
A Current Encyclopedia of Bioinformatics Tools, Data Formats and Resources for Mass Spectrometry Lipidomics

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Materials and Methods
3. Data Standards and Formats
4. Software for Lipid Identification from Mass Spectrometry
4.1. Targeted Workflow
4.2. Untargeted Workflow
4.3. Targeted and Untargeted Workflow
5. Data Post-Processing, Statistical Analysis, Visualization and Pathway Integration
6. Databases, Repositories and Other Resources
7. Discussion
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CCS | Collisional cross section |
| CE | Capillary electrophoresis |
| ChEBI | Chemical entities of biological interest |
| CSV | Comma-separated values, spreadsheet/table data format |
| CV | Controlled vocabulary |
| DDA | Data-dependent acquisition |
| DI | Direct infusion |
| DI-MS | Direct infusion/shotgun mass spectrometry |
| DIA | Data-independent acquisition |
| DTIMS | Drift tube ion mobility spectrometry |
| FAIMS | High-field asymmetric-waveform ion mobility spectrometry |
| FAIR | Findable, accessible, indexable and retrievable |
| FDR | False discovery rate |
| GC | Gas chromatography |
| GL | Glycerolipids |
| GO | Gene ontology |
| GP | Glycerophospholipids |
| HILIC | Hydrophilic interaction liquid chromatography |
| HMDB | Human metabolome database |
| HTML | Hypertext markup language |
| HUPO | Human proteome organization |
| IMS | Ion mobility spectrometry |
| LC | Liquid chromatography |
| LSI | Lipidomics standards initiative |
| MALDI | Matrix-assisted laser desorption/ionization |
| MRM | Multiple reaction monitoring |
| MS | Mass spectrometry or mass spectrum |
| MS/MS | Tandem mass spectrometry or mass spectrum |
| MS1 | First order mass spectrum, single fragmentation |
| MS2 | Second order mass spectrum, fragmentation of ions from MS1, MS/MS |
| MSE | DIA with alternating low- and high-energy collision-induced dissociation |
| MSI | Metabolomics standards initiative |
| MSn | Higher (nth) order mass spectrometry or mass spectrum |
| NMR | Nuclear magnetic resonance |
| PRIDE | Proteomics identification database |
| PRM | Parallel reaction monitoring |
| PSI | HUPO Proteomics standards initiative |
| QA | Quality Assurance |
| QC | Quality Control |
| RPLC | Reversed-phase liquid chromatography |
| RT | Retention time |
| SFC | Supercritical fluid chromatography |
| SMILES | Simplified molecular input line entry system |
| SPLASH | Spectral hash |
| SRM | Selective reaction monitoring |
| TOF | Time-of-flight |
| TWIMS | Traveling wave ion mobility spectrometry |
| TXT | Semi-structured, text-based file format |
| VBA | Visual Basic for Applications |
| XLSX | MS Excel spreadsheet format |
| XML | Extensible markup language |
References
- Züllig, T.; Trötzmüller, M.; Köfeler, H.C. Lipidomics from Sample Preparation to Data Analysis: A Primer. Anal. Bioanal. Chem. 2020, 412, 2191–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paglia, G.; Smith, A.J.; Astarita, G. Ion Mobility Mass Spectrometry in the Omics Era: Challenges and Opportunities for Metabolomics and Lipidomics. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- But Is the Code (Re)Usable? Nat. Comput. Sci. 2021, 1, 449. [CrossRef]
- Rampler, E.; Abiead, Y.E.; Schoeny, H.; Rusz, M.; Hildebrand, F.; Fitz, V.; Koellensperger, G. Recurrent Topics in Mass Spectrometry-Based Metabolomics and Lipidomics—Standardization, Coverage, and Throughput. Anal. Chem. 2021, 93, 519–545. [Google Scholar] [CrossRef]
- Köfeler, H.C.; Ahrends, R.; Baker, E.S.; Ekroos, K.; Han, X.; Hoffmann, N.; Holčapek, M.; Wenk, M.R.; Liebisch, G. Recommendations for Good Practice in MS-Based Lipidomics. J. Lipid Res. 2021, 62, 100138. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.E.; Aimo, L.; Bridge, A.J.; Clair, G.; Fedorova, M.; Helms, J.B.; Molenaar, M.R.; Ni, Z.; Orešič, M.; Slenter, D.; et al. Interpreting the Lipidome: Bioinformatic Approaches to Embrace the Complexity. Metabolomics 2021, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, K.; Misra, B.B. Software Tools, Databases and Resources in Metabolomics: Updates from 2018 to 2019. Metabolomics 2020, 16, 36. [Google Scholar] [CrossRef]
- Stanstrup, J.; Broeckling, C.D.; Helmus, R.; Hoffmann, N.; Mathé, E.; Naake, T.; Nicolotti, L.; Peters, K.; Rainer, J.; Salek, R.M.; et al. The MetaRbolomics Toolbox in Bioconductor and Beyond. Metabolites 2019, 9, 200. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, E.W.; Orchard, S.; Binz, P.-A.; Bittremieux, W.; Eisenacher, M.; Hermjakob, H.; Kawano, S.; Lam, H.; Mayer, G.; Menschaert, G.; et al. Proteomics Standards Initiative: Fifteen Years of Progress and Future Work. J. Proteome Res. 2017, 16, 4288–4298. [Google Scholar] [CrossRef]
- Liebisch, G.; Ahrends, R.; Arita, M.; Arita, M.; Bowden, J.A.; Ejsing, C.S.; Griffiths, W.J.; Holčapek, M.; Köfeler, H.; Mitchell, T.W.; et al. Lipidomics Needs More Standardization. Nat. Metab. 2019, 1, 745–747. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A Resource Evolving in Response to the Needs of Its Scientific Community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An International Repository for Metabolomics Data and Metadata, Metabolite Standards, Protocols, Tutorials and Training, and Analysis Tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Bai, M.; da Veiga Leprevost, F.; Squizzato, S.; Park, Y.M.; Haug, K.; Carroll, A.J.; Spalding, D.; Paschall, J.; Wang, M.; et al. Discovering and Linking Public Omics Data Sets Using the Omics Discovery Index. Nat. Biotechnol. 2017, 35, 406–409. [Google Scholar] [CrossRef] [Green Version]
