2.2.1. SHFM3 Cases Reported in PubMed
PubMed literature review on reported cases was conducted up to 23 July 2015, and there were 14 publications regarding duplication at 10q24 in SHFM3, including seven publications using microarray to define the boundary of the duplication. The genes and exons in the duplicated region are presented from centromeric to telomeric direction (
Figure S1a, and Table S1).
Tandem duplication at 10q24 in patients with SHFM3 was first reported by de Mollerat
et al. [
1] in 2003 using Southern, pulsed field gel electrophoresis and dosage analyses. This duplicated region contained a disrupted extra copy of the
FBXW4 gene and the entire
LBX1 and
BTRC genes, known to be involved in limb development. The smallest duplicated region (440 kb) defines the minimal duplicated region common to all patients, which narrows the number of genes included in the duplicated region to genes from
LBX1 to a portion of the
FBXW4 gene involving a region of about 440–570 kb (
Table S1, Patient 26–32). The proximal and distal breakpoints were within a 130 and 80 kb region, respectively. The proximal breakpoints are located at the intergenic region centromeric to
LOC159673 (two cases), centromeric to
HUG1 (
TLX1NB1) (one case), telomeric to
TBX1 (one case), telomeric to
LOC203696 (centromeric to
LBX1) (three cases), respectively. Therefore, the majority of the proximal breakpoints are located at the intergenic region centromeric to
LBX1. The distal breakpoints located at 5′ untranslated region (UTR) (two cases), intron 2 (one case) and intron 5 (four cases) of
FBXW4 gene, respectively. Therefore, the majority of the distal breakpoints are located at intron 5 of
FBXW4 gene.
To refine the minimum duplicated region and to further characterize the SHFM3 locus, Kano
et al. [
2] in 2005 screened 28 non-syndromic SHFM families for tandem genomic duplication of 10q24 by Southern blot and sequence analysis of the
FBXW4 gene. Of 28 families, only two showed genomic rearrangements. Representative patients from the two families exhibit typical SHFM phenotypes with symmetrically affected hands and feet. One patient is a familial case with a 512 kb tandem duplication containing genes from
LBX1 to
FBXW4 (
Table S1, Patient 33), which is similar to the proband in this study. The other patient is a sporadic case arising from a
de novo, 447 kb duplication of maternal origin, containing genes from
LBX1 to a portion of
FBXW4 (exon 9–6) (
Table S1, Patient 34). Therefore, the proximal breakpoints in both cases are located at the intergenic region centromeric to
LBX1, while the distal breakpoints are located at intron 5 of
FBXW4 gene in the smaller duplication case, and at 5′UTR of
FBXW4 gene in the longer duplication case.
Using FISH and genomic DNA quantitative PCR, Lyle
et al. [
3] in 2006 showed duplication at 10q24 locus in 12 patients with SHFM3, in which seven patients have one duplicated segment, while five patients have two discontinuous duplicated segments. The proximal and distal breakpoint clusters are maximally about 160 kb and about 60 kb, respectively. In patients with only one duplicated segment, the distal breakpoint is between exon 3 and 7 of
FBXW4 in all patients, while the proximal breakpoint is between
KAZALD1 exon 5 and
TLX1NB1 (
HUG1) intron 2 in one patient, between
TLX1NB1 (
HUG1) intron 2 and
LBX1 exon 1 in four patients including the daughter in the RK040 family, and from
LBX1 exon 1 to the intergenic region between
AX747408 and
BTRC in two patients including the father in the RK040 family. Therefore, the proximal breakpoint changes when it transmits from the father to the daughter. The duplicated segment contains genes from
TLX1NB1 to a portion of
FBXW4 (exon 9–7) in one patient; from
LBX1 to a portion of
FBXW4 (exon 9–7) in four patients including the daughter in the RK040 family; from
BTRC to a portion of
FBXW4 (exon 9–7) in two patients including the father in the RK040 family (
Table S1, Patient 28, and 35 to 40).
