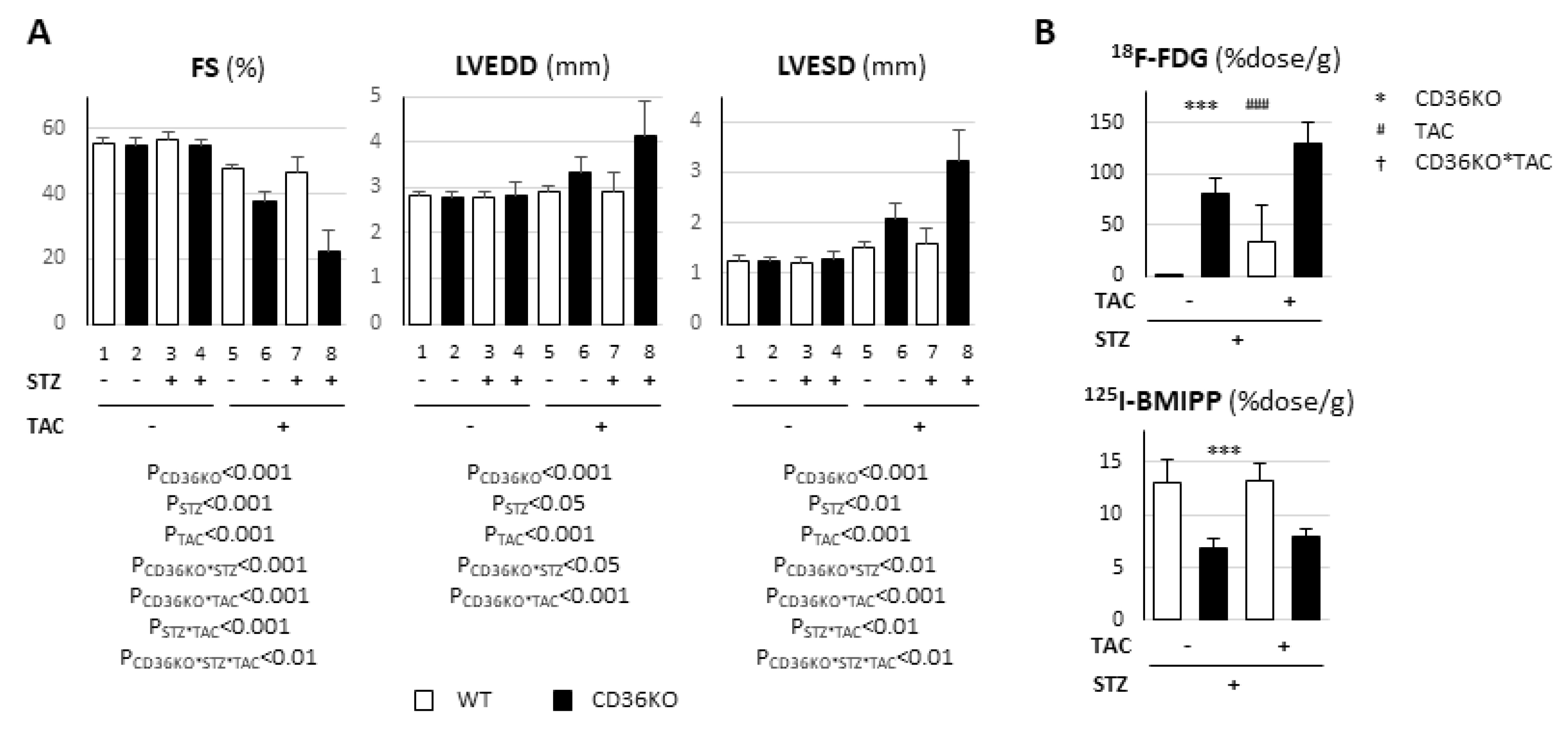
2.1. Pronounced Left Ventricular Contractile Dysfunction in STZ-Treated CD36KO Mice
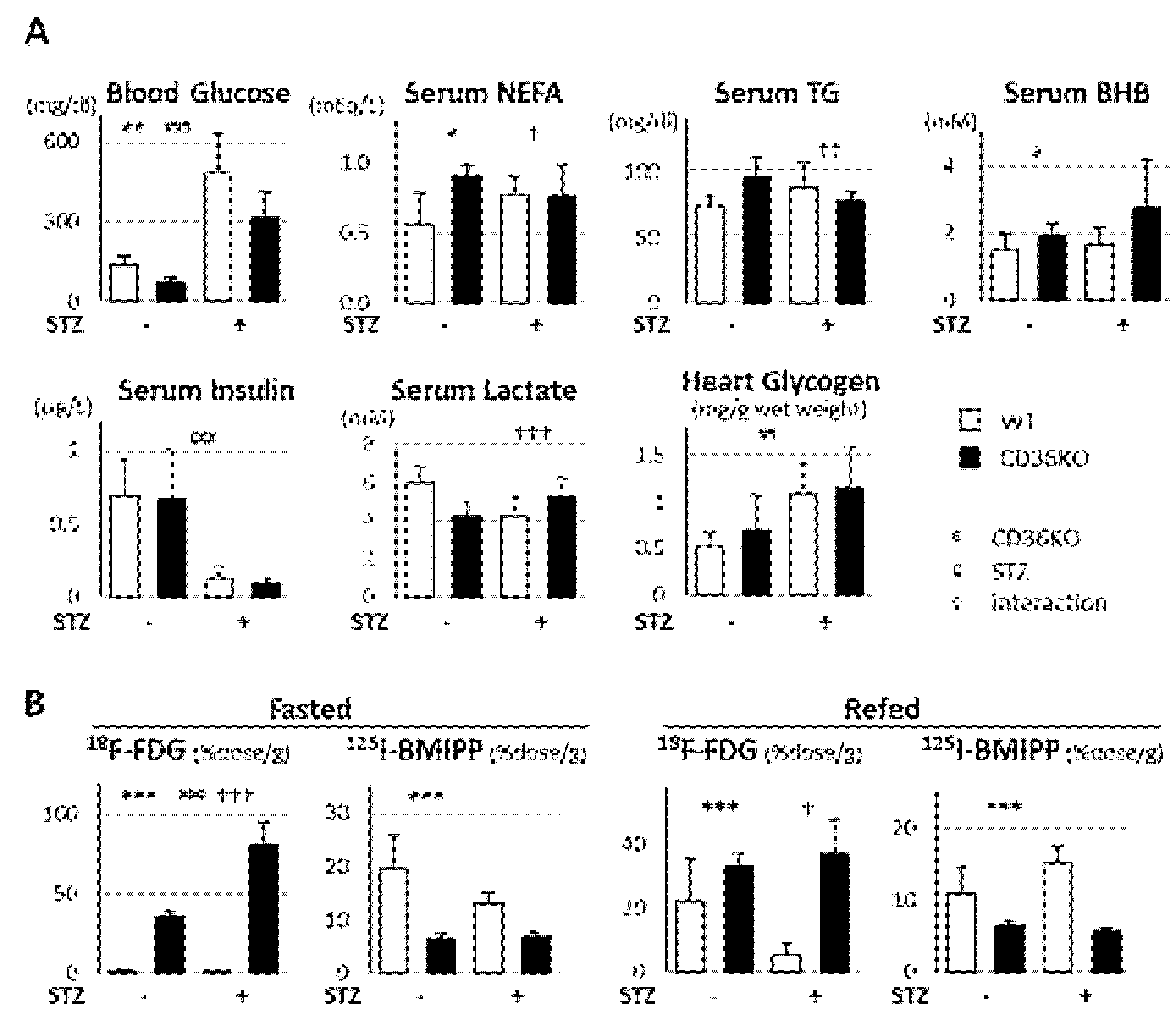
2.2. Accelerated Glucose Uptake and Low Fatty Acid Uptake in CD36KO-STZ Hearts
2.3. STZ Treatment Suppresses Enhanced Glycolytic Rate in CD36KO Hearts
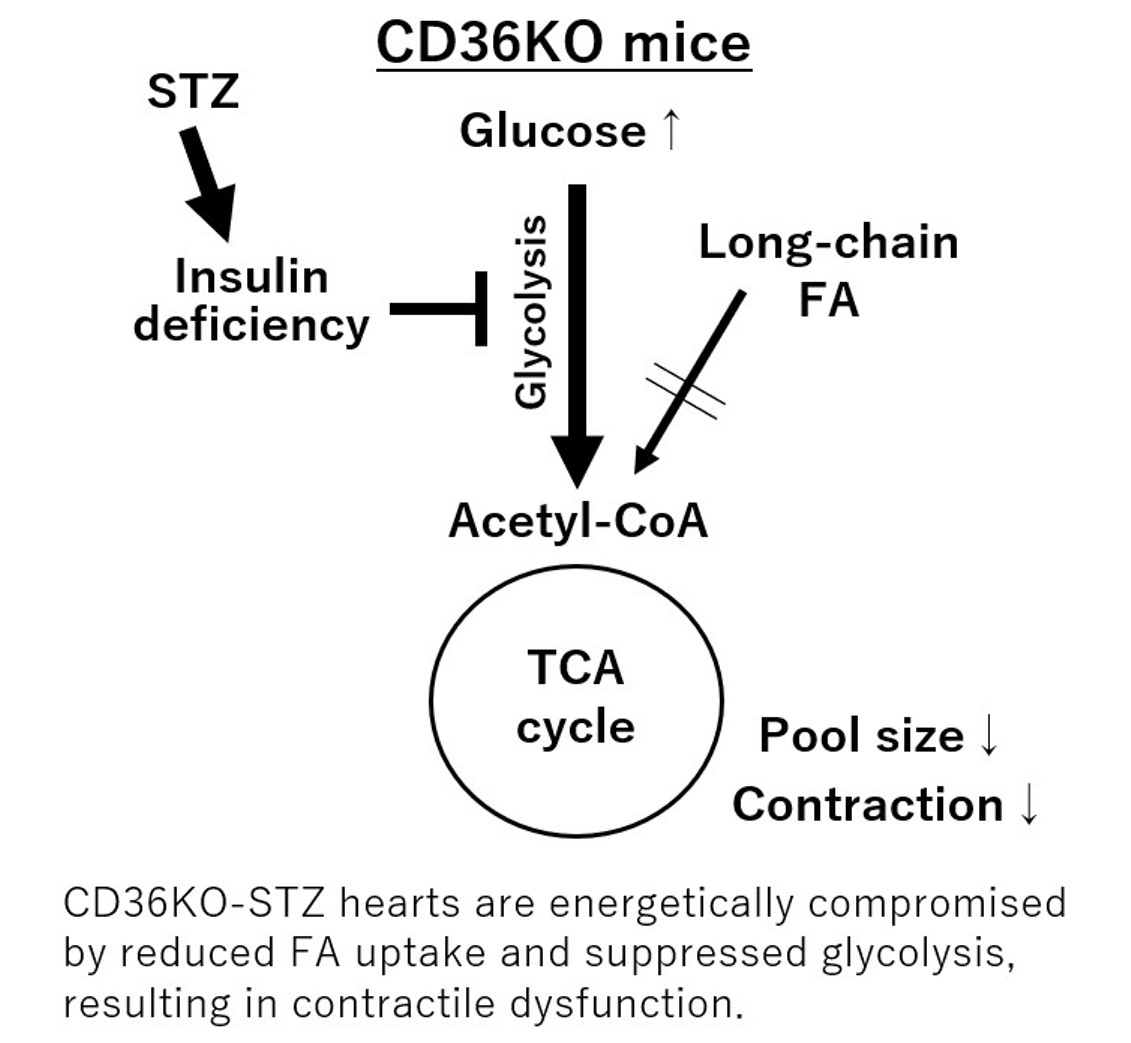
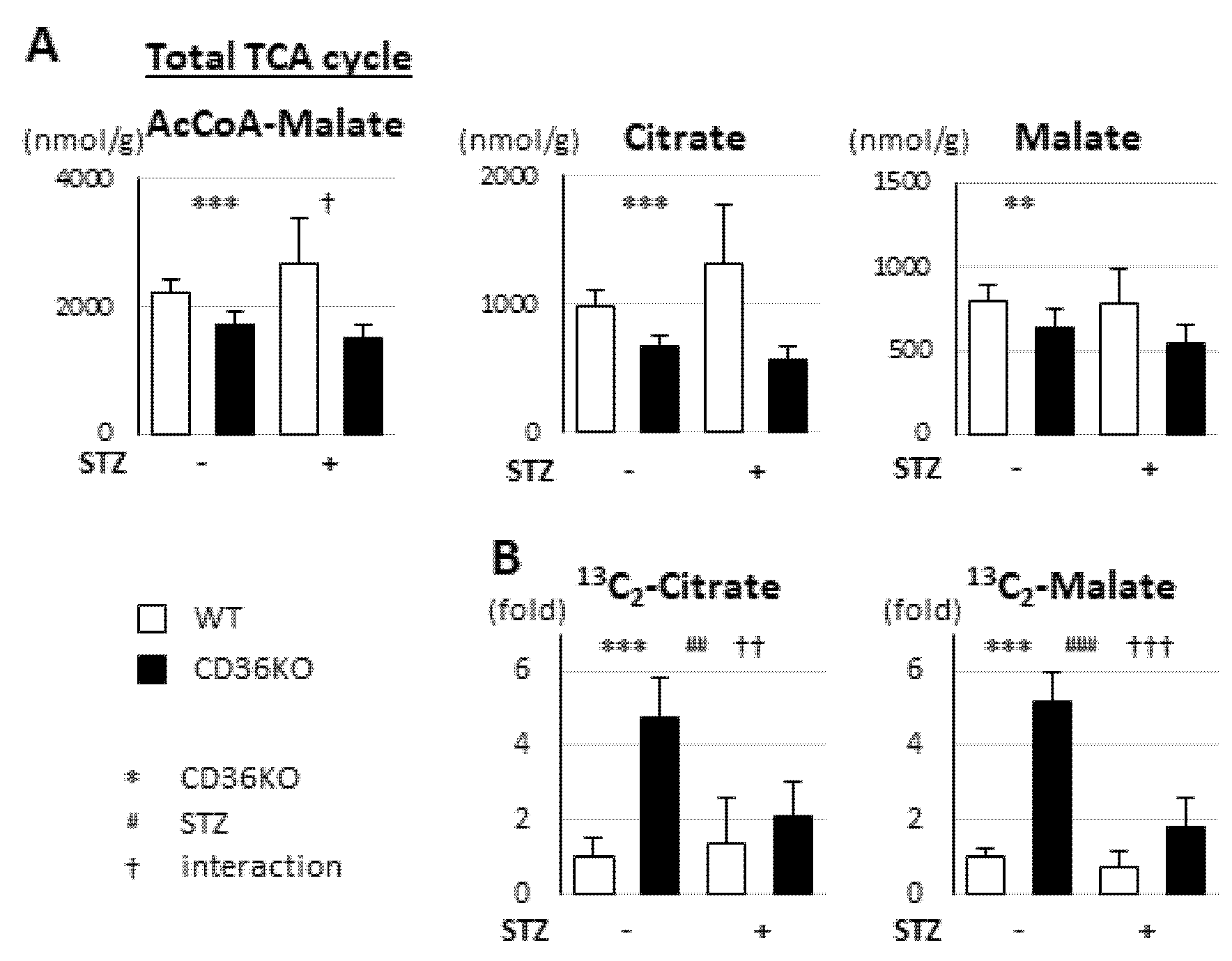
2.4. Reduced FA Uptake and Suppressed Glycolysis Diminish Total Energy Supply in CD36KO-STZ Hearts
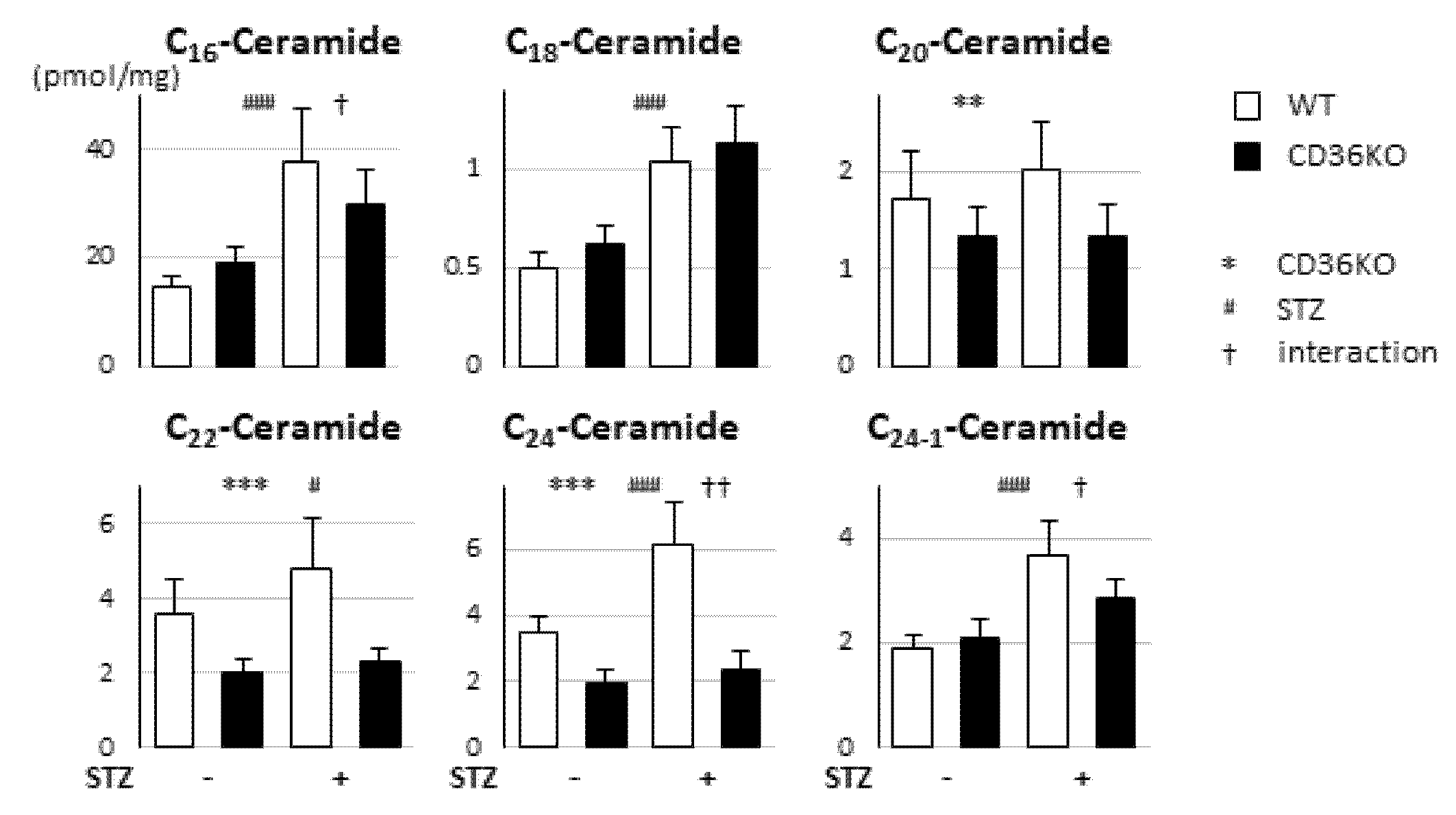
2.5. No Enhancement of Ceramide Levels in CD36KO-STZ Hearts Compared to WT-STZ Hearts
2.6. Synergistic Exacerbation of Contractile Dysfunction by Increased Workload in CD36KO-STZ Hearts
3. Discussion
3.1. Is CD36 a Detrimental Factor for Diabetic Cardiomyopathy In Vivo?
3.2. Association between Cardiac Metabolism and Contractile Function
4. Materials and Methods
4.1. Mice
4.2. STZ-Induced Diabetic Model, TAC and Sample Preparation
4.3. Cardiac Function and Hemodynamic Parameters
4.4. Measurement of Blood Metabolites and Glycogen in Hearts
4.5. Biodistribution of 125I-BMIPP (15-(p-iodophenyl)-3-(R,S)-methyl pentadecanoic acid) and 18F-FDG (2-fluorodeoxyglucose)
4.6. Metabolome Analysis by Capillary Electrophoresis–Mass Spectrometry
4.7. Tracing Study with 13C6-Glucose
