
Small-Molecule Inhibition of Rho/MKL/SRF Transcription in Prostate Cancer Cells: Modulation of Cell Cycle, ER Stress, and Metastasis Gene Networks

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Culture
2.3. Microarray Analysis
2.4. Bioinformatic Analysis
2.5. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
3. Results
3.1. Microarray Analysis
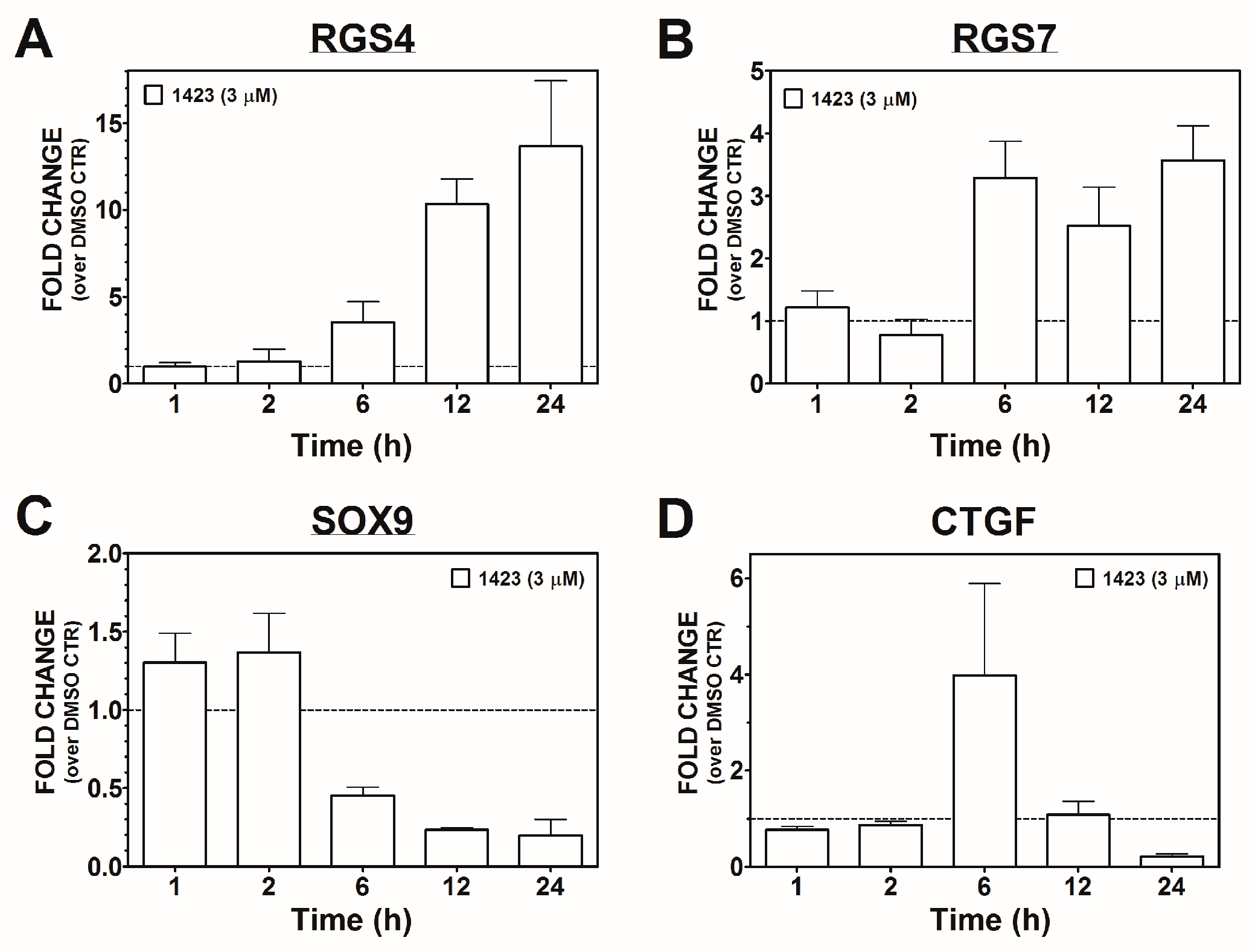
3.2. Candidate Gene Analysis
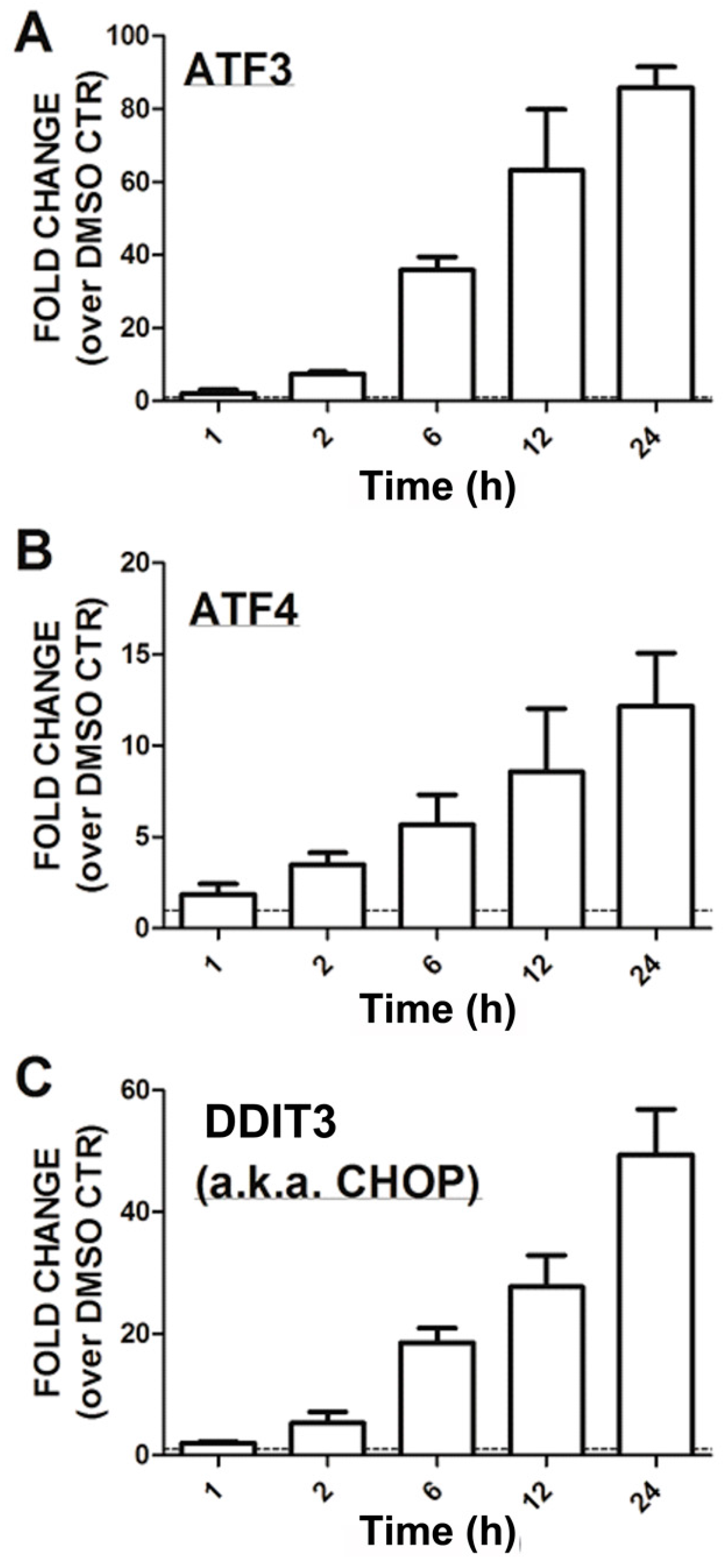
3.3. Time-Course of CCG-1423 Regulated Gene Expression
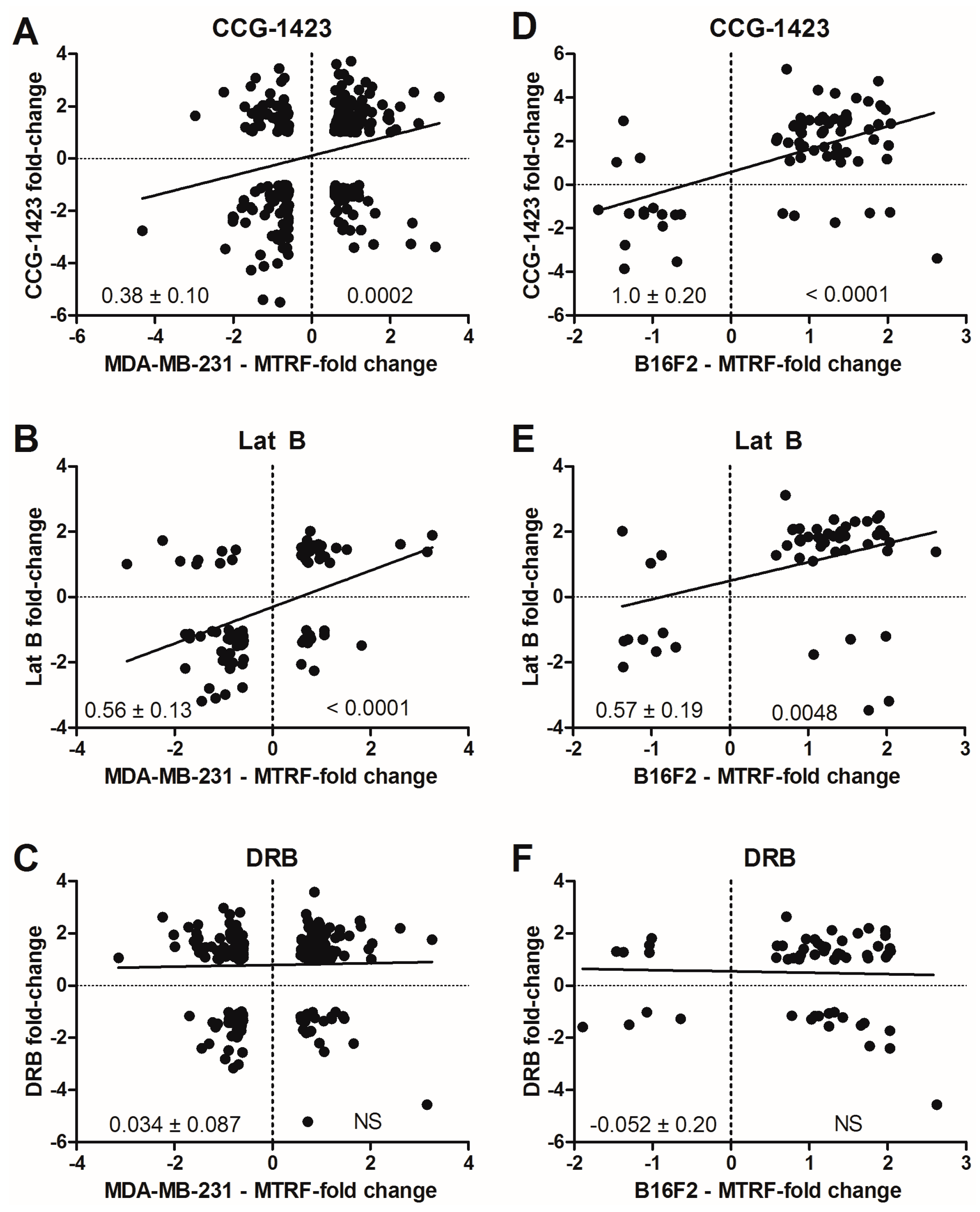
3.4. Global Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MKL | Megakaryocytic Leukemia |
| SRF | Serum Response Factor |
| Lat B | Latrunculin B |
| DRB | 5,6-dichloro-1-β-d-ribofuranosyl-1H-benzimidazole |
| ER | Endoplasmic reticulum |
References
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, T.K. Cancer metastasis therapeutic targets and drug discovery: Emerging small-molecule protein kinase inhibitors. Expert Opin. Investig. Drugs 2004, 13, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Winnepenninckx, V.; Lazar, V.; Michiels, S.; Dessen, P.; Stas, M.; Alonso, S.R.; Avril, M.F.; Ortiz Romero, P.L.; Robert, T.; Balacescu, O.; et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J. Natl. Cancer Inst. 2006, 98, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.A.; Golub, T.R.; Lander, E.S.; Hynes, R.O. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000, 406, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Chong, R.A.; Yang, Q.; Wei, Y.; Blanco, M.A.; Li, F.; Reiss, M.; Au, J.L.; Haffty, B.G.; Kang, Y. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell 2009, 15, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massague, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Zhang, X.H.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massague, J. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massague, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits pthrp secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. Rho-GTPases and cancer. Nat. Rev. Cancer 2002, 2, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Tanji, M.; Ishizaki, T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009, 28, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Berenjeno, I.M.; Nunez, F.; Bustelo, X.R. Transcriptomal profiling of the cellular transformation induced by Rho subfamily GTPases. Oncogene 2007, 26, 4295–4305. [Google Scholar] [CrossRef] [PubMed]
- Cen, B.; Selvaraj, A.; Burgess, R.C.; Hitzler, J.K.; Ma, Z.; Morris, S.W.; Prywes, R. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of srf target genes. Mol. Cell. Biol. 2003, 23, 6597–6608. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, A.; Prywes, R. Expression profiling of serum inducible genes identifies a subset of SRF target genes that are MKL dependent. BMC Mol. Biol. 2004, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, T.; Mayanagi, T.; Sobue, K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 2007, 179, 1027–1042. [Google Scholar] [CrossRef] [PubMed]
- Medjkane, S.; Perez-Sanchez, C.; Gaggioli, C.; Sahai, E.; Treisman, R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat. Cell Biol. 2009, 11, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Evelyn, C.R.; Wade, S.M.; Wang, Q.; Wu, M.; Iniguez-Lluhi, J.A.; Merajver, S.D.; Neubig, R.R. CCG-1423: A small-molecule inhibitor of rhoa transcriptional signaling. Mol. Cancer Ther. 2007, 6, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Dashner, E.J.; van Golen, C.M.; van Golen, K.L. RhoC GTPase is required for PC-3 prostate cancer cell invasion but not motility. Oncogene 2006, 25, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Volsky, D.J. Page: Parametric analysis of gene set enrichment. BMC Bioinform. 