In patients with two discontinuous duplicated segments, the centromeric duplicated segment contains
TLX1NB1 and the telomeric duplicated segment contains genes from
LBX1 to a portion of
FBXW4 (exon 9–7) in two patients; the centromeric duplicated segment contains
TLX1NB1 and
TLX1 and the telomeric duplicated segment contains genes from
BTRC to a portion of
FBXW4 (exon 9–7) in two patients; the centromeric duplicated segment contains genes from
LBX1 to a portion of
FBXW4 (exon 9–3) and telomeric duplicated segment contains
FGF8 in one patient. For the centromeric duplicated region, the proximal breakpoint is between
KAZALD1 exon 5 and
TLX1NB1 intron 2, and the distal breakpoint is between
TLX1 exon 3 and
LBX1 exon 1 in four patients; the proximal breakpoint is between
TLX1 exon 3 and
LBX1 exon 1, and the distal breakpoint is between
FBXW4 exon 3 and
FGF8 exon 2 in one patient. For the telomeric duplicated region, the proximal breakpoint is between
TLX1 exon 3 and
LBX1 exon 1, and the distal breakpoint is between exon 3 and 7 of
FBXW4 in two patients; the proximal breakpoint is from
LBX1 exon 1 to the intergenic region between
AX747408 and
BTRC, and the distal breakpoint is between exon 3 and 7 of
FBXW4 in two patients; the proximal breakpoint is between
FBXW4 exon 3 and
FGF8 exon 2, and the distal breakpoint is between
FGF8 exon 2 and
MGEA5 exon 2 in one patient (
Table S1, Patient 26, and 29–32).
In addition, using RNA quantitative PCR, expression analysis of 13 candidate genes within and flanking the duplicated region showed that
BTRC (present in three copies) and
SUFU (present in two copies) were overexpressed in SHFM3 patients compared to controls, suggesting that SHFM3 may be caused by overexpression of
BTRC and
SUFU, both of which are involved in beta-catenin signaling [
3].
Abnormal bands were observed using pulsed-field gel electrophoresis in eight cases, all but one being non-syndromic reported by Everman
et al. in 2006 [
4]. In all but one case, abnormal bands were observed with probes to the genes
FBXW4 and
BTRC and to the centromeric region approximately 400-kb upstream of
FBXW4. In the remaining case, abnormal bands were observed with probes to
FBXW4 and
BTRC, but not with a probe to the centromeric region.
Array comparative genomic hybridization was first performed by Dimitrov
et al. [
5] in 2010 on seven individuals from four families with SHFM. In the first family, two siblings have distal limb deficiency with micrognathia syndrome (DLDMS). A 532.77 kb triplication at 10q24 was found in the more severely affected brother (
Table S1, Patient 2), while duplication of the same region was present in the mildly affected sister (
Table S1, Patient 3). This region contains genes from LBX1 to FBXW4. The patient in the second family with a
de novo DLDMS has 528.72 kb duplication at 10q24. There are genes from
LBX1 to
FBXW4 in the duplicated region (
Table S1, Patient 4). In the third family, a 597.29 kb duplication at 10q24 was found in the sister with SHFM, the brother with DLDMS, and shown as somatic mosaicism for this duplication in the phenotypically normal mother. The duplicated region contains genes from
LINC01514 to
FBXW4 (
Table S1, Patient 5–7). The patient in the fourth family has syndromic SHFM, who has 658.43 kb duplication at 10q24, which contains genes from a portion of
TLX1NB (exon 2–1, breakpoint at intron 2) to a portion of
FGF8 (exon 6–2, breakpoint at intron 1) (
Table S1, Patient 8). FISH was performed in Patients 2, 3 and 5, while qPCR was conducted in all cases to confirm the array comparative genomic hybridization results.
Two affected brothers were found to have a small duplication of approximately 539 kb at 10q24.31-q24.32 through array-based comparative genomic hybridization by Filho
et al. [
6] in 2011. The duplicated region contains genes from a portion of
LINC01514 (exon 6, breakpoint at intron 5) to
FBXW4. The patients’ sister and father do not have the duplication, but qPCR showed that the mother’s DNA carries the duplication in 20% of blood lymphocytes (
Table S1, Patient 9–11). The microarray aberrations found in both affected brothers were confirmed by qPCR.