4.8. Measurement of Ceramides
4.9. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2020, cvaa319. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Gibb, A.A.; Hill, B.G. Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ. Res. 2018, 123, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Clarke, K.; Levelt, E. Metabolic Remodeling in Diabetic Cardiomyopathy. Cardiovasc. Res. 2017, 113, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatz, J.F.C.; Luiken, J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, N.H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342. [Google Scholar] [CrossRef] [Green Version]
- Greenwalt, D.E.; Scheck, S.H.; Rhinehart-Jones, T. Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J. Clin. Investig. 1995, 96, 1382–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbarawan, Y.; Syamsunarno, M.; Koitabashi, N.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Sano, M.; Sunaga, H.; et al. Myocardial fatty acid uptake through CD36 is indispensable for sufficient bioenergetic metabolism to prevent progression of pressure overload-induced heart failure. Sci. Rep. 2018, 8, 12035. [Google Scholar] [CrossRef]
- Nagendran, J.; Pulinilkunnil, T.; Kienesberger, P.C.; Sung, M.M.; Fung, D.; Febbraio, M.; Dyck, J.R. Cardiomyocyte-specific ablation of CD36 improves post-ischemic functional recovery. J. Mol. Cell Cardiol. 2013, 63, 180–188. [Google Scholar] [CrossRef]