2005, 6. [Google Scholar] [CrossRef] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Adams, L.D.; Pabon, L.M.; Mahoney, W.M., Jr.; Beaudry, D.; Gunaje, J.; Geary, R.L.; Deblois, D.; Schwartz, S.M. RGS5, RGS4, and RGS2 expression and aortic contractibility are dynamically co-regulated during aortic banding-induced hypertrophy. J. Mol. Cell. Cardiol. 2008, 44, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Chertkow, Y.; Weinreb, O.; Youdim, M.B.; Silver, H. Gene expression changes in peripheral mononuclear cells from schizophrenic patients treated with a combination of antipsychotic with fluvoxamine. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Sanchez, T.; Pappalardo, A.; Lynch, K.R.; Hla, T.; Ferrer, F. Induction of antiproliferative connective tissue growth factor expression in wilms’ tumor cells by sphingosine-1-phosphate receptor 2. Mol. Cancer Res. 2008, 6, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Vincourt, J.B.; Vignaud, J.M.; Lionneton, F.; Sirveaux, F.; Kawaki, H.; Marchal, S.; Lomazzi, S.; Plenat, F.; Guillemin, F.; Netter, P.; et al. Increased expression of matrilin-3 not only in osteoarthritic articular cartilage but also in cartilage-forming tumors, and down-regulation of SOX9 via epidermal growth factor domain 1-dependent signaling. Arthritis Rheumatol. 2008, 58, 2798–2808. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.H.; Mendpara, N.; Hooks, S.B. Regulator of g-protein signalling expression and function in ovarian cancer cell lines. Cell. Mol. Biol. Lett. 2009, 14, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wolff, D.W.; Wei, T.; Wang, B.; Deng, C.; Kirui, J.K.; Jiang, H.; Qin, J.; Abel, P.W.; Tu, Y. Breast cancer migration and invasion depend on proteasome degradation of regulator of G-protein signaling 4. Cancer Res. 2009, 69, 5743–5751. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.T.; Horvath, C.M.; Iyengar, R. STAT3-mediated transformation of NIH-3T3 cells by the constitutively active q205l GALPHAO protein. Science 2000, 287, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Champion Chip Transcription Factor Search Portal. Available online: http://www.sabiosciences.com/chipqpcrsearch.php?app=TFBS (accessed on 27 March 2016).
- Mullis, T.C.; Tang, X.; Chong, K.T. Expression of connective tissue growth factor (CTGF/CCN2) in head and neck squamous cell carcinoma. J. Clin. Pathol. 2008, 61, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Bennewith, K.L.; Huang, X.; Ham, C.M.; Graves, E.E.; Erler, J.T.; Kambham, N.; Feazell, J.; Yang, G.P.; Koong, A.; Giaccia, A.J. The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res. 2009, 69, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Z.; Feng, G.; You, W.; Li, J. Expression of connective tissue growth factor is in agreement with the expression of VEGF, VEGF-C, -D and associated with shorter survival in gastric cancer. Pathol. Int. 2007, 57, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Boag, J.M.; Beesley, A.H.; Firth, M.J.; Freitas, J.R.; Ford, J.; Brigstock, D.R.; de Klerk, N.H.; Kees, U.R. High expression of connective tissue growth factor in PRE-B acute lymphoblastic leukaemia. Br. J. Haematol. 2007, 138, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Fang, Y.; Xu, J.; Wang, L.; Xu, F.; Xu, E.; Huang, Q.; Lai, M. Analysis of SOX9 expression in colorectal cancer. Am. J. Clin. Pathol. 2008, 130, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; McKnight, N.C.; Zhang, T.; Lu, M.L.; Balk, S.P.; Yuan, X. SOX9 is expressed in normal prostate basal cells and regulates androgen receptor expression in prostate cancer cells. Cancer Res. 2007, 67, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Leav, I.; Ibaragi, S.; Wegner, M.; Hu, G.F.; Lu, M.L.; Balk, S.P.; Yuan, X. SOX9 is expressed in human fetal prostate epithelium and enhances prostate cancer invasion. Cancer Res. 2008, 68, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. David: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [PubMed]