- Mayer, G.; Müller, W.; Schork, K.; Uszkoreit, J.; Weidemann, A.; Wittig, U.; Rey, M.; Quast, C.; Felden, J.; Glöckner, F.O.; et al. Implementing FAIR Data Management within the German Network for Bioinformatics Infrastructure (de.NBI) Exemplified by Selected Use Cases. Brief. Bioinform. 2021, 22, bbab010. [Google Scholar] [CrossRef]
- Turewicz, M.; Kohl, M.; Ahrens, M.; Mayer, G.; Uszkoreit, J.; Naboulsi, W.; Bracht, T.; Megger, D.A.; Sitek, B.; Marcus, K.; et al. BioInfra.Prot: A Comprehensive Proteomics Workflow Including Data Standardization, Protein Inference, Expression Analysis and Data Publication. J. Biotechnol. 2017, 261, 116–125. [Google Scholar] [CrossRef]
- Martínez-Bartolomé, S.; Binz, P.-A.; Albar, J.P. The Minimal Information About a Proteomics Experiment (MIAPE) from the Proteomics Standards Initiative. In Plant Proteomics: Methods and Protocols; Jorrin-Novo, J.V., Komatsu, S., Weckwerth, W., Wienkoop, S., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; pp. 765–780. ISBN 978-1-62703-631-3. [Google Scholar]
- Deutsch, E.W.; Albar, J.P.; Binz, P.-A.; Eisenacher, M.; Jones, A.R.; Mayer, G.; Omenn, G.S.; Orchard, S.; Vizcaíno, J.A.; Hermjakob, H. Development of Data Representation Standards by the Human Proteome Organization Proteomics Standards Initiative. J. Am. Med. Inf. Assoc. 2015, 22, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Mayer, G.; Jones, A.R.; Binz, P.-A.; Deutsch, E.W.; Orchard, S.; Montecchi-Palazzi, L.; Vizcaíno, J.A.; Hermjakob, H.; Oveillero, D.; Julian, R.; et al. Controlled Vocabularies and Ontologies in Proteomics: Overview, Principles and Practice. Biochim. Biophys. Acta 2014, 1844, 98–107. [Google Scholar] [CrossRef]
- Martens, L.; Chambers, M.; Sturm, M.; Kessner, D.; Levander, F.; Shofstahl, J.; Tang, W.H.; Römpp, A.; Neumann, S.; Pizarro, A.D.; et al. MzML—A Community Standard for Mass Spectrometry Data. Mol. Cell. Proteom. 2011, 10, R110.000133. [Google Scholar] [CrossRef] [Green Version]
- Turewicz, M.; Deutsch, E.W. Spectra, Chromatograms, Metadata: MzML-the Standard Data Format for Mass Spectrometer Output. Methods Mol. Biol. 2011, 696, 179–203. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.R.; Eisenacher, M.; Mayer, G.; Kohlbacher, O.; Siepen, J.; Hubbard, S.J.; Selley, J.N.; Searle, B.C.; Shofstahl, J.; Seymour, S.L.; et al. The MzIdentML Data Standard for Mass Spectrometry-Based Proteomics Results. Mol. Cell. Proteom. 2012, 11, M111-014381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizcaíno, J.A.; Mayer, G.; Perkins, S.; Barsnes, H.; Vaudel, M.; Perez-Riverol, Y.; Ternent, T.; Uszkoreit, J.; Eisenacher, M.; Fischer, L.; et al. The MzIdentML Data Standard Version 1.2, Supporting Advances in Proteome Informatics. Mol. Cell. Proteom. 2017, 16, 1275–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walzer, M.; Qi, D.; Mayer, G.; Uszkoreit, J.; Eisenacher, M.; Sachsenberg, T.; Gonzalez-Galarza, F.F.; Fan, J.; Bessant, C.; Deutsch, E.W.; et al. The MzQuantML Data Standard for Mass Spectrometry-Based Quantitative Studies in Proteomics. Mol. Cell Proteom. 2013, 12, 2332–2340. [Google Scholar] [CrossRef] [Green Version]
- ARömpp, A.; Schramm, T.; Hester, A.; Klinkert, I.; Both, J.P.; Heeren, R.; Stöckli, M.; Spengler, B. ImzML: Imaging Mass Spectrometry Markup Language: A Common Data Format for Mass Spectrometry Imaging. Methods Mol. Biol. 2011, 696, 205–224. [Google Scholar] [CrossRef]
- Mayer, G.; Montecchi-Palazzi, L.; Ovelleiro, D.; Jones, A.R.; Binz, P.-A.; Deutsch, E.W.; Chambers, M.; Kallhardt, M.; Levander, F.; Shofstahl, J.; et al. The HUPO Proteomics Standards Initiative- Mass Spectrometry Controlled Vocabulary. Database 2013, 2013, bat009. [Google Scholar] [CrossRef]
- Ghali, F.; Krishna, R.; Lukasse, P.; Martínez-Bartolomé, S.; Reisinger, F.; Hermjakob, H.; Vizcaíno, J.A.; Jones, A.R. Tools (Viewer, Library and Validator) That Facilitate Use of the Peptide and Protein Identification Standard Format, Termed MzIdentML. Mol. Cell. Proteom. 2013, 12, 3026–3035. [Google Scholar] [CrossRef] [Green Version]
- Griss, J.; Jones, A.R.; Sachsenberg, T.; Walzer, M.; Gatto, L.; Hartler, J.; Thallinger, G.G.; Salek, R.M.; Steinbeck, C.; Neuhauser, N.; et al. The MzTab Data Exchange Format: Communicating Mass-Spectrometry-Based Proteomics and Metabolomics Experimental Results to a Wider Audience. Mol. Cell. Proteom. 2014, 13, 2765–2775. [Google Scholar] [CrossRef] [Green Version]
- Sansone, S.-A.; Fan, T.; Goodacre, R.; Griffin, J.L.; Hardy, N.W.; Kaddurah-Daouk, R.; Kristal, B.S.; Lindon, J.; Mendes, P.; Morrison, N.; et al. The Metabolomics Standards Initiative. Nat. Biotechnol. 2007, 25, 846–848. [Google Scholar] [CrossRef]
- Spicer, R.A.; Salek, R.; Steinbeck, C. Compliance with Minimum Information Guidelines in Public Metabolomics Repositories. Sci. Data 2017, 4, 170137. [Google Scholar] [CrossRef] [Green Version]