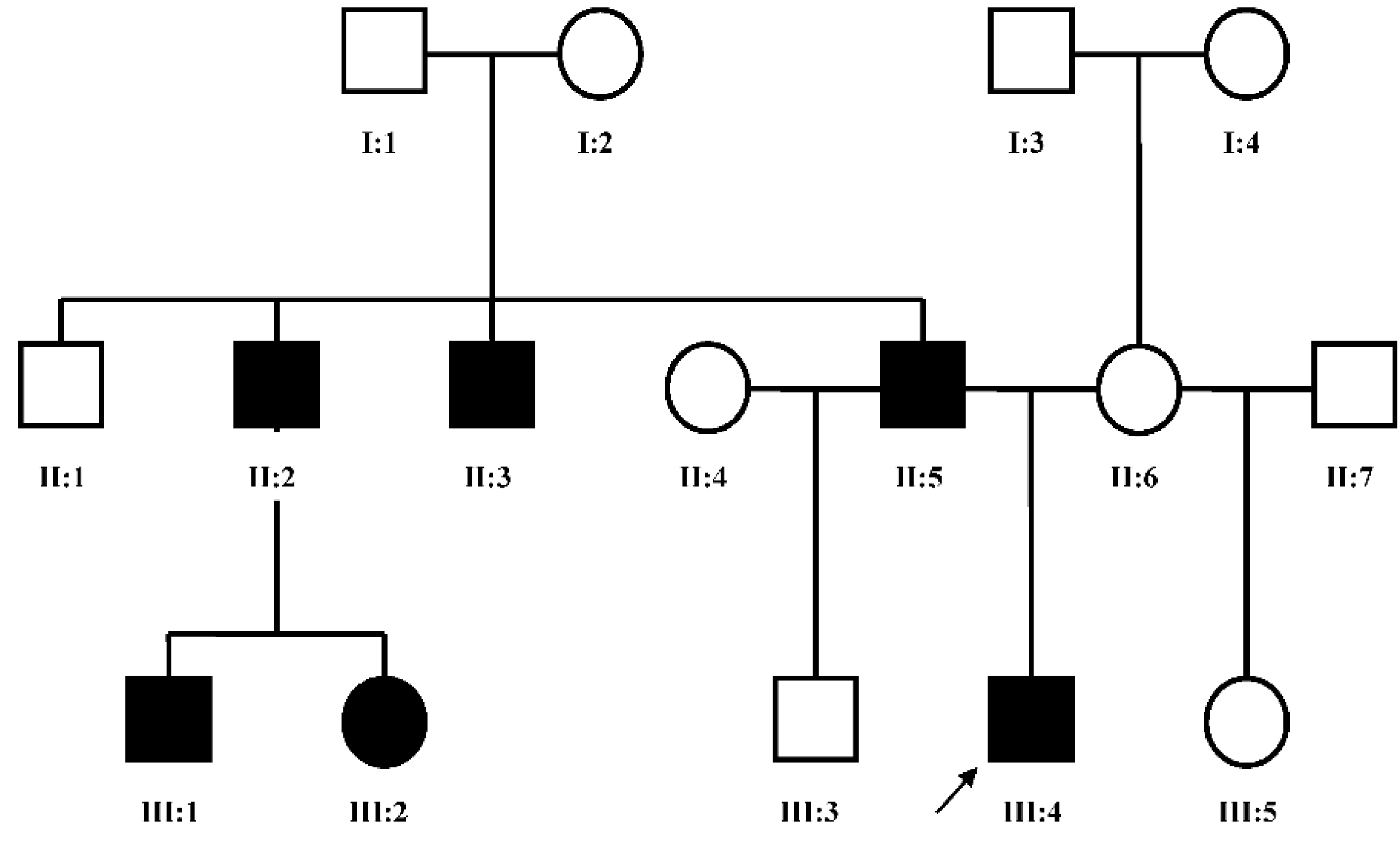
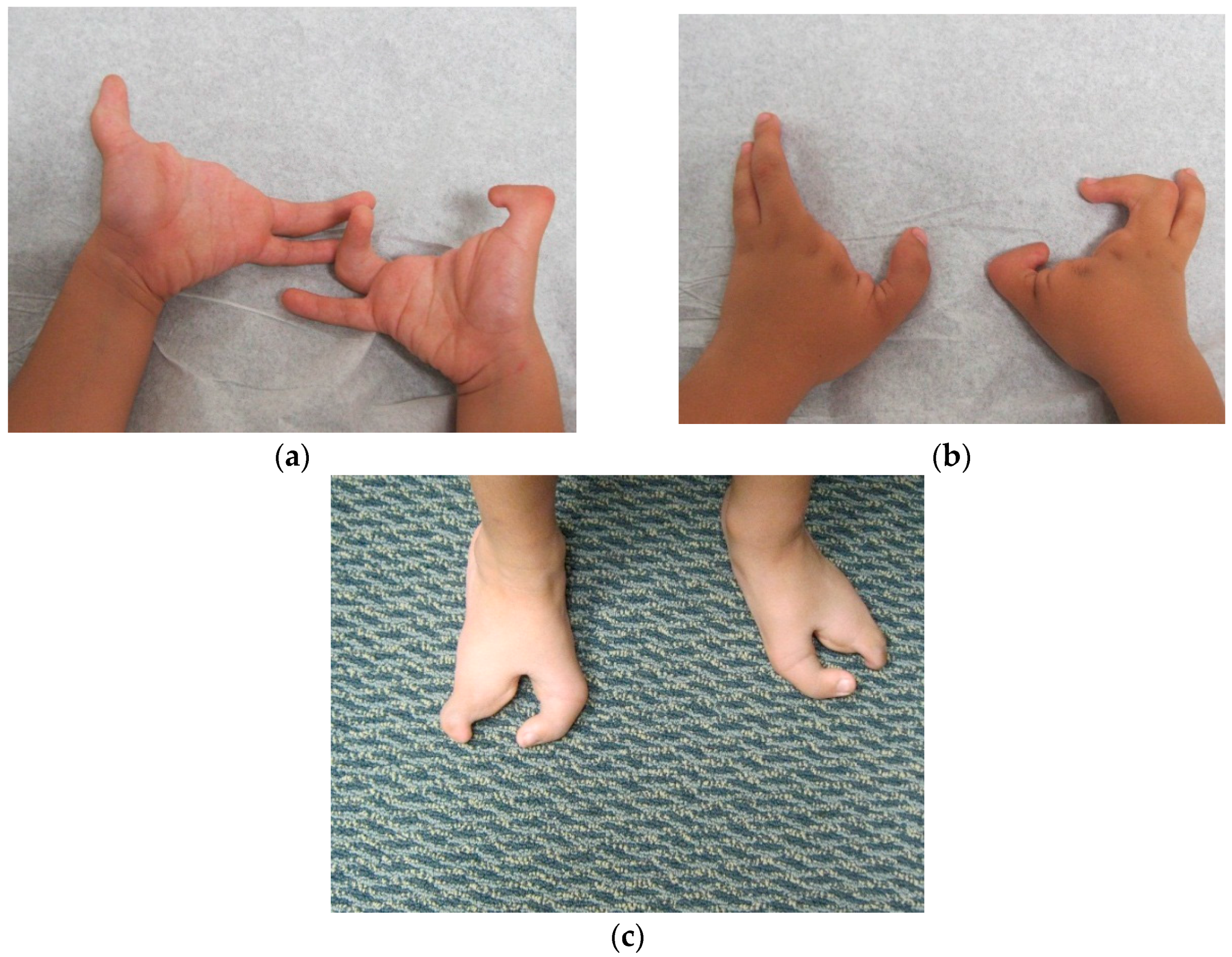
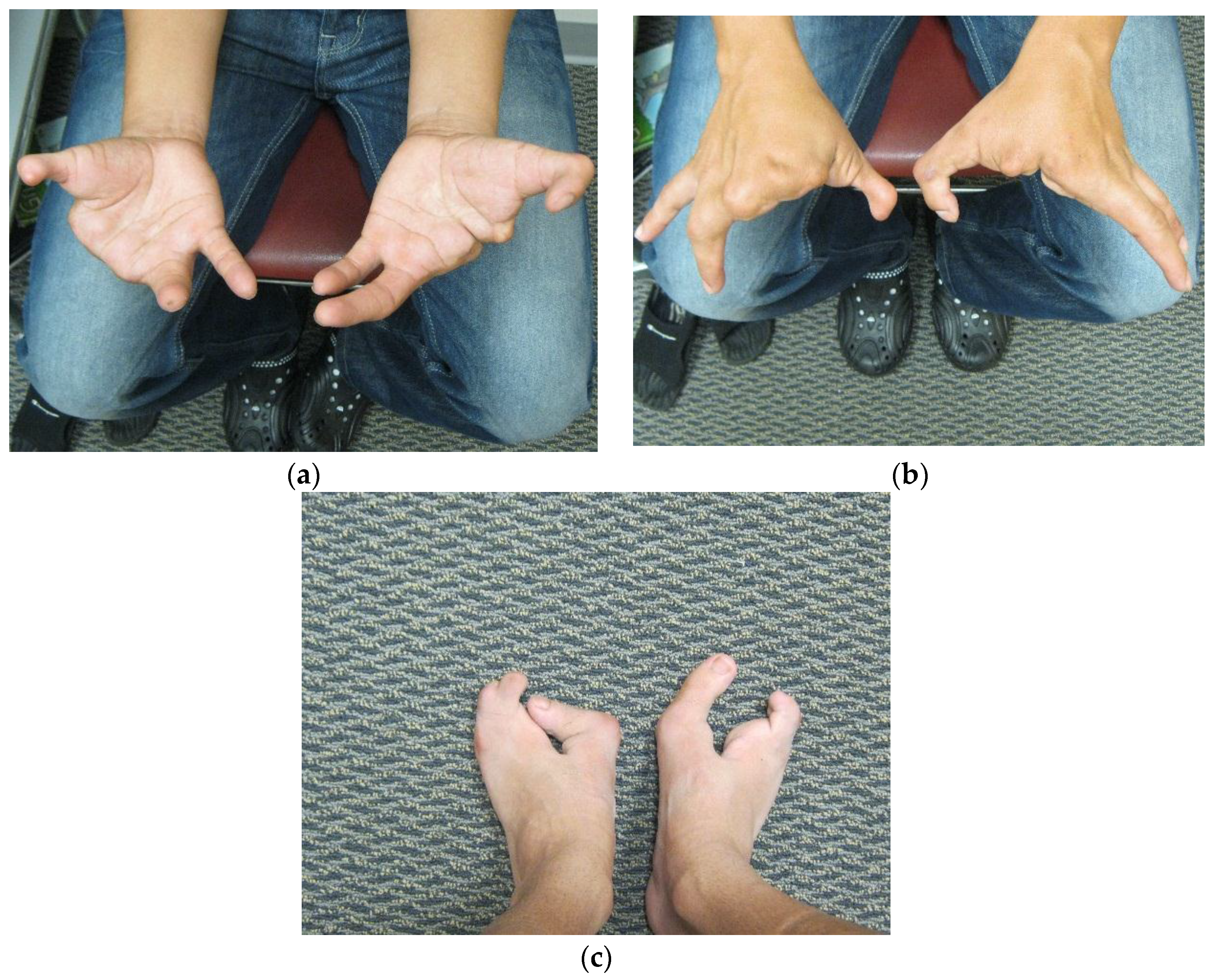
Dai
et al. [
7] in 2013 found the duplication at 10q24.31-q24.32 containing two discontinuous DNA fragments using Affymetrix cytogenetic 2.7M array in a SHFM family with four-generation-span. The proband (IV:3) was a three-year-old boy with severe distal deficiency affecting all four limbs. The other three patients exhibited similar phenotypes, although phenotypic variations were observed between affected family members. Triphalangeal thumb was only identified in the boy’s paternal grandfather (II:5), and complete 1/2 toe syndactyly in the boy (IV:3). Similar hypoplasia/agenesis of 1st ray existed in the boy, his father (III:9) and paternal