- Zohn, I.M.; Campbell, S.L.; Khosravi-Far, R.; Rossman, K.L.; Der, C.J. Rho family proteins and RAS transformation: The rhoad less traveled gets congested. Oncogene 1998, 17, 1415–1438. [Google Scholar] [CrossRef] [PubMed]
- Evelyn, C.R.; Bell, J.L.; Ryu, J.G.; Wade, S.M.; Kocab, A.; Harzdorf, N.L.; Hollis Showalter, H.D.; Neubig, R.R.; Larsen, S.D. Design, synthesis and prostate cancer cell-based studies of analogs of the Rho/MKL1 transcriptional pathway inhibitor, CCG-1423. Bioorg. Med. Chem. Lett. 2010, 20, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, L.; Dubyk, C.W.; Riesenberger, T.A.; Cooper, C.R.; van Golen, K.L. Rho gtpases in PC-3 prostate cancer cell morphology, invasion and tumor cell diapedesis. Clin. Exp. Metastasis 2008, 25, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2FS in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann, A.; Rosselli, F.; Lazar, V.; Winnepenninckx, V.; Mansuet-Lupo, A.; Dessen, P.; van den Oord, J.J.; Spatz, A.; Sarasin, A. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2008, 27, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Van Golen, K.L.; Wu, Z.F.; Qiao, X.T.; Bao, L.W.; Merajver, S.D. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000, 60, 5832–5838. [Google Scholar] [PubMed]
- Hakem, A.; Sanchez-Sweatman, O.; You-Ten, A.; Duncan, G.; Wakeham, A.; Khokha, R.; Mak, T.W. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005, 19, 1974–1979. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | 2 h | 24 h | ||||
|---|---|---|---|---|---|---|
| Stimulated | Inhibited | Total | Stimulated | Inhibited | Total | |
| CCG-1423 | 48 | 1 | 49 | 837 | 1307 | 2142 |
| Lat B | 15 | 3 | 18 | 225 | 383 | 608 |
| DRB | 5 | 714 | 719 | 1058 | 2026 | 3020 * |
