Probiotic Supplementation in a Clostridium difficile-Infected Gastrointestinal Model Is Associated with Restoring Metabolic Function of Microbiota

 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Simulation of Gastrointestinal Conditions
2.1.1. Fecal Slurry Preparation
2.1.2. Probiotic Treatment Preparation
2.1.3. Batch Culture Fermentation
2.2. FW Metabolite Analysis
2.2.1. Short Chain Fatty Acids (SCFA) Analysis
2.2.2. Hydrogen Sulfide (H2S) Determination Assay
2.2.3. Ammonium (NH4) Determination Assay
2.3. DNA Extraction and Analysis
2.3.1. DNA Extraction
2.3.2. Detection of Probiotic Strains by Real-Time Polymerase Chain Reaction (qPCR)
2.3.3. 16S rRNA Gene Amplicon Sequencing and Bioinformatics
2.4. Statistical Analyses
2.5. Availability of Data
3. Results
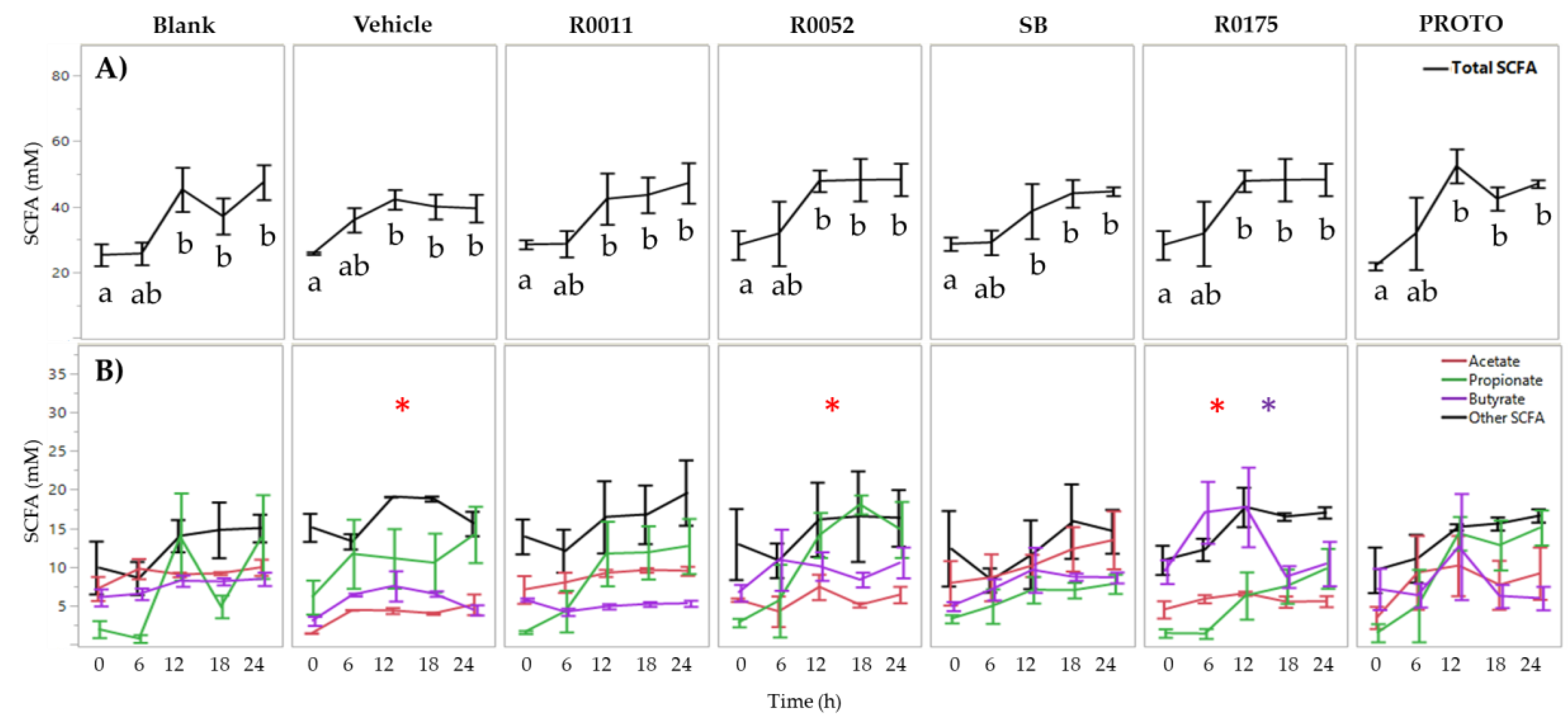
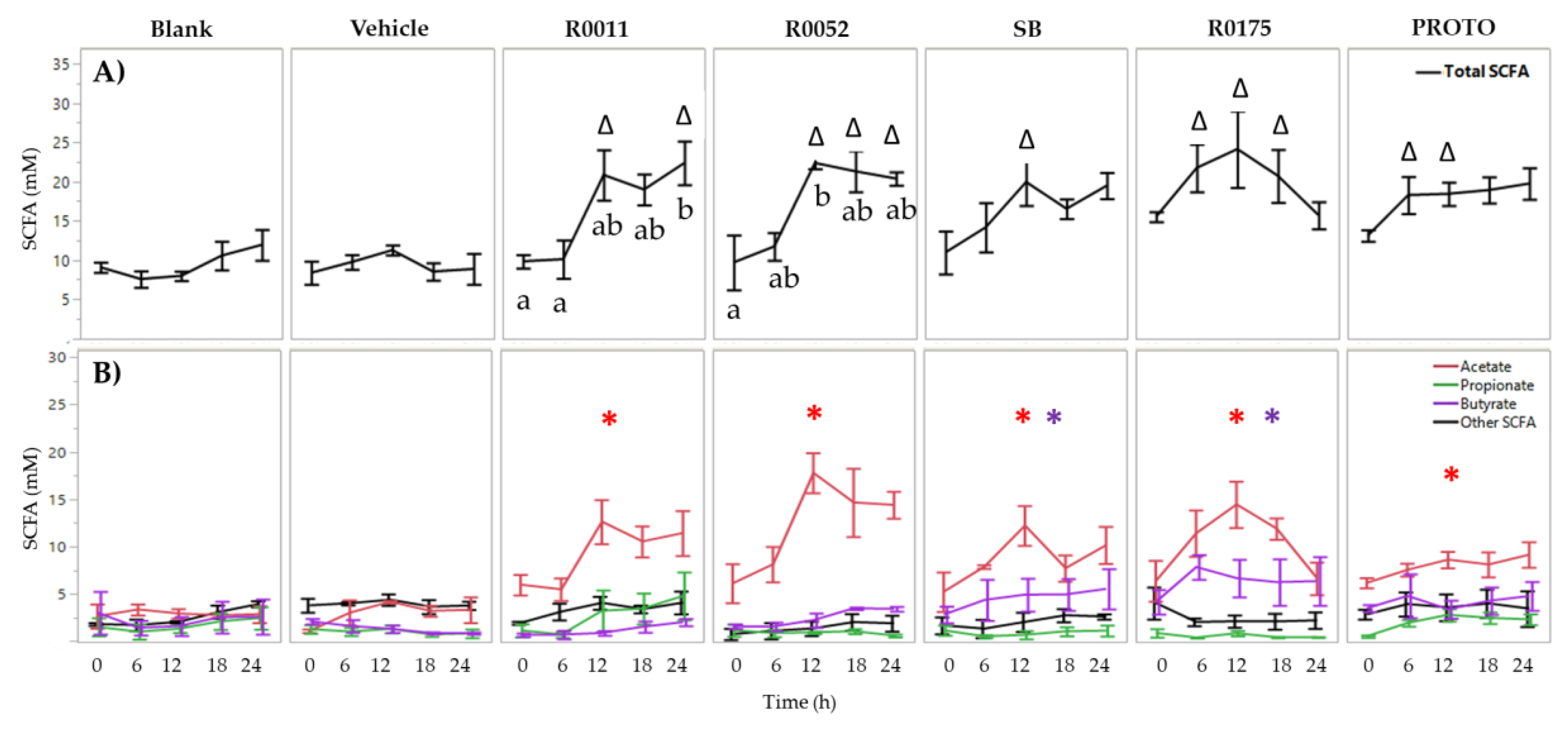
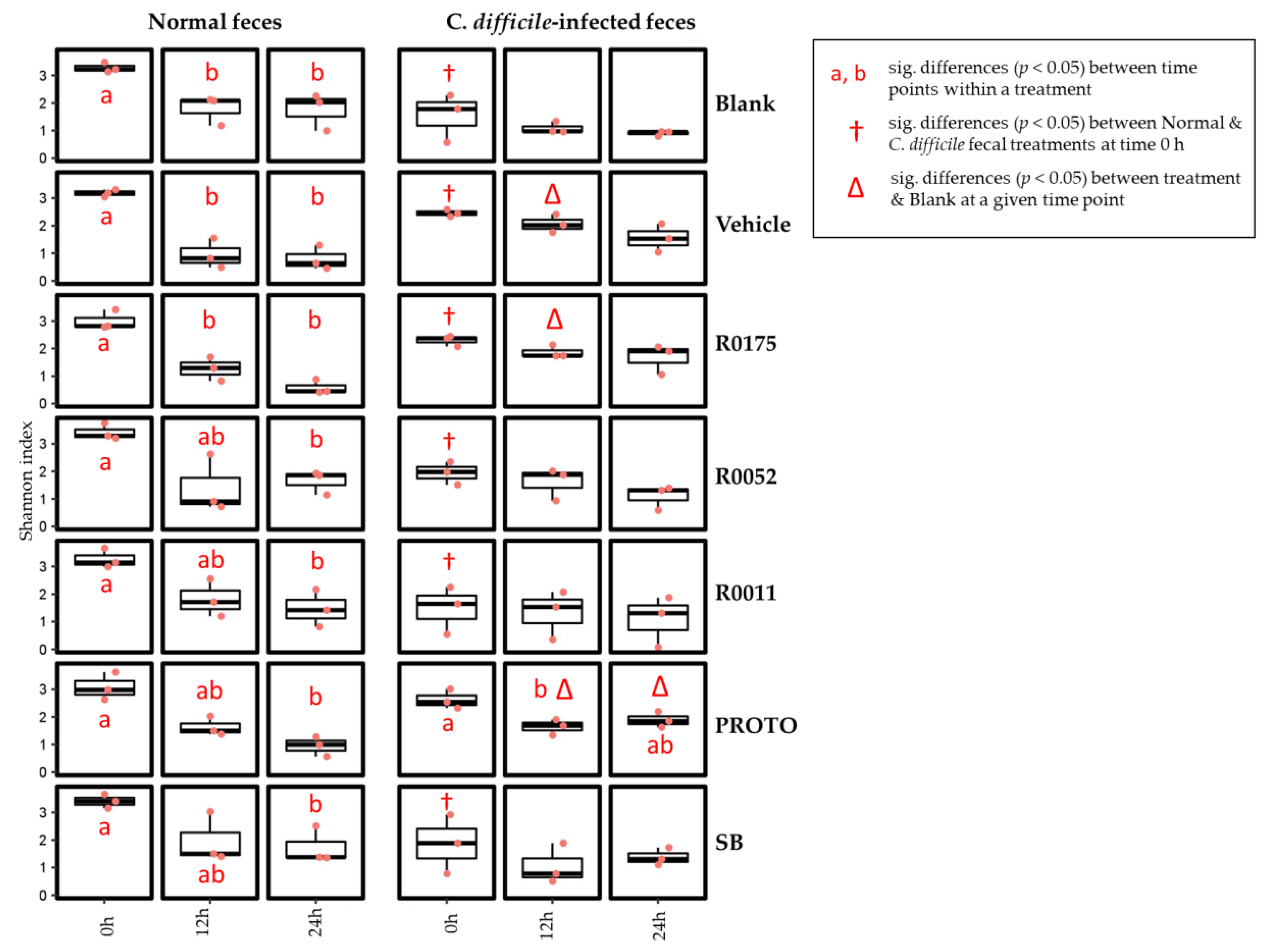
3.1. SCFA Determination in FW
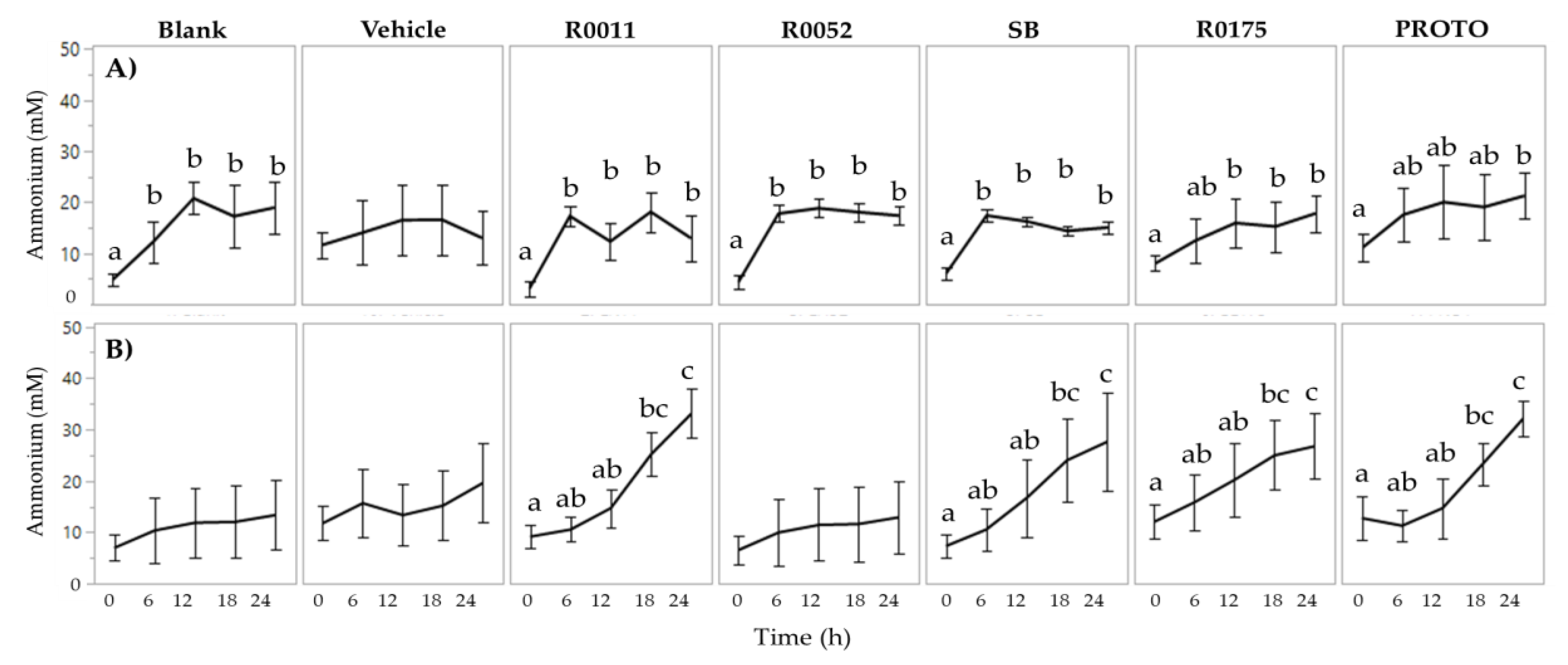
