Divergent Microbiota Dynamics along the Coastal Marine Ecosystem of Puerto Rico



Abstract
:1. Introduction
2. Materials and Methods
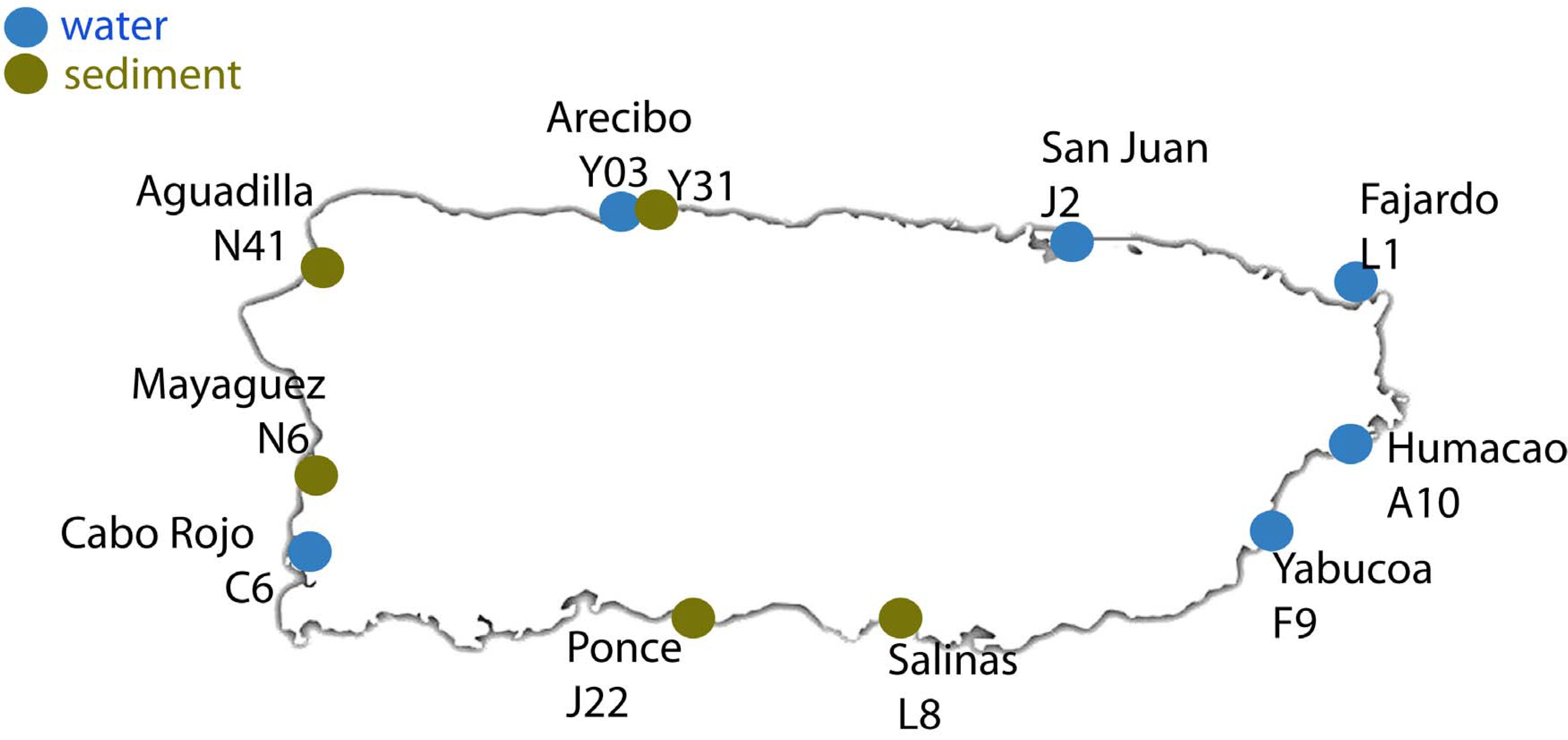
2.1. Sample Collection and Processing
2.2. 16S rRNA Gene Amplicons and Sequence Analysis
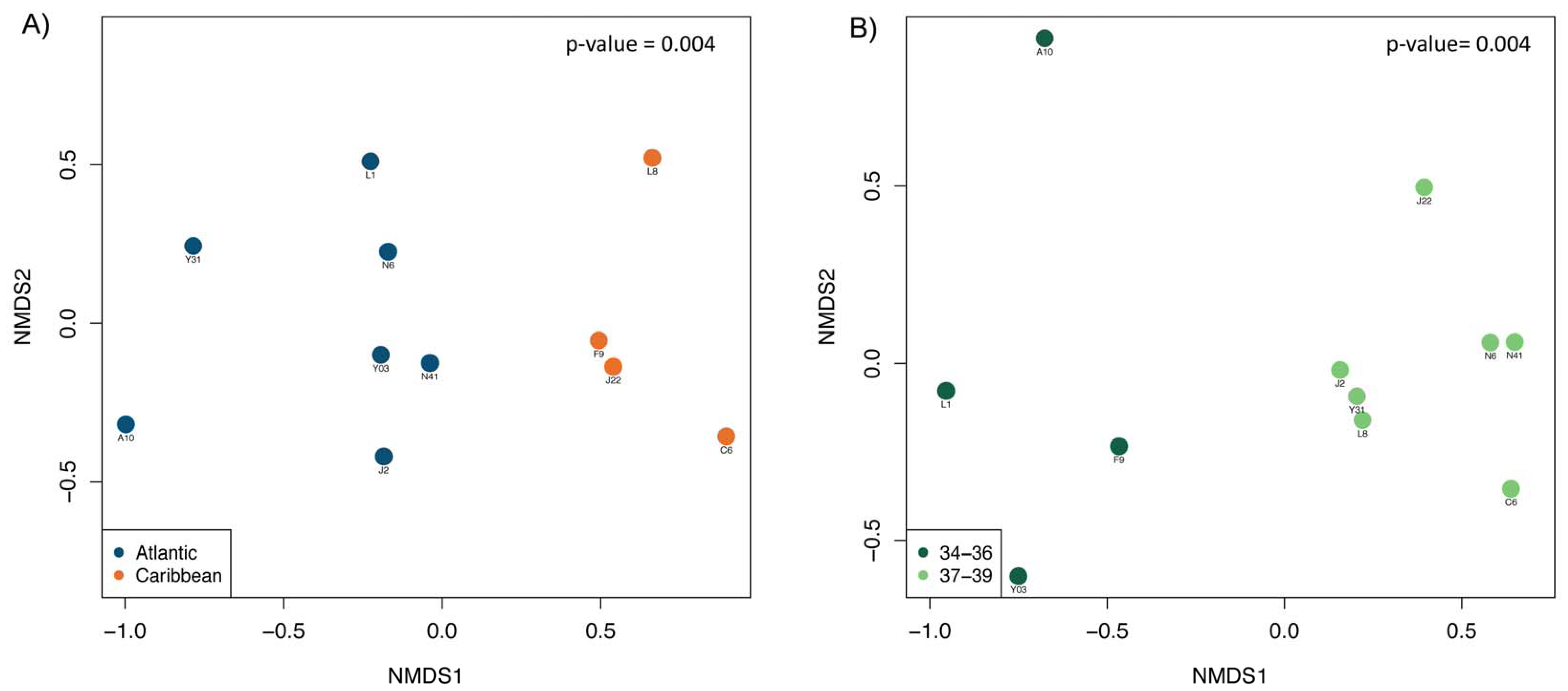
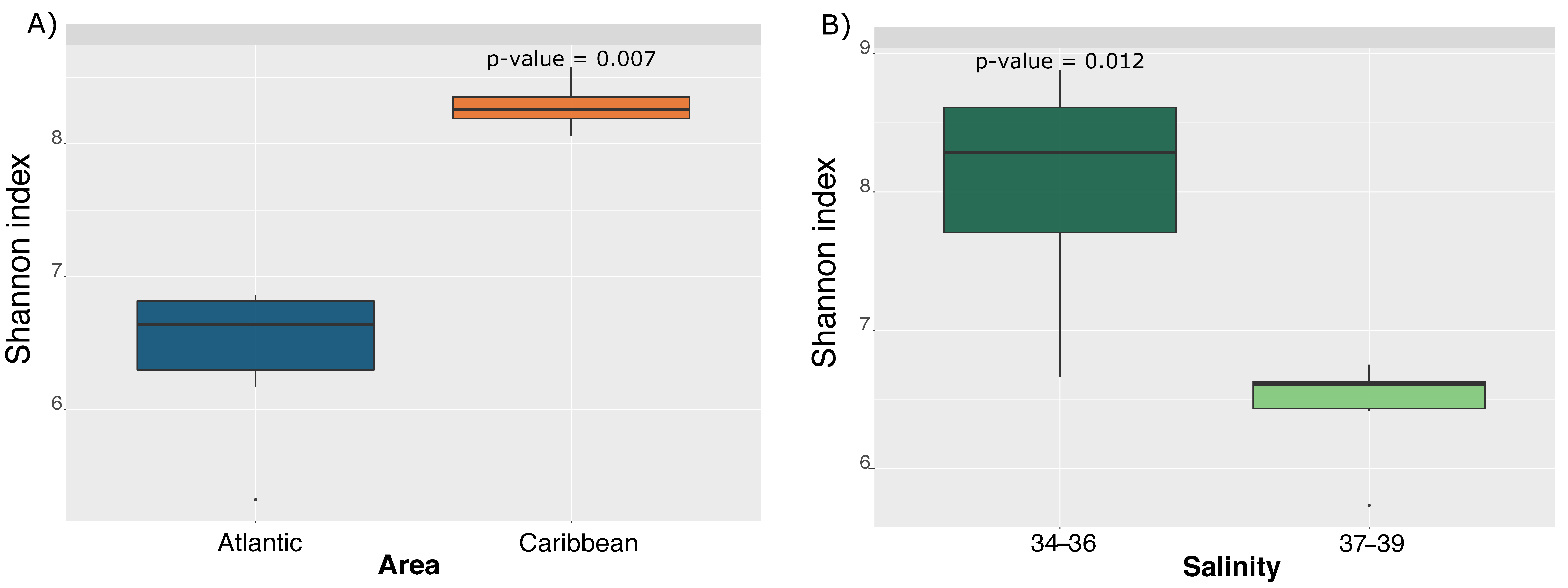
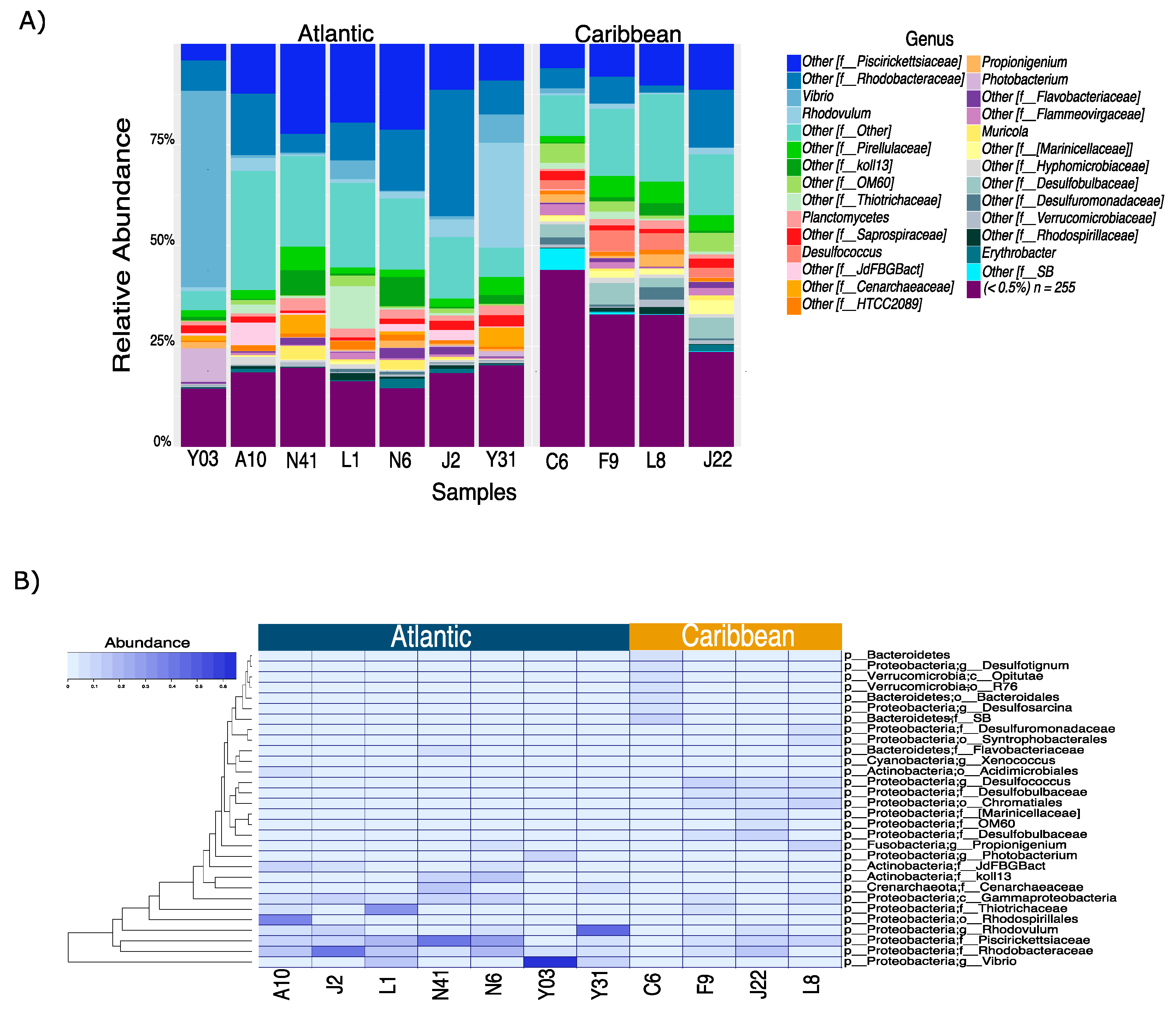
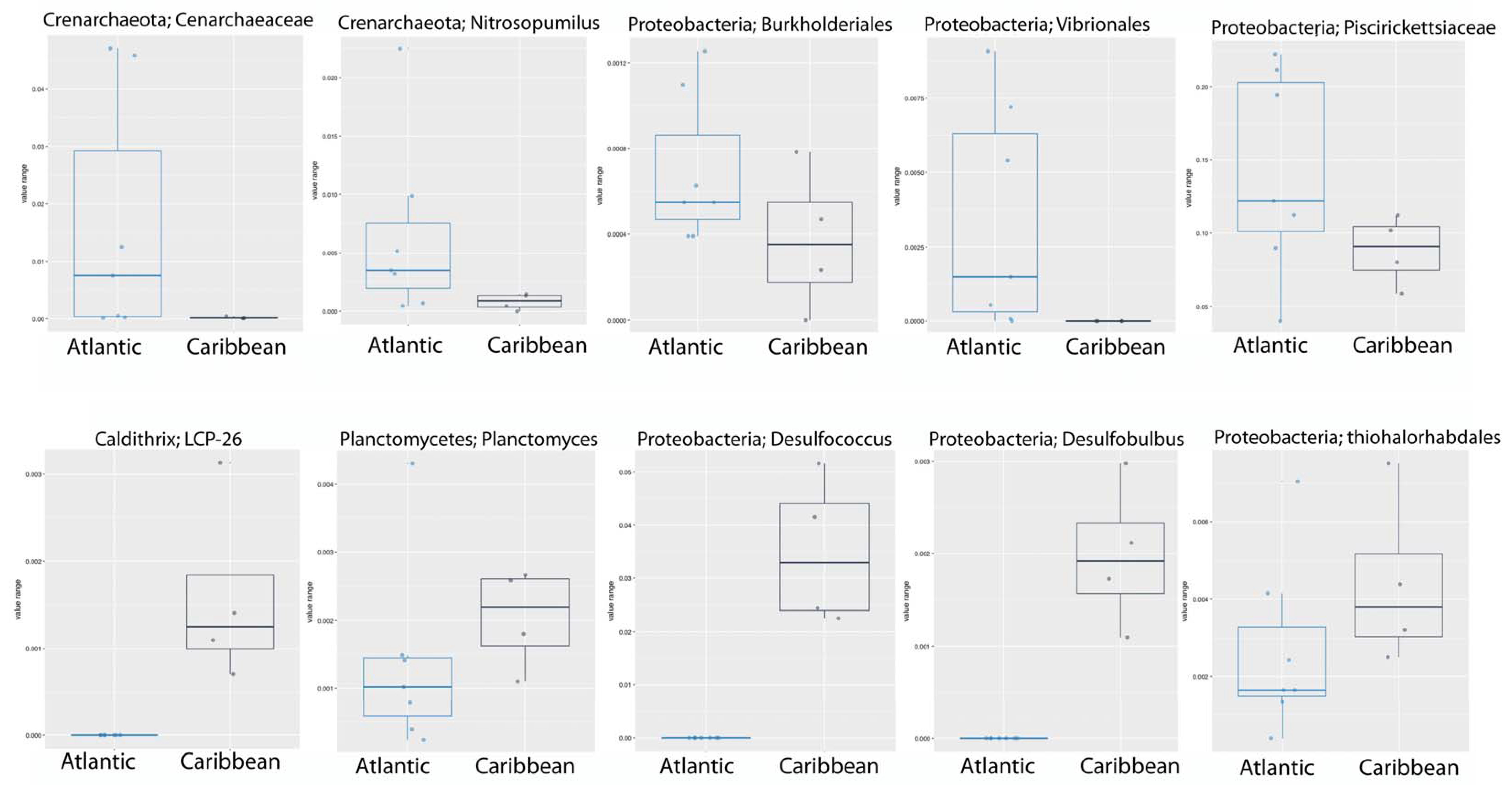
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Ocean plankton. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [Green Version]
- Godoy-Vitorino, F.; Ruiz-Diaz, C.P.; Rivera-Seda, A.; Ramirez-Lugo, J.S.; Toledo-Hernandez, C. The microbial biosphere of the coral Acropora cervicornis in Northeastern Puerto Rico. PeerJ 2017, 5, e3717. [Google Scholar] [CrossRef]
- Lawler, S.N.; Kellogg, C.A.; France, S.C.; Clostio, R.W.; Brooke, S.D.; Ross, S.W. Coral-Associated Bacterial Diversity Is Conserved across Two Deep-Sea Anthothela Species. Front. Microbiol. 2016, 7, 458. [Google Scholar] [CrossRef]
- Sun, W.; Anbuchezhian, R.; Li, Z. (Eds.) Association of Coral-Microbes, and the Ecological Roles of Microbial Symbionts in Corals. In The Cnidaria, Past, Present and Future; Springer: Cham, Switzerland, 2016; pp. 347–357. [Google Scholar]
- Ainsworth, T.D.; Fordyce, A.J.; Camp, E.F. The Other Microeukaryotes of the Coral Reef Microbiome. Trends Microbiol. 2017, 25, 980–991. [Google Scholar] [CrossRef]