- Rocca-Serra, P.; Salek, R.M.; Arita, M.; Correa, E.; Dayalan, S.; Gonzalez-Beltran, A.; Ebbels, T.; Goodacre, R.; Hastings, J.; Haug, K.; et al. Data Standards Can Boost Metabolomics Research, and If There Is a Will, There Is a Way. Metabolomics 2016, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salek, R.M.; Neumann, S.; Schober, D.; Hummel, J.; Billiau, K.; Kopka, J.; Correa, E.; Reijmers, T.; Rosato, A.; Tenori, L.; et al. COordination of Standards in MetabOlomicS (COSMOS): Facilitating Integrated Metabolomics Data Access. Metabolomics 2015, 11, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, N.; Rein, J.; Sachsenberg, T.; Hartler, J.; Haug, K.; Mayer, G.; Alka, O.; Dayalan, S.; Pearce, J.T.M.; Rocca-Serra, P.; et al. MzTab-M: A Data Standard for Sharing Quantitative Results in Mass Spectrometry Metabolomics. Anal. Chem. 2019, 91, 3302–3310. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.D.; Moseley, H.N.B. The Mwtab Python Library for RESTful Access and Enhanced Quality Control, Deposition, and Curation of the Metabolomics Workbench Data Repository. Metabolites 2021, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Sansone, S.-A.; Rocca-Serra, P.; Brandizi, M.; Brazma, A.; Field, D.; Fostel, J.; Garrow, A.G.; Gilbert, J.; Goodsaid, F.; Hardy, N.; et al. The First RSBI (ISA-TAB) Workshop: “Can a Simple Format Work for Complex Studies?”. OMICS 2008, 12, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocca-Serra, P.; Brandizi, M.; Maguire, E.; Sklyar, N.; Taylor, C.; Begley, K.; Field, D.; Harris, S.; Hide, W.; Hofmann, O.; et al. ISA Software Suite: Supporting Standards-Compliant Experimental Annotation and Enabling Curation at the Community Level. Bioinformatics 2010, 26, 2354–2356. [Google Scholar] [CrossRef] [PubMed]
- Psaroudakis, D.; Liu, F.; König, P.; Scholz, U.; Junker, A.; Lange, M.; Arend, D. Isa4j: A Scalable Java Library for Creating ISA-Tab Metadata. F1000Research 2020, 9, ELIXIR-1388. [Google Scholar] [CrossRef]
- Hoffmann, N.; Hartler, J.; Ahrends, R. JmzTab-M: A Reference Parser, Writer, and Validator for the Proteomics Standards Initiative MzTab 2.0 Metabolomics Standard. Anal. Chem. 2019, 91, 12615–12618. [Google Scholar] [CrossRef]
- O’Donnell, V.B.; FitzGerald, G.A.; Murphy, R.C.; Liebisch, G.; Dennis, E.A.; Quehenberger, O.; Subramaniam, S.; Wakelam, M.J.O. Steps Toward Minimal Reporting Standards for Lipidomics Mass Spectrometry in Biomedical Research Publications. Circ. Genom. Precis. Med. 2020, 13, e003019. [Google Scholar] [CrossRef]
- Stein, S.E.; Scott, D.R. Optimization and Testing of Mass Spectral Library Search Algorithms for Compound Identification. J. Am. Soc. Mass Spectrom. 1994, 5, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, Ş.; Vandermarliere, E.; Martens, L. Methods to Calculate Spectrum Similarity. In Proteome Bioinformatics; Keerthikumar, S., Mathivanan, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 75–100. ISBN 978-1-4939-6740-7. [Google Scholar]
- McDonald, W.H.; Tabb, D.L.; Sadygov, R.G.; MacCoss, M.J.; Venable, J.; Graumann, J.; Johnson, J.R.; Cociorva, D.; Yates, J.R., III. MS1, MS2, and SQT—Three Unified, Compact, and Easily Parsed File Formats for the Storage of Shotgun Proteomic Spectra and Identifications. Rapid Commun. Mass Spectrom. 2004, 18, 2162–2168. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.G.; Paton, N.W.; Taylor, C.F. A Common Open Representation of Mass Spectrometry Data and Its Application to Proteomics Research. Nat. Biotechnol. 2004, 22, 1459–1466. [Google Scholar]
- Orchard, S.; Montechi-Palazzi, L.; Deutsch, E.W.; Binz, P.-A.; Jones, A.R.; Paton, N.; Pizarro, A.; Creasy, D.M.; Wojcik, J.; Hermjakob, H. Five Years of Progress in the Standardization of Proteomics Data 4th Annual Spring Workshop of the HUPO-Proteomics Standards Initiative April 23–25, 2007 Ecole Nationale Supérieure (ENS), Lyon, France. Proteomics 2007, 7, 3436–3440. [Google Scholar] [CrossRef] [PubMed]
- Haimi, P.; Uphoff, A.; Hermansson, M.; Somerharju, P. Software Tools for Analysis of Mass Spectrometric Lipidome Data. Anal. Chem. 2006, 78, 8324–8331. [Google Scholar] [CrossRef] [PubMed]
- Haimi, P.; Chaithanya, K.; Kainu, V.; Hermansson, M.; Somerharju, P. Instrument-Independent Software Tools for the Analysis of MS-MS and LC-MS Lipidomics Data. Methods Mol. Biol. 2009, 580, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Marepally, S.R.; Nune, D.S.; Pallakollu, P.; Ragan, G.; Roth, M.R.; Wang, L.; Lushington, G.H.; Visvanathan, M.; Welti, R. LipidomeDB Data Calculation Environment: Online Processing of Direct-Infusion Mass Spectral Data for Lipid Profiles. Lipids 2011, 46, 879–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruehan, C.; Johnson, D.; Welti, R. LipidomeDB Data Calculation Environment Has Been Updated to Process Direct-Infusion Multiple Reaction Monitoring Data. Lipids 2018, 53, 1019–1020. [Google Scholar] [CrossRef]
- Wolrab, D.; Cífková, E.; Čáň, P.; Lísa, M.; Peterka, O.; Chocholoušková, M.; Jirásko, R.; Holčapek, M. LipidQuant 1.0: Automated Data Processing in Lipid Class Separation-Mass Spectrometry Quantitative Workflows. Bioinformatics 2021, 37, 4591–4592. [Google Scholar] [CrossRef]
- Pauling, J.K.; Hermansson, M.; Hartler, J.; Christiansen, K.; Gallego, S.F.; Peng, B.; Ahrends, R.; Ejsing, C.S. Proposal for a Common Nomenclature for Fragment Ions in Mass Spectra of Lipids. PLoS ONE 2017, 12, e0188394. [Google Scholar] [CrossRef] [Green Version]
- Husen, P.; Tarasov, K.; Katafiasz, M.; Sokol, E.; Vogt, J.; Baumgart, J.; Nitsch, R.; Ekroos, K.; Ejsing, C.S. Analysis of Lipid Experiments (ALEX): A Software Framework for Analysis of High-Resolution Shotgun Lipidomics Data. PLoS ONE 2013, 8, e79736. [Google Scholar] [CrossRef] [Green Version]
- Kochen, M.A.; Chambers, M.C.; Holman, J.D.; Nesvizhskii, A.I.; Weintraub, S.T.; Belisle, J.T.; Islam, M.N.; Griss, J.; Tabb, D.L. Greazy: Open-Source Software for Automated Phospholipid Tandem Mass Spectrometry Identification. Anal. Chem. 2016, 88, 5733–5741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kind, T.; Liu, K.-H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in Silico Tandem Mass Spectrometry Database for Lipid Identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kind, T.; Okazaki, Y.; Saito, K.; Fiehn, O. LipidBlast Templates as Flexible Tools for Creating New In-Silico Tandem Mass Spectral Libraries. Anal. Chem. 2014, 86, 11024–11027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cajka, T.; Fiehn, O. LC-MS-Based Lipidomics and Automated Identification of Lipids Using the LipidBlast In-Silico MS/MS Library. Methods Mol. Biol. 2017, 1609, 149–170. [Google Scholar] [CrossRef]