- Kim, T.T.; Dyck, J.R. The role of CD36 in the regulation of myocardial lipid metabolism. Biochim. Biophys. Acta 2016, 1861, 1450–1460. [Google Scholar] [CrossRef]
- Koonen, D.P.; Febbraio, M.; Bonnet, S.; Nagendran, J.; Young, M.E.; Michelakis, E.D.; Dyck, J.R. CD36 expression contributes to age-induced cardiomyopathy in mice. Circulation 2007, 116, 2139–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Kovacs, A.; Febbraio, M.; Finck, B.N.; Kelly, D.P. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ. Res. 2007, 100, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Umbarawan, Y.; Syamsunarno, M.; Koitabashi, N.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Nagahata-Naito, Y.; Obinata, H.; Sano, M.; Sunaga, H.; et al. Glucose is preferentially utilized for biomass synthesis in pressure-overloaded hearts: Evidence from fatty acid-binding protein-4 and -5 knockout mice. Cardiovasc. Res. 2018, 114, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Bonen, A.; Jain, S.S.; Snook, L.A.; Han, X.X.; Yoshida, Y.; Buddo, K.H.; Lally, J.S.; Pask, E.D.; Paglialunga, S.; Beaudoin, M.S.; et al. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 2015, 58, 2381–2391. [Google Scholar] [CrossRef] [Green Version]