aunt (III:10). No non-limb malformations were identified in any patient. The centromeric duplicated segment is 247 kb in the boy, 257 kb in the boy’s paternal grandfather and paternal aunt, and 259 kb in the boy’s father. The centromeric duplicated segment involves genes from
LINC01514 to a portion of
BTRC (exon 1, breakpoint at intron 1) in all patients. The proximal breakpoint is at the same location for all patients, while the distal breakpoint is different among the boy, his father, and his paternal grandfather, although it is the same in the paternal grandfather and paternal aunt. Therefore, the distal breakpoint for the centromeric duplicated segment changes when it transmits from the father to the son, however, it stays the same when it transmits from the father to the daughter. The telomeric duplication is 114 kb in the boy and his paternal grandfather, 116 kb in his paternal aunt, and 125 kb in his father. The telomeric duplicated segment encompasses genes from
POLL to a portion of
FBXW4 (exon 9–2, breakpoint at intron 1) in all patients. The distal breakpoint is at the same location for all patients, while the proximal breakpoint is different among his paternal grandfather, his father and his paternal aunt, but it is the same between the boy and his paternal grandfather. Therefore, the proximal breakpoint for the telomeric duplicated segment changes when it transmits from the father to both his son and daughter (
Table S1, Patient 12–15). The microarray aberrations found in all cases were confirmed by qPCR.