- Hutchins, P.D.; Russell, J.D.; Coon, J.J. LipiDex: An Integrated Software Package for High-Confidence Lipid Identification. Cell Syst. 2018, 6, 621–625.e5. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, A.; Brasher, C.J.; Slatter, D.A.; Meckelmann, S.W.; Hawksworth, J.I.; Allen, S.M.; O’Donnell, V.B. LipidFinder: A Computational Workflow for Discovery of Lipids Identifies Eicosanoid-Phosphoinositides in Platelets. JCI Insight 2017, 2, e91634. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Alvarez-Jarreta, J.; Brasher, C.J.; Nguyen, A.; Hawksworth, J.I.; Rodrigues, P.; Meckelmann, S.; Allen, S.M.; O’Donnell, V.B. LipidFinder on LIPID MAPS: Peak Filtering, MS Searching and Statistical Analysis for Lipidomics. Bioinformatics 2019, 35, 685–687. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Jarreta, J.; Rodrigues, P.R.S.; Fahy, E.; O’Connor, A.; Price, A.; Gaud, C.; Andrews, S.; Benton, P.; Siuzdak, G.; Hawksworth, J.I.; et al. LipidFinder 2.0: Advanced Informatics Pipeline for Lipidomics Discovery Applications. Bioinformatics 2021, 37, 1478–1479. [Google Scholar] [CrossRef]
- Ni, Z.; Angelidou, G.; Lange, M.; Hoffmann, R.; Fedorova, M. LipidHunter Identifies Phospholipids by High-Throughput Processing of LC-MS and Shotgun Lipidomics Datasets. Anal. Chem. 2017, 89, 8800–8807. [Google Scholar] [CrossRef]
- Zhou, Z.; Shen, X.; Chen, X.; Tu, J.; Xiong, X.; Zhu, Z.-J. LipidIMMS Analyzer: Integrating Multi-Dimensional Information to Support Lipid Identification in Ion Mobility-Mass Spectrometry Based Lipidomics. Bioinformatics 2019, 35, 698–700. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, Z.; Zhu, Z.-J. The Use of LipidIMMS Analyzer for Lipid Identification in Ion Mobility-Mass Spectrometry-Based Untargeted Lipidomics. Methods Mol. Biol. 2020, 2084, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Koelmel, J.P.; Kroeger, N.M.; Ulmer, C.Z.; Bowden, J.A.; Patterson, R.E.; Cochran, J.A.; Beecher, C.W.W.; Garrett, T.J.; Yost, R.A. LipidMatch: An Automated Workflow for Rule-Based Lipid Identification Using Untargeted High-Resolution Tandem Mass Spectrometry Data. BMC Bioinform. 2017, 18, 331. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Zhang, Q.; Gao, X.; Wu, S.; Lin, G. LipidMiner: A Software for Automated Identification and Quantification of Lipids from Multiple Liquid Chromatography-Mass Spectrometry Data Files. Rapid Commun. Mass Spectrom. 2014, 28, 981–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Z.; Mayr, M.; Zeeshan, S.; Dandekar, T.; Mueller, M.J.; Fekete, A. Lipid-Pro: A Computational Lipid Identification Solution for Untargeted Lipidomics on Data-Independent Acquisition Tandem Mass Spectrometry Platforms. Bioinformatics 2015, 31, 1150–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, R.; Schuhmann, K.; Schwudke, D.; Sampaio, J.L.; Bornstein, S.R.; Schroeder, M.; Shevchenko, A. LipidXplorer: A Software for Consensual Cross-Platform Lipidomics. PLoS ONE 2012, 7, e29851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, R.; Schwudke, D.; Shevchenko, A. LipidXplorer: Software for Quantitative Shotgun Lipidomics Compatible with Multiple Mass Spectrometry Platforms. Curr. Protoc. Bioinform. 2013, 43, 14.12.1–14.12.30. [Google Scholar] [CrossRef]
- Ross, D.H.; Cho, J.H.; Zhang, R.; Hines, K.M.; Xu, L. LiPydomics: A Python Package for Comprehensive Prediction of Lipid Collision Cross Sections and Retention Times and Analysis of Ion Mobility-Mass Spectrometry-Based Lipidomics Data. Anal. Chem. 2020, 92, 14967–14975. [Google Scholar] [CrossRef]
- Kyle, J.E.; Crowell, K.L.; Casey, C.P.; Fujimoto, G.M.; Kim, S.; Dautel, S.E.; Smith, R.D.; Payne, S.H.; Metz, T.O. LIQUID: An-Open Source Software for Identifying Lipids in LC-MS/MS-Based Lipidomics Data. Bioinformatics 2017, 33, 1744–1746. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.R.; Edwards, B.R.; Fredricks, H.F.; Van Mooy, B.A.S. LOBSTAHS: An Adduct-Based Lipidomics Strategy for Discovery and Identification of Oxidative Stress Biomarkers. Anal. Chem. 2016, 88, 7154–7162. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Bond, N.J.; Koulman, A.; Griffin, J.L.; Hall, Z. MassPix: An R Package for Annotation and Interpretation of Mass Spectrometry Imaging Data for Lipidomics. Metabolomics 2017, 13, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-Independent MS/MS Deconvolution for Comprehensive Metabolome Analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A Lipidome Atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Benton, H.P.; Wong, D.M.; Trauger, S.A.; Siuzdak, G. XCMS2: Processing Tandem Mass Spectrometry Data for Metabolite Identification and Structural Characterization. Anal. Chem. 2008, 80, 6382–6389. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Ahrends, R. Adaptation of Skyline for Targeted Lipidomics. J. Proteome Res. 2016, 15, 291–301. [Google Scholar] [CrossRef]