- Hernandez-Agreda, A.; Gates, R.D.; Ainsworth, T.D. Defining the Core Microbiome in Corals’ Microbial Soup. Trends Microbiol. 2017, 25, 125–140. [Google Scholar] [CrossRef]
- Fan, L.; Liu, M.; Simister, R.; Webster, N.S.; Thomas, T. Marine microbial symbiosis heats up: The phylogenetic and functional response of a sponge holobiont to thermal stress. ISME J. 2013, 7, 991–1002. [Google Scholar] [CrossRef]
- Karlinska-Batres, K.; Worheide, G. Phylogenetic diversity and community structure of the symbionts associated with the coralline sponge Astrosclera willeyana of the Great Barrier Reef. Microb. Ecol. 2013, 65, 740–752. [Google Scholar] [CrossRef]
- Steinert, G.; Taylor, M.W.; Deines, P.; Simister, R.L.; de Voogd, N.J.; Hoggard, M.; Schupp, P.J. In four shallow and mesophotic tropical reef sponges from Guam the microbial community largely depends on host identity. PeerJ 2016, 4, e1936. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Steele, J.A.; Caporaso, J.G.; Steinbruck, L.; Reeder, J.; Temperton, B.; Huse, S.; McHardy, A.C.; Knight, R.; Joint, I.; et al. Defining seasonal marine microbial community dynamics. ISME J. 2012, 6, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Harley, C.D.; Anderson, K.M.; Demes, K.W.; Jorve, J.P.; Kordas, R.L.; Coyle, T.A.; Graham, M.H. Effects of Climate Change on Global Seaweed Communities. J. Phycol. 2012, 48, 1064–1078. [Google Scholar] [CrossRef]