| Gene Categories | CCG-1423 | Lat B | DRB | References |
|---|---|---|---|---|
| Total Genes Regulated | 2142 | 608 | 3020 | |
| FBS-MKL1-Dependent Genes (#) (NIH3T3) | 5 out of 28 | 0 out of 28 | 7 out of 28 | [20] |
| FBS-MKL1-Dependent Genes (%) (NIH3T3) | 17.8% | 0.0% | 25.0% | [20] |
| MKL1-Dependent Genes (#) (MDA-MB-231) | 273 out of 1070 | 83 out of 1070 | 271 out of 1070 | [22] |
| MKL1-Dependent Genes (%) (MDA-MB-231) | 25.5% | 7.8% | 25.3% | [22] |
| MKL1-Dependent Genes (#) (B16F2) | 73 out of 323 | 54 out of 323 | 58 out of 323 | [22] |
| MKL1-Dependent Genes (%) (B16F2) | 22.6% | 16.7% | 18.0% | [22] |
| Gene | Microarray | qRT-PCR |
|---|---|---|
| (Fold-change) | (Fold-change) | |
| RGS4 | 21.7, 21.6, 19.4 * | 12.4 |
| RGS7 | 9.6 | 9.9 |
| CTGF | 0.05 | 0.12 |
| SOX9 | 0.12, 0.13 * | 0.19 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evelyn, C.R.; Lisabeth, E.M.; Wade, S.M.; Haak, A.J.; Johnson, C.N.; Lawlor, E.R.; Neubig, R.R. Small-Molecule Inhibition of Rho/MKL/SRF Transcription in Prostate Cancer Cells: Modulation of Cell Cycle, ER Stress, and Metastasis Gene Networks. Microarrays 2016, 5, 13. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays5020013
Evelyn CR, Lisabeth EM, Wade SM, Haak AJ, Johnson CN, Lawlor ER, Neubig RR. Small-Molecule Inhibition of Rho/MKL/SRF Transcription in Prostate Cancer Cells: Modulation of Cell Cycle, ER Stress, and Metastasis Gene Networks. Microarrays. 2016; 5(2):13. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays5020013
Chicago/Turabian StyleEvelyn, Chris R., Erika M. Lisabeth, Susan M. Wade, Andrew J. Haak, Craig N. Johnson, Elizabeth R. Lawlor, and Richard R. Neubig. 2016. "Small-Molecule Inhibition of Rho/MKL/SRF Transcription in Prostate Cancer Cells: Modulation of Cell Cycle, ER Stress, and Metastasis Gene Networks" Microarrays 5, no. 2: 13. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays5020013