- Peng, B.; Kopczynski, D.; Pratt, B.S.; Ejsing, C.S.; Burla, B.; Hermansson, M.; Benke, P.I.; Tan, S.H.; Chan, M.Y.; Torta, F.; et al. LipidCreator Workbench to Probe the Lipidomic Landscape. Nat. Commun. 2020, 11, 2057. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Ulmer, C.Z.; Koelmel, J.P.; Ragland, J.M.; Garrett, T.J.; Bowden, J.A. LipidPioneer: A Comprehensive User-Generated Exact Mass Template for Lipidomics. J. Am. Soc. Mass Spectrom. 2017, 28, 562–565. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Hsu, F.-F.; Ladenson, J.; Turk, J. Algorithm for Processing Raw Mass Spectrometric Data to Identify and Quantitate Complex Lipid Molecular Species in Mixtures by Data-Dependent Scanning and Fragment Ion Database Searching. J. Am. Soc. Mass Spectrom. 2007, 18, 1848–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goracci, L.; Tortorella, S.; Tiberi, P.; Pellegrino, R.M.; Di Veroli, A.; Valeri, A.; Cruciani, G. Lipostar, a Comprehensive Platform-Neutral Cheminformatics Tool for Lipidomics. Anal. Chem. 2017, 89, 6257–6264. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, S.; Tiberi, P.; Bowman, A.P.; Claes, B.S.R.; Ščupáková, K.; Heeren, R.M.A.; Ellis, S.R.; Cruciani, G. LipostarMSI: Comprehensive, Vendor-Neutral Software for Visualization, Data Analysis, and Automated Molecular Identification in Mass Spectrometry Imaging. J. Am. Soc. Mass Spectrom. 2020, 31, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Kutuzova, S.; Colaianni, P.; Röst, H.; Sachsenberg, T.; Alka, O.; Kohlbacher, O.; Burla, B.; Torta, F.; Schrübbers, L.; Kristensen, M.; et al. SmartPeak Automates Targeted and Quantitative Metabolomics Data Processing. Anal. Chem. 2020, 92, 15968–15974. [Google Scholar] [CrossRef]
- Röst, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A Flexible Open-Source Software Platform for Mass Spectrometry Data Analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef]
- Martano, G.; Leone, M.; D’Oro, P.; Matafora, V.; Cattaneo, A.; Masseroli, M.; Bachi, A. SMfinder: Small Molecules Finder for Metabolomics and Lipidomics Analysis. Anal. Chem. 2020, 92, 8874–8882. [Google Scholar] [CrossRef]
- Hastings, J.; de Matos, P.; Dekker, A.; Ennis, M.; Harsha, B.; Kale, N.; Muthukrishnan, V.; Owen, G.; Turner, S.; Williams, M.; et al. The ChEBI Reference Database and Ontology for Biologically Relevant Chemistry: Enhancements for 2013. Nucleic. Acids Res. 2013, 41, D456–D463. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 Years and Still GOing Strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Baker, C.J.; Kanagasabai, R.; Ang, W.T.; Veeramani, A.; Low, H.-S.; Wenk, M.R. Towards Ontology-Driven Navigation of the Lipid Bibliosphere. BMC Bioinform. 2008, 9, S5. [Google Scholar] [CrossRef] [Green Version]
- Chepelev, L.L.; Riazanov, A.; Kouznetsov, A.; Low, H.S.; Dumontier, M.; Baker, C.J.O. Prototype Semantic Infrastructure for Automated Small Molecule Classification and Annotation in Lipidomics. BMC Bioinform. 2011, 12, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.; Low, H.S.; Zhou, H.; Wenk, M.R.; Wong, L. LipidGO: Database for Lipid-Related GO Terms and Applications. Bioinformatics 2014, 30, 1043–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J. Cheminformatics 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clair, G.; Reehl, S.; Stratton, K.G.; Monroe, M.E.; Tfaily, M.M.; Ansong, C.; Kyle, J.E. Lipid Mini-On: Mining and Ontology Tool for Enrichment Analysis of Lipidomic Data. Bioinformatics 2019, 35, 4507–4508. [Google Scholar] [CrossRef]
- Molenaar, M.R.; Jeucken, A.; Wassenaar, T.A.; van de Lest, C.H.A.; Brouwers, J.F.; Helms, J.B. LION/Web: A Web-Based Ontology Enrichment Tool for Lipidomic Data Analysis. Gigascience 2019, 8, giz061. [Google Scholar] [CrossRef] [Green Version]
- More, P.; Bindila, L.; Wild, P.; Andrade-Navarro, M.; Fontaine, J.-F. LipiDisease: Associate Lipids to Diseases Using Literature Mining. Bioinformatics 2021, 37, 3981–3982. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Flight, R.M.; Moseley, H.N.B. Deriving Lipid Classification Based on Molecular Formulas. Metabolites 2020, 10, 122. [Google Scholar] [CrossRef] [Green Version]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Ehmki, E.S.R.; Schmidt, R.; Ohm, F.; Rarey, M. Comparing Molecular Patterns Using the Example of SMARTS: Applications and Filter Collection Analysis. J. Chem. Inf. Model. 2019, 59, 2572–2586. [Google Scholar] [CrossRef]
- Taylor, R.; Miller, R.H.; Miller, R.D.; Porter, M.; Dalgleish, J.; Prince, J.T. Automated Structural Classification of Lipids by Machine Learning. Bioinformatics 2015, 31, 621–625. [Google Scholar] [CrossRef] [Green Version]
- Gaud, C.; Sousa, B.C.; Nguyen, A.; Fedorova, M.; Ni, Z.; O’Donnell, V.B.; Wakelam, M.J.O.; Andrews, S.; Lopez-Clavijo, A.F. BioPAN: A Web-Based Tool to Explore Mammalian Lipidome Metabolic Pathways on LIPID MAPS. F1000Res 2021, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Kopczynski, D.; Hoffmann, N.; Peng, B.; Ahrends, R. Goslin: A Grammar of Succinct Lipid Nomenclature. Anal. Chem. 2020, 92, 10957–10960. [Google Scholar] [CrossRef] [PubMed]
- Kopczynski, D.; Hoffmann, N.; Peng, B.; Liebisch, G.; Spener, F.; Ahrends, R. Goslin 2.0 Implements the Recent Lipid Shorthand Nomenclature for MS-Derived Lipid Structures. Anal. Chem. 2022, 94, 6097–6101. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: LIPID MAPS Structure Database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aimo, L.; Liechti, R.; Hyka-Nouspikel, N.; Niknejad, A.; Gleizes, A.; Götz, L.; Kuznetsov, D.; David, F.P.A.; van der Goot, F.G.; Riezman, H.; et al. The SwissLipids Knowledgebase for Lipid Biology. Bioinformatics 2015, 31, 2860–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Z.; Fedorova, M. LipidLynxX: A Data Transfer Hub to Support Integration of Large Scale Lipidomics Datasets. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Subramaniam, S. RefMet: A Reference Nomenclature for Metabolomics. Nat. Methods 2020, 17, 1173–1174. [Google Scholar] [CrossRef]