- Sung, M.M.; Koonen, D.P.; Soltys, C.L.; Jacobs, R.L.; Febbraio, M.; Dyck, J.R. Increased CD36 expression in middle-aged mice contributes to obesity-related cardiac hypertrophy in the absence of cardiac dysfunction. J. Mol. Med. 2011, 89, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.J.; Arumugam, Y.; Bell, R.C.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Bonen, A. Changes in fatty acid transport and transporters are related to the severity of insulin deficiency. Am. J. Physiology. Endocrinol. Metab. 2002, 283, E612–E621. [Google Scholar] [CrossRef] [Green Version]
- Warren, J.S.; Oka, S.I.; Zablocki, D.; Sadoshima, J. Metabolic reprogramming via PPARalpha signaling in cardiac hypertrophy and failure: From metabolomics to epigenetics. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H584–H596. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Shen, W.J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-alpha. Future Cardiol. 2017, 13, 259–278. [Google Scholar] [CrossRef]
- Lee, T.I.; Kao, Y.H.; Tsai, W.C.; Chung, C.C.; Chen, Y.C.; Chen, Y.J. HDAC Inhibition Modulates Cardiac PPARs and Fatty Acid Metabolism in Diabetic Cardiomyopathy. PPAR Res. 2016, 2016, 5938740. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.I.; Kao, Y.H.; Chen, Y.C.; Tsai, W.C.; Chung, C.C.; Chen, Y.J. Cardiac metabolism, inflammation, and peroxisome proliferator-activated receptors modulated by 1,25-dihydroxyvitamin D3 in diabetic rats. Int. J. Cardiol. 2014, 176, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Young, M.E.; Guthrie, P.H.; Razeghi, P.; Leighton, B.; Abbasi, S.; Patil, S.; Youker, K.A.; Taegtmeyer, H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes 2002, 51, 2587–2595. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [Green Version]
- Depre, C.; Young, M.E.; Ying, J.; Ahuja, H.S.; Han, Q.; Garza, N.; Davies, P.J.; Taegtmeyer, H. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J. Mol. Cell Cardiol. 2000, 32, 985–996. [Google Scholar] [CrossRef]
- Umbarawan, Y.; Kawakami, R.; Syamsunarno, M.; Koitabashi, N.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Sunaga, H.; et al. Reduced fatty acid uptake aggravates cardiac contractile dysfunction in streptozotocin-induced diabetic cardiomyopathy. Sci. Rep. 2020, 10, 20809. [Google Scholar] [CrossRef]
- Iso, T.; Kurabayashi, M. Association between Cardiac Metabolism and Contractile Function in vivo in Mice with Reduced Fatty Acid Uptake. Metabolites 2021, in press. [Google Scholar]