- Harley, C.D.; Randall Hughes, A.; Hultgren, K.M.; Miner, B.G.; Sorte, C.J.; Thornber, C.S.; Rodriguez, L.F.; Tomanek, L.; Williams, S.L. The impacts of climate change in coastal marine systems. Ecol. Lett. 2006, 9, 228–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortell, P.D.; Maldonado, M.T.; Granger, J.; Price, N.M. Marine bacteria and biogeochemical cycling of iron in the oceans. FEMS Microbiol. Ecol. 2006, 29. [Google Scholar] [CrossRef]
- Piganeau, G.; Desdevises, Y.; Derelle, E.; Moreau, H. Picoeukaryotic sequences in the Sargasso sea metagenome. Genome Biol. 2008, 9, R5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yooseph, S.; Nealson, K.H.; Rusch, D.B.; McCrow, J.P.; Dupont, C.L.; Kim, M.; Johnson, J.; Montgomery, R.; Ferriera, S.; Beeson, K.; et al. Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature 2010, 468, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Williamson, S.J.; Rusch, D.B.; Yooseph, S.; Halpern, A.L.; Heidelberg, K.B.; Glass, J.I.; Andrews-Pfannkoch, C.; Fadrosh, D.; Miller, C.S.; Sutton, G.; et al. The Sorcerer II Global Ocean Sampling Expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE 2008, 3, e1456. [Google Scholar] [CrossRef]