- Gao, L.; Ji, S.; Burla, B.; Wenk, M.R.; Torta, F.; Cazenave-Gassiot, A. LICAR: An Application for Isotopic Correction of Targeted Lipidomic Data Acquired with Class-Based Chromatographic Separations Using Multiple Reaction Monitoring. Anal. Chem. 2021, 93, 3163–3171. [Google Scholar] [CrossRef]
- Mohamed, A.; Molendijk, J.; Hill, M.M. Lipidr: A Software Tool for Data Mining and Analysis of Lipidomics Datasets. J. Proteome Res. 2020, 19, 2890–2897. [Google Scholar] [CrossRef]
- Mohamed, A.; Hill, M.M. LipidSuite: Interactive Web Server for Lipidomics Differential and Enrichment Analysis. Nucleic Acids Res. 2021, 49, W346–W351. [Google Scholar] [CrossRef]
- Manzini, S.; Busnelli, M.; Colombo, A.; Kiamehr, M.; Chiesa, G. Liputils: A Python Module to Manage Individual Fatty Acid Moieties from Complex Lipids. Sci. Rep. 2020, 10, 13368. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Lerno, L.A.; German, J.B.; Lebrilla, C.B. Method for the Identification of Lipid Classes Based on Referenced Kendrick Mass Analysis. Anal. Chem. 2010, 82, 4236–4245. [Google Scholar] [CrossRef] [Green Version]
- Korf, A.; Vosse, C.; Schmid, R.; Helmer, P.O.; Jeck, V.; Hayen, H. Three-Dimensional Kendrick Mass Plots as a Tool for Graphical Lipid Identification. Rapid Commun. Mass Spectrom. 2018, 32, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Marella, C.; Torda, A.E.; Schwudke, D. The LUX Score: A Metric for Lipidome Homology. PLoS Comput. Biol. 2015, 11, e1004511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggers, L.F.; Müller, J.; Marella, C.; Scholz, V.; Watz, H.; Kugler, C.; Rabe, K.F.; Goldmann, T.; Schwudke, D. Lipidomes of Lung Cancer and Tumour-Free Lung Tissues Reveal Distinct Molecular Signatures for Cancer Differentiation, Age, Inflammation, and Pulmonary Emphysema. Sci. Rep. 2017, 7, 11087. [Google Scholar] [CrossRef] [Green Version]
- Wohlgemuth, G.; Mehta, S.S.; Mejia, R.F.; Neumann, S.; Pedrosa, D.; Pluskal, T.; Schymanski, E.L.; Willighagen, E.L.; Wilson, M.; Wishart, D.S.; et al. SPLASH, a Hashed Identifier for Mass Spectra. Nat. Biotechnol. 2016, 34, 1099–1101. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS Comprehensive Classification System for Lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS Online Tools for Lipid Research. Nucleic Acids Res. 2007, 35, W606–W612. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, V.B.; Dennis, E.A.; Wakelam, M.J.O.; Subramaniam, S. LIPID MAPS: Serving the next Generation of Lipid Researchers with Tools, Resources, Data, and Training. Sci. Signal. 2019, 12, eaaw2964. [Google Scholar] [CrossRef] [Green Version]
- Liebisch, G.; Vizcaíno, J.A.; Köfeler, H.; Trötzmüller, M.; Griffiths, W.J.; Schmitz, G.; Spener, F.; Wakelam, M.J.O. Shorthand Notation for Lipid Structures Derived from Mass Spectrometry. J. Lipid. Res. 2013, 54, 1523–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, J.M.; Moreno, P.; Fabregat, A.; Hermjakob, H.; Steinbeck, C.; Apweiler, R.; Wakelam, M.J.O.; Vizcaíno, J.A. LipidHome: A Database of Theoretical Lipids Optimized for High Throughput Mass Spectrometry Lipidomics. PLoS ONE 2013, 8, e61951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, P.; Morgat, A.; Axelsen, K.B.; Muthukrishnan, V.; Coudert, E.; Aimo, L.; Hyka-Nouspikel, N.; Gasteiger, E.; Kerhornou, A.; Neto, T.B.; et al. Rhea, the Reaction Knowledgebase in 2022. Nucleic Acids Res. 2022, 50, D693–D700. [Google Scholar] [CrossRef]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Köfeler, H.; et al. Update on LIPID MAPS Classification, Nomenclature, and Shorthand Notation for MS-Derived Lipid Structures. J. Lipid Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The Human Metabolome Database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A Public Repository for Sharing Mass Spectral Data for Life Sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Leaptrot, K.L.; May, J.C.; Dodds, J.N.; McLean, J.A. Ion Mobility Conformational Lipid Atlas for High Confidence Lipidomics. Nat. Commun. 2019, 10, 985. [Google Scholar] [CrossRef]
- Zheng, X.; Aly, N.A.; Zhou, Y.; Dupuis, K.T.; Bilbao, A.; Paurus, V.L.; Orton, D.J.; Wilson, R.; Payne, S.H.; Smith, R.D.; et al. A Structural Examination and Collision Cross Section Database for over 500 Metabolites and Xenobiotics Using Drift Tube Ion Mobility Spectrometry. Chem. Sci. 2017, 8, 7724–7736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ara, T.; Enomoto, M.; Arita, M.; Ikeda, C.; Kera, K.; Yamada, M.; Nishioka, T.; Ikeda, T.; Nihei, Y.; Shibata, D.; et al. Metabolonote: A Wiki-Based Database for Managing Hierarchical Metadata of Metabolome Analyses. Front. Bioeng. Biotechnol. 2015, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haug, K.; Salek, R.M.; Steinbeck, C. Global Open Data Management in Metabolomics. Curr. Opin. Chem. Biol. 2017, 36, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.; Phapale, P.; Chernyavsky, I.; Lavigne, R.; Fay, D.; Tarasov, A.; Kovalev, V.; Fuchser, J.; Nikolenko, S.; Pineau, C.; et al. FDR-Controlled Metabolite Annotation for High-Resolution Imaging Mass Spectrometry. Nat. Methods 2017, 14, 57–60. [Google Scholar] [CrossRef]
- Nishi, A.; Ohbuchi, K.; Kaifuchi, N.; Shimobori, C.; Kushida, H.; Yamamoto, M.; Kita, Y.; Tokuoka, S.M.; Yachie, A.; Matsuoka, Y.; et al. LimeMap: A Comprehensive Map of Lipid Mediator Metabolic Pathways. NPJ Syst. Biol. Appl. 2021, 7, 1–6. [Google Scholar] [CrossRef]
- Christie, W.W. The LipidWeb. Available online: https://lipidmaps.org/resources/lipidweb/lipidweb_html/index.html (accessed on 15 February 2022).
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Data Analysis, Machine Learning and Applications; Preisach, C., Burkhardt, H., Schmidt-Thieme, L., Decker, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 319–326. [Google Scholar]