- Putri, M.; Syamsunarno, M.R.; Iso, T.; Yamaguchi, A.; Hanaoka, H.; Sunaga, H.; Koitabashi, N.; Matsui, H.; Yamazaki, C.; Kameo, S.; et al. CD36 is indispensable for thermogenesis under conditions of fasting and cold stress. Biochem. Biophys. Res. Commun. 2015, 457, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Febbraio, M.; Abumrad, N.A.; Hajjar, D.P.; Sharma, K.; Cheng, W.; Pearce, S.F.; Silverstein, R.L. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J. Biol. Chem. 1999, 274, 19055–19062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbarawan, Y.; Syamsunarno, M.; Obinata, H.; Yamaguchi, A.; Sunaga, H.; Matsui, H.; Hishiki, T.; Matsuura, T.; Koitabashi, N.; Obokata, M.; et al. Robust suppression of cardiac energy catabolism with marked accumulation of energy substrates during lipopolysaccharide-induced cardiac dysfunction in mice. Metabolism 2017, 77, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Iso, T.; Maeda, K.; Hanaoka, H.; Suga, T.; Goto, K.; Syamsunarno, M.R.; Hishiki, T.; Nagahata, Y.; Matsui, H.; Arai, M.; et al. Capillary endothelial fatty acid binding proteins 4 and 5 play a critical role in fatty acid uptake in heart and skeletal muscle. Arter. Thromb. Vasc. Biol. 2013, 33, 2549–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, K.; Iso, T.; Hanaoka, H.; Yamaguchi, A.; Suga, T.; Hattori, A.; Irie, Y.; Shinagawa, Y.; Matsui, H.; Syamsunarno, M.R.; et al. Peroxisome proliferator-activated receptor-gamma in capillary endothelia promotes fatty acid uptake by heart during long-term fasting. J. Am. Heart Assoc. 