A two-year-old boy having SHFM of all four limbs with the hands more severely affected than the feet was reported by Ockeloen
et al. [
8] in 2013. A 250K Affymetrix SNP array analysis showed a 600 kb gain at 10q24.31–q24.32. The duplicated region contains genes from a portion of
TLX1NB (part of exon 3–1, breakpoint at exon 3) to a portion of
FBXW4 (exon 9–2, breakpoint at intron 1) (
Table S1, Patient 16).
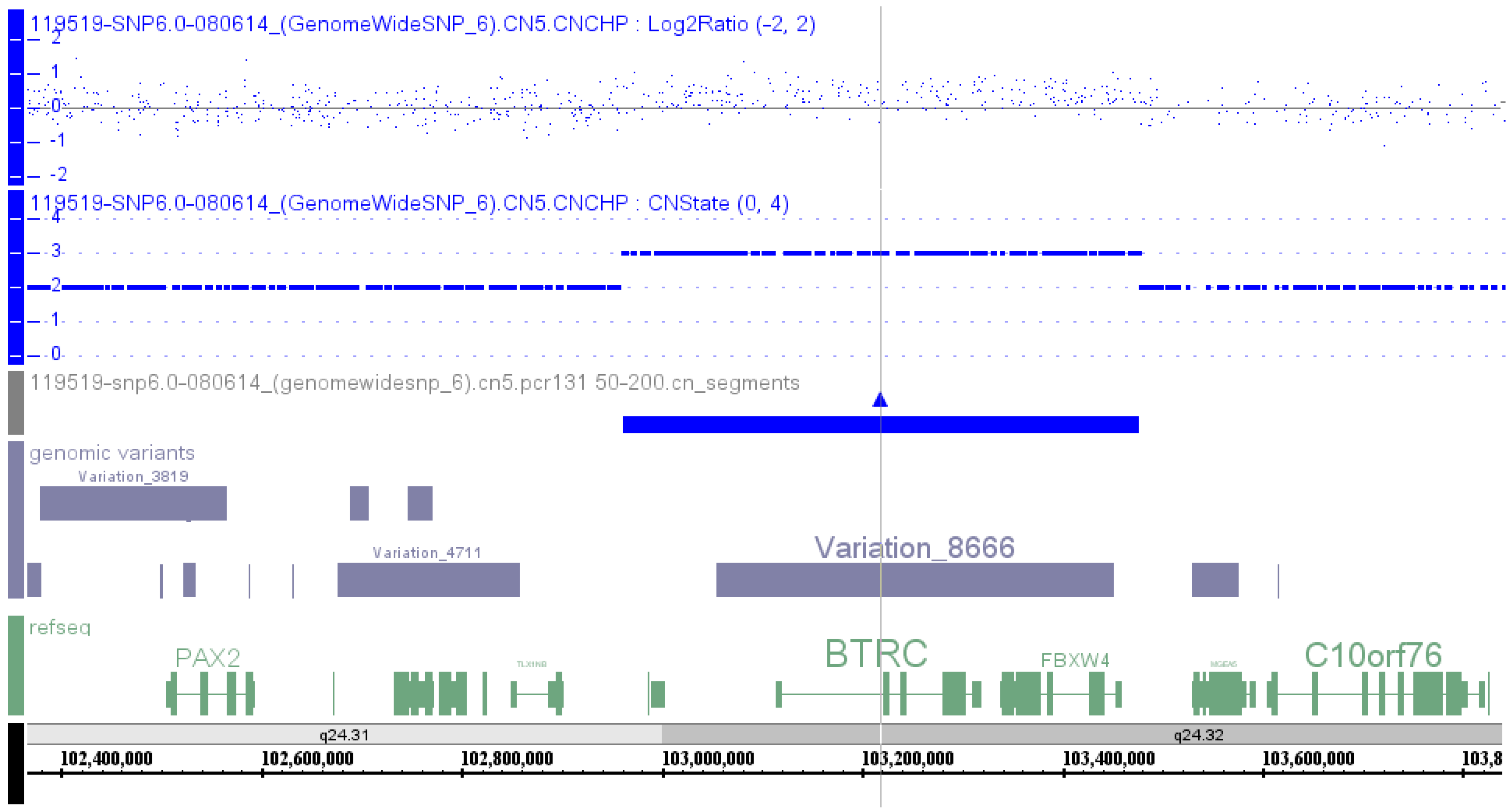
Affymetrix SNP 6.0 array was used to perform a genome-wide copy number variation scan, and quantitative real-time PCR (qPCR) was applied to validate the identified genomic duplication by Liu
et al. [
9] in 2014 in order to identify the potential pathogenic mutation in a Chinese family with SHFM. A microduplication of about 560 kb on the chromosome 10q24 was identified, and the qPCR assay confirmed the presence of this microduplication in all the available affected family members (
Table S1, Patient 47).