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable Data Analysis with Snakemake. F1000Research 2021, 10, PMC8114187. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow Enables Reproducible Computational Workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Amstutz, P.; Crusoe, M.R.; Tijanić, N.; Chapman, B.; Chilton, J.; Heuer, M.; Kartashov, A.; Leehr, D.; Ménager, H.; Nedeljkovich, M.; et al. Common Workflow Language, version 1.0; Figshare; Digital Science: London, UK, 2016. [Google Scholar] [CrossRef]
- Eisenacher, M.; Kohl, M.; Turewicz, M.; Koch, M.-H.; Uszkoreit, J.; Stephan, C. Search and Decoy: The Automatic Identification of Mass Spectra. Methods Mol. Biol. 2012, 893, 445–488. [Google Scholar] [CrossRef]
- Fujimoto, G.M.; Kyle, J.E.; Lee, J.-Y.; Metz, T.O.; Payne, S.H. A Generalizable Method for False-Discovery Rate Estimation in Mass Spectrometry-Based Lipidomics. bioRxiv 2020. bioRxiv:2020.02.18.946483. [Google Scholar]
- Dai, C.; Füllgrabe, A.; Pfeuffer, J.; Solovyeva, E.M.; Deng, J.; Moreno, P.; Kamatchinathan, S.; Kundu, D.J.; George, N.; Fexova, S.; et al. A Proteomics Sample Metadata Representation for Multiomics Integration and Big Data Analysis. Nat. Commun. 2021, 12, 5854. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
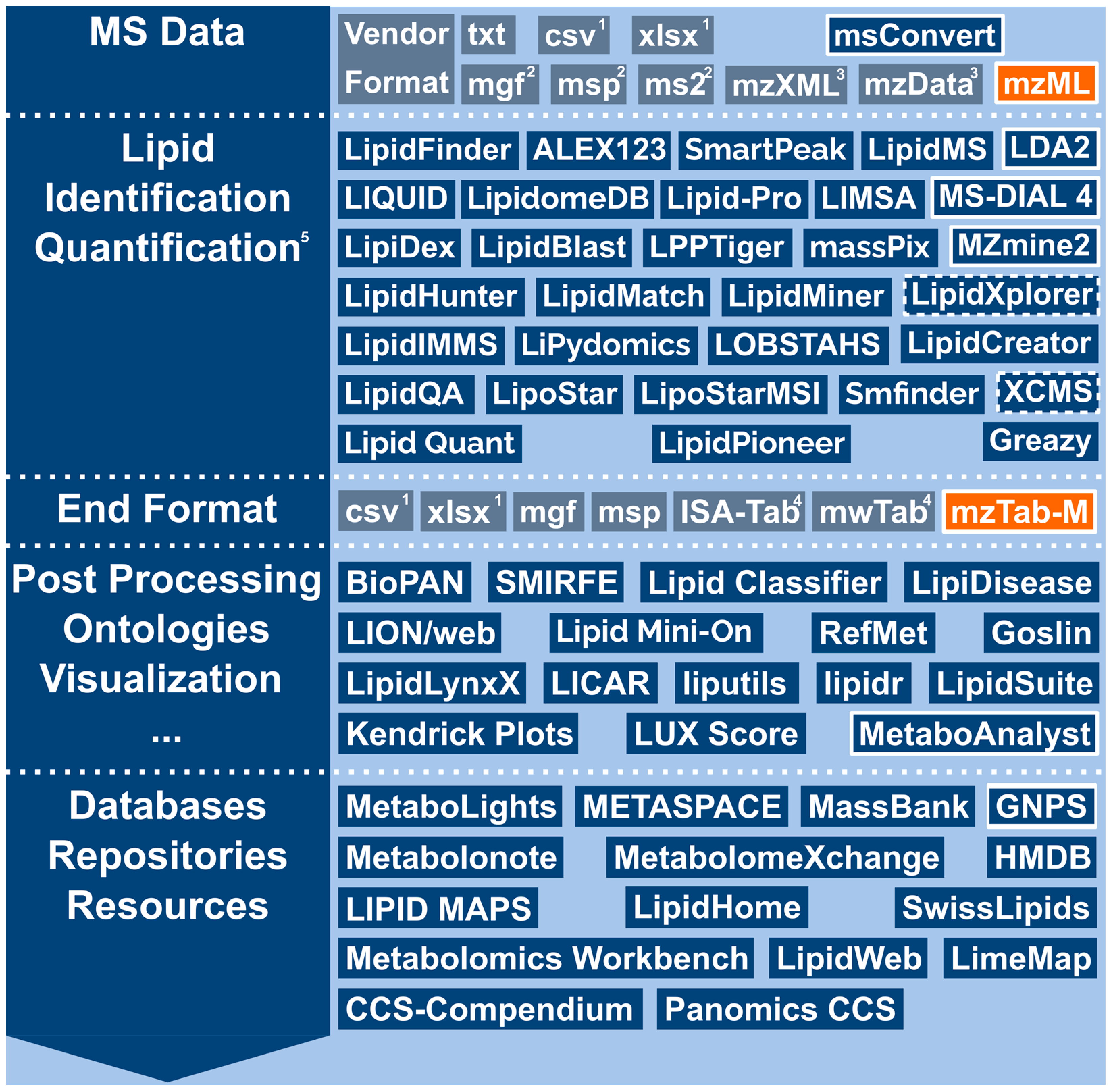{kind=link}
| Workflow $ | Name | Handling | MS * | Identification # | Quant | Input | Output | Last Release | Open-Source | License | Programming Language |
|---|---|---|---|---|---|---|---|---|---|---|---|
| T | LIMSA | C, DI | MS1, MS2 | Compound/Fragment library | yes | XLSX, CSV, HTML | NA | 2006 | NA (1) | GPL v3 | C++, VBA, Excel |
| T | LipidomeDB | DI, C | MS1, MS2 | m/z Library + Transitions + rule-based | yes | XLSX | XLSX, HTML | 2019 | no | NA | Java |
| T | LipidQuant | C (2) | MS1 | m/z library + rule-based | yes | TXT | XLSX | 2021 | yes | CC-BY 4 | VBA, Excel |
| U | ALEX and ALEX 123 | DI | MS1, MS2, MS3 | Manual | no | manual input of parameters | HTML | 2017 | no | NA | NA (3) |
| U | Greazy (4) | C, DI | MS1, MS2 | Fragment/Spectral Library + score | no | vendor, mzML | mzTab (via LipidLama) | 2022 | yes | Apache v2 | C# |
| U | LDA2 | C | MS1, MS2 | Rule-based | yes | mzML, TXT | XLSX, mzTab-M | 2021 | yes | GPL v3 | Java |
| U | LipidBlast | C | MS1, MS2 | Spectral Library + score | no | MSP, MGF, XLSX | MGF, XLSX | 2014 | yes | CC-BY | EXCEL |
| U | LipiDex | C | MS1, MS2 | Spectral Library + rule-based | yes | MGF, mzXML, CSV | CSV | 2018 | yes | MIT | Java |
| U | LipidFinder | C | MS1 | Rule-based, LMSD | no | CSV, JSON (5) | 2021 | yes | MIT | Python | |
| U | LipidHunter (4) | C, DI | MS1, MS2 | Rule-based | yes | mzML, XLSX, TXT | XLSX, HTML, TXT | 2020 | yes | GPL v2, Proprietary | Python |
| U | LipidIMMS | C, IM | MS1 + CCS, MS2 | CCS Library + Spectral Library + score | no | MSP, MGF | CSV, HTML | 2020 | no | NA | NA (3) |
| U | LipidMatch (6) | C, I, DI | MS1, MS2, MSE/DIA | Compound/Fragment library + rule-based | yes | CSV, MS2 (ProteoWizard) | CSV | 2020 | yes | CC BY 4.0 | R |
| U | LipidMiner | C | MS1, MS2 | Compound/Fragment library + rule-based | yes | raw | XLSX, CSV | 2014 | no | NA | C#, Python |
| U | LipidMS | C | MS1, MS2, MSE/DIA | Compound/Fragment library + rule-based | yes | mzXML, CSV | CSV | 2022 | yes | GPL v3 | R |
| U | Lipid-Pro | C | MSE/DIA | Compound/Fragment library | yes | CSV | XLSX, TXT | 2015 | no | Proprietary | C# |
| U | LipidXplorer | DI | MS1, MS2, MS3 | Rule based | no | mzML (MS1 + MS2) | CSV, HTML | 2019 | yes | GPL v2 | Python |
| U | LiPydomics | C, IM | MS1 | CCS Library + m/z Library + HILIC RT Library + rule-based | yes | CSV | XLSX | 2021 | yes | MIT | Python |
| U | LIQUID | C | MS1, MS2 | Spectral Library + rule-based | yes | RAW, mzML | TSV, mzTab, MSP | 2021 | yes | Apache v2 | C# |
| U | LOBSTAHS | C | MS1 | Spectral Library + rule-based | yes | mzML, mzXML, mzData, CSV | XLSX, CSV | 2021 | yes | GPL v3 | R |
| U | LPPTiger (7) | C | MS1, MS2 | Spectral Library + score | yes | mzML, XLSX, TXT | XLSX, HTML | 2021 | yes | GPL v2, Proprietary | Python |
| U | MassPix | I | MS1 | m/z Library + rule-based | no | imzML | CSV | 2017 | yes | NA | R |
| U | MS-DIAL 4 | C, CE, IM | MS1, MS2, MSE/DIA | Spectral Library + rule-based | yes | vendor, mzML | CSV, mzTab-M, XLSX | 2022 | yes | GPL v3 | C# |
| U | MZmine 2 | C | MS1, MS2 | Spectral Library + rule-based | yes | vendor, mzML, mzXML, mzData, CSV, mzTab, XML | CSV, mzTab, XML | 2019 | yes | GPL v2 | Java |
| U | XCMS | C | MS1, MS2 | Spectral Library + score | yes | mzML, mzXML, netCDF | CSV | 2021 | yes | GPL v2 | R, C |
| T + U | LipidCreator and Skyline | C | MS1, MS2, MSE/DIA | Fragment/Spectral Library + score (8) | yes (8) | vendor, mzML (MS1 + MS2) | XLSX, CSV, BLIB | 2021 | yes | MIT | C# |
| T + U | LipidPioneer | C | MS1, MS2 | Compound/m/z Library (8) | yes (8) | XLSX | XLSX | 2017 | yes (9) | NA | VBA, Excel |
| T + U | LipidQA | DI | MS1, MS2 | Spectral Library + score | yes | vendor (Thermo, Waters) | CSV | 2007 | NA (1) | NA | Visual C++ |
| T + U | LipoStar | C, IM | MS1, MS2, MSE/DIA | Compound/Fragment library + rule-based validation | yes | vendor | CSV | 2022 | no | Proprietary | C# |
| T + U | LipoStarMSI | DI, I | MS1, MS2 | Spectral Library + rule based | yes | vendor (Bruker, Waters), imzML | CSV | 2020 | no | Proprietary | C# |
| T + U | SmartPeak | C | MS1, MS2 | Transitions + rule-based | yes | mzML, CSV | mzTab, XML, CSV | 2022 | yes | MIT | C++, Python |
| T + U | Smfinder | C | MS1, MS2 | Spectral Library + score | yes | mzML, mzXML | XLSX, TXT | 2020 | yes (9) | NA | Python, R, C++ |
| Category | Name | Type | Open Source | License | Programming Language | Last Release | Version |
|---|---|---|---|---|---|---|---|
| Ontology, Enrichment | Lipid Mini-On | Web application, Library (1) | yes | BSD 2-Clause | R | 2019 | 0.1.43 |
| Ontology, Enrichment | LION/web | Web application | yes | GPL v3 | R | 2020 | NA |
| Ontology, Enrichment | LipiDisease | Web application | no | NA | R | 2021 | NA |
| Ontology, Classification (2) | SMIRFE | Library | yes | NA | Python | 2020 | 187eb261983b6d0aca1c (3) |
| Ontology, Classification (4) | Lipid Classifier | Library | yes | A-GPL v3 | Ruby | 2014 | 0.0.0.1 |
| Ontology, Enrichment, Pathway Analysis | BioPAN | Web application | no | GPL v3 | PHP, R, HTML, JavaScript | 2020 | NA |
| Post-Processing | Goslin | Web application, Library | yes | MIT, Apache v2 | C++, C#, Java, Python, R | 2022 | 2.0 |
| Post-Processing | LipidLynxX | Web application, Library | yes | GPL v3 | Python | 2020 | 0.9.24 |
| Post-Processing | RefMet | Web application | no | NA | PHP, R | 2021 | NA |
| Post-Processing | LICAR | Web application | yes | MIT | R | 2021 | 1.0 |
| Statistical Analysis, Visualization | lipidr | Library | yes | MIT | R | 2021 | 2.8.1 (5) |
| Statistical Analysis, Visualization | LipidSuite | Web application | no | NA | R | 2021 | 1 |
| Statistical Analysis, Visualization | liputils | Library | yes | GPL v3 | Python | 2021 | 0.16.2 |
| Statistical Analysis, Visualization | MetaboAnalyst | Web application, Library | no (6) | GPL v2 | Java, R (7) | 2021 | 5.0 |
| Visualization | Kendrick mass-defect plots | Library (8) | yes | GPL v2 | Java | 2019 (9) | 2.53 |
| Statistical Analysis, Visualization | LUX Score | Web application, application | yes | Apache v2 | Perl, R, Python | 2018 | 1.0.1 |
| Category | Name | Main Purpose | Lipid Specific | Lipid Structures | Structural Levels | Ontology | Spectral Data | Biochemical Reaction Data | Curation |
|---|---|---|---|---|---|---|---|---|---|
| Database | CCS-Compendium | Compendium of experimentally acquired Collisional Cross Section (Ion Mobility) data from molecular standards acquired on drift tube instruments | yes | yes | yes (1) | ClassyFire/ChemOnt | no | no | manual |
| Database | Panomics CCS | Collisional Cross Section (Ion Mobility) Database for Metabolites and Xenobiotics acquired on drift tube instruments | no | yes | no | no | no | yes | manual |
| Database | GNPS | Knowledge base for raw, processed or annotated fragmentation mass spectrometry data | no | yes | no | - | yes (2) | yes (3) | no (4) |
| Database | HMDB | Curated database of small molecule metabolites found in the human body | no | yes | yes (5) | ClassyFire/ChemOnt | yes (6) | yes | manual |
| Database | LIPID MAPS | Curated portal for LIPID MAPS lipid classification, experimentally determined structures, in-silico combinatorial structures and other lipid resources | yes | yes | yes (7) | LIPID MAPS (8) | yes (9) | yes | manual |
| Database | LipidHome | In-silico generated theoretical lipid structures | yes | yes (10) | no | Liebisch 2013 | no | no | manual |
| Database | SwissLipids | Curated database of lipid structures with experimental evidence and integration with biological knowledge and models | yes | yes | yes | Liebisch 2013 | no | yes | manual |
| Repository | MassBank | Curated database of mass spectrometry reference spectra | no | no | no | - | yes | no | manual (11) |
| Repository | MetaboLights | Repository for metabolomics data (MS and Nuclear Magnetic Resonance (NMR)) and metadata | no | yes | no | ChEBI | yes (12) | no | manual (13) |
| Repository | Metabolomics Workbench | Repository for metabolomics data (MS and NMR) and metadata | no | yes | yes | RefMet | yes | no | manual (14) |
| Repository | Metabolonote | Wiki-based repository for metabolomics metadata | no | no | no | - | yes (12) | no | manual |
| Repository | MetabolomeXchange | Aggregator of metabolomics metadata from MetaboLights, Metabolomics Workbench, Metabolonote and Metabolomic Repository Bordeaux | no | no | no | - | no | no | no |
| Repository | METASPACE | Repository for imaging mass spectrometry for metabolomics | no | yes | no | HMDB/ClassyFire/ChemOnt (15) | yes | no | manual |
| Resource | LimeMap | Curated CellDesigner XML and Vanted GML graph of lipid mediator pathways | yes | no | no (15) | - | no | yes (16) | manual |
| Resource | LipidWeb | Literature review and biochemistry of lipids | yes | yes (17) | no | - | yes (17) | yes (17) | manual |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, N.; Mayer, G.; Has, C.; Kopczynski, D.; Al Machot, F.; Schwudke, D.; Ahrends, R.; Marcus, K.; Eisenacher, M.; Turewicz, M. A Current Encyclopedia of Bioinformatics Tools, Data Formats and Resources for Mass Spectrometry Lipidomics. Metabolites 2022, 12, 584. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12070584
Hoffmann N, Mayer G, Has C, Kopczynski D, Al Machot F, Schwudke D, Ahrends R, Marcus K, Eisenacher M, Turewicz M. A Current Encyclopedia of Bioinformatics Tools, Data Formats and Resources for Mass Spectrometry Lipidomics. Metabolites. 2022; 12(7):584. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12070584
Chicago/Turabian StyleHoffmann, Nils, Gerhard Mayer, Canan Has, Dominik Kopczynski, Fadi Al Machot, Dominik Schwudke, Robert Ahrends, Katrin Marcus, Martin Eisenacher, and Michael Turewicz. 2022. "A Current Encyclopedia of Bioinformatics Tools, Data Formats and Resources for Mass Spectrometry Lipidomics" Metabolites 12, no. 7: 584. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12070584