2013, 2, e004861. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umbarawan, Y.; Kawakami, R.; Syamsunarno, M.R.A.A.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Koitabashi, N.; Sunaga, H.; et al. Reduced Fatty Acid Use from CD36 Deficiency Deteriorates Streptozotocin-Induced Diabetic Cardiomyopathy in Mice. Metabolites 2021, 11, 881. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11120881
Umbarawan Y, Kawakami R, Syamsunarno MRAA, Obinata H, Yamaguchi A, Hanaoka H, Hishiki T, Hayakawa N, Koitabashi N, Sunaga H, et al. Reduced Fatty Acid Use from CD36 Deficiency Deteriorates Streptozotocin-Induced Diabetic Cardiomyopathy in Mice. Metabolites. 2021; 11(12):881. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11120881
Chicago/Turabian StyleUmbarawan, Yogi, Ryo Kawakami, Mas Rizky A. A. Syamsunarno, Hideru Obinata, Aiko Yamaguchi, Hirofumi Hanaoka, Takako Hishiki, Noriyo Hayakawa, Norimichi Koitabashi, Hiroaki Sunaga, and et al. 2021. "Reduced Fatty Acid Use from CD36 Deficiency Deteriorates Streptozotocin-Induced Diabetic Cardiomyopathy in Mice" Metabolites 11, no. 12: 881. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11120881