- Seeleuthner, Y.; Mondy, S.; Lombard, V.; Carradec, Q.; Pelletier, E.; Wessner, M.; Leconte, J.; Mangot, J.F.; Poulain, J.; Labadie, K.; et al. Single-cell genomics of multiple uncultured stramenopiles reveals underestimated functional diversity across oceans. Nat. Commun. 2018, 9, 310. [Google Scholar] [CrossRef]
- Hubbard, D.; Burke, R.; Gill, P.; Ramirez, W.; Sherman, C. Coral-reef Geology: Puerto Rico and the US Virgin Islands. In Coral Reefs of the USA; Springer Science: New York, NY, USA, 1970. [Google Scholar]
- Suh, S.S.; Park, M.; Hwang, J.; Kil, E.J.; Jung, S.W.; Lee, S.; Lee, T.K. Seasonal Dynamics of Marine Microbial Community in the South Sea of Korea. PLoS ONE 2015, 10, e0131633. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Meyer, F.; Jansson, J.; Gordon, J.; Pace, N.; Tiedje, J.; Ley, R.; Fierer, N.; Field, D.; Kyrpides, N.; et al. The Earth Microbiome Project: Meeting report of the “1 EMP meeting on sample selection and acquisition” at Argonne National Laboratory October 6 2010. Stand. Genom. Sci. 2010, 3, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; Gonzalez, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. 2011, 21, 1E.5.1–1E.5.20. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Derilus, D.; Forestil, A.; Fortuné, J.; Polyanska, O.; Louime, C.; Gervais, G.; Massey, S.E. Functional Metagenomics Characterization of an Anaerobic Saltwater Bioreactor. J. Renew. Energy 2019, 1–15. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.R.; Chai, B.; Farris, R.J.; Wang, Q.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Bandela, A.M.; Cardenas, E.; Garrity, G.M.; Tiedje, J.M. The ribosomal database project (RDP-II): Introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007, 35, D169–D172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008; Available online: http://www.R-project.org (accessed on 31 May 2020).
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.4-4; R Development Core Team: Vienna, Austria, 2010. [Google Scholar]
- Zhao, S.; Guo, Y.; Sheng, Q.; Shyr, Y. Advanced heat map and clustering analysis using heatmap3. Biomed. Res. Int. 2014, 986048. [Google Scholar] [CrossRef] [Green Version]
- Biers, E.J.; Sun, S.; Howard, E.C. Prokaryotic genomes and diversity in surface ocean waters: Interrogating the global ocean sampling metagenome. Appl. Environ. Microbiol. 2009, 75, 2221–2229. [Google Scholar] [CrossRef] [Green Version]
- Gajigan, A.P.; Yniguez, A.T.; Villanoy, C.L.; San Diego-McGlone, M.L.; Jacinto, G.S.; Conaco, C. Diversity and community structure of marine microbes around the Benham Rise underwater plateau, northeastern Philippines. PeerJ 2018, 6, e4781. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.; Müller, O.; Nordmann, E.-L.; Seuthe, L.; Bratbak, G.; Øvreås, L. Changes in Marine Prokaryote Composition with Season and Depth Over an Arctic Polar Year. Front. Mar. Sci. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.J.; Kirchman, D.L. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J. 2013, 7, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Sekar, R.; Mills, D.K.; Remily, E.R.; Voss, J.D.; Richardson, L.L. Microbial communities in the surface mucopolysaccharide layer and the black band microbial mat of black band-diseased Siderastrea siderea. Appl. Environ. Microbiol. 2006, 72, 5963–5973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy-Vitorino, F.; Toledo-Hernandez, C. Reef-Building Corals as a Tool for Climate Change Research in the Genomics Era. Results Probl. Cell Differ. 2018, 65, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Gutierrez, T. Evaluating the Detection of Hydrocarbon-Degrading Bacteria in 16S rRNA Gene Sequencing Surveys. Front. Microbiol. 2017, 8, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondoso, J.; Godoy-Vitorino, F.; Balague, V.; Gasol, J.M.; Harder, J.; Lage, O.M. Epiphytic Planctomycetes communities associated with three main groups of macroalgae. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yu, D.; Wang, Y.; Chen, G.; Tang, P.; Huang, S. A novel control strategy for the partial nitrification and anammox process (PN/A) of immobilized particles: Using salinity as a factor. Bioresour. Technol. 2020, 302, 122864. [Google Scholar] [CrossRef] [PubMed]
- Corredor, J.; Morell, J. Seasonal variation of physical and biogeochemical features in eastern Caribbean surface water. J. Geophys. Res. 2001, 106, 4517–4525. [Google Scholar] [CrossRef]
- Kirchman, D.L.; Cottrell, M.T.; Lovejoy, C. The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ. Microbiol. 2010, 12, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Rudiak-Gould, P. Promiscuous corroboration and climate change translation: A case study from the Marshall Islands. Glob. Environ. Chang. 2012, 22, 46–54. [Google Scholar] [CrossRef]
- Gifford, S.; Sharma, S.; Booth, M.; Moran, M. Expression patterns reveal niche diversification in a marine microbial assemblage. ISME J. 2013, 7, 281–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S. Climate science: A sinking feeling. Nature 2006, 440, 734–736. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Site | Coordinates | Sample | Region | Number of Sequences | Number of OTUs |
|---|---|---|---|---|---|---|
| C6 | Cabo Rojo | Lat: 18° 05′ 15″ N, Lon: 067° 08′ 49″ W | Water | Caribbean | 21,493 | 2268 |
| F9 | Yabucoa | Lat: 18° 03′ 01″ N, Lon: 066° 52′ 45″ W | 25,022 | 2716 | ||
| A10 | Humacao | Lat: 18° 09′ 18″ N, Lon: 065° 49′ 13″ W | Atlantic | 25,366 | 2162 | |
| L1 | Fajardo | Lat: 18° 19′ 32″ N, Lon: 065° 39′ 08″ W | 22,788 | 2461 | ||
| J2 | San Juan | Lat: 18° 27′ 55″ N, Lon: 066° 06′ 20″ W | 25,583 | 2618 | ||
| Y03 | Arecibo | Lat: 18° 20′ 47″ N, Lon: 066° 45′ 10″ W | 57,110 | 2268 | ||
| L8 | Salinas | Lat: 17° 58′ 39″ N, Lon: 066° 17′ 452″ W | Sediment | Caribbean | 29,605 | 2027 |
| J22 | Ponce | Lat: 18° 00′ 39″ N, Lon: 066° 36′ 50″ W | 27,670 | 2239 | ||
| N6 | Mayagüez | Lat: 18° 12′ 04″ N, Lon: 067° 08′ 42″ W | Atlantic | 29,066 | 2231 | |
| N41 | Aguadilla | Lat: 18° 25′ 38″ N, Lon: 067° 09′ 14″ W | 29,537 | 2102 | ||
| Y31 | Arecibo | Lat: 18° 20′ 47″ N, Lon: 066° 45′ 10″ W | 31,134 | 1052 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louime, C.J.; Vazquez-Sanchez, F.; Derilus, D.; Godoy-Vitorino, F. Divergent Microbiota Dynamics along the Coastal Marine Ecosystem of Puerto Rico. Microbiol. Res. 2020, 11, 45-55. https://0-doi-org.brum.beds.ac.uk/10.3390/microbiolres11020009
Louime CJ, Vazquez-Sanchez F, Derilus D, Godoy-Vitorino F. Divergent Microbiota Dynamics along the Coastal Marine Ecosystem of Puerto Rico. Microbiology Research. 2020; 11(2):45-55. https://0-doi-org.brum.beds.ac.uk/10.3390/microbiolres11020009
Chicago/Turabian StyleLouime, Clifford Jaylen, Frances Vazquez-Sanchez, Dieunel Derilus, and Filipa Godoy-Vitorino. 2020. "Divergent Microbiota Dynamics along the Coastal Marine Ecosystem of Puerto Rico" Microbiology Research 11, no. 2: 45-55. https://0-doi-org.brum.beds.ac.uk/10.3390/microbiolres11020009