To identify genomic aberrations underlying pathogenesis of SHFM in two Chinese families and to provide genetic counseling and prenatal diagnosis for them, Wang
et al. [
10] used array-based comparative genomic hybridization to analyze both blood and amniotic fluid samples from one of the families and showed a 662.3 kb duplication at 10q24.31–q24.32 (
Table S1, Patient 48).
The most recent publication was from Chen
et al. [
11] in 2014 for a patient with SHFM using genome-wide copy number variation SNP microarray, and the tiny copy number variations were verified by real-time fluorescent quantitative PCR. The results of SNP microarray has revealed that the patient has carried a 394 kb duplication at 10q24.31–q24.32, which contains genes from
LBX1 to a portion of
DPCD (exon 1, breakpoint at intron 1). By real-time fluorescent quantitative PCR, the duplicate area encompassing the pathogenic genes was verified, and duplication in exon 9 of the nearby
FBXW4 gene was detected (
Table S1, Patient 17).
2.2.2. SHFM3 Cases Reported in DECIPHER Database
Searching of DECIPHER database using SHFM3 as a keyword was performed on 23 July 2015, and there were 16 entries. Clinical information was available in 11 cases, and limb anomalies were present in nine cases. Seven patients had duplication at 10q24.31-q24.32. Clinical information was available in five patients, all having limb anomalies with classical SHFM phenotype, except one patient having short palm, small feet and duplication of genes from a portion of BTRC (exon 2–14, breakpoint at intron 1) to a portion of
FBXW4 (exon 9–2, breakpoint at intron 1) (
Table S1, Patient 18). The inheritance was unknown. In four cases with classical SHFM phenotype, the duplicated segment contains genes from a portion of
LINC01514 (exon 3–6, breakpoint at intron 2) to a portion of
FBXW4 (exon 9–5, breakpoint at intron 4) having SHFM in the hands only (
Table S1, Patient 19), while from LBX1 to a portion of
FBXW4 (exon 9–6, breakpoint at intron 5) (
Table S1, Patient 20), from a portion of
LINC01514 (exon 4 to 6, breakpoint at intron 3) to a portion of
FBXW4 (exon 9–5, breakpoint at intron 4) (
Table S1, Patient 21) and from
LBX1 to a portion of
FBXW4 (exon 9 to part of exon 1, breakpoint at exon 1) (
Table S1, Patient 22) having SHFM in both hands and feet.
One patient with SHFM phenotype had a 1687 kb duplication at 10q23.33-q24.31, which contains 43 genes from a portion of
DNMBP (exon 4 to 1, breakpoint at intron 4) to a portion of
FBXW4 (exon 9–6, breakpoint at intron 5) (
Table S1, Patient 23). The inheritance was unknown. Four patients had the same 3112 kb duplicated region at 10q23.33-q25.1, which contains 82 genes from a portion of
DNMBP (exon 4–1, breakpoint at intron 4) to a portion of
CNNM2 (exon 1–4, breakpoint at intron 4). SHFM phenotype was found in two patients (
Table S1, Patient 24 and 25), but it was unknown in the other two cases. The inheritance was unknown for all four cases.


{kind=link}
{kind=link}
{kind=link}
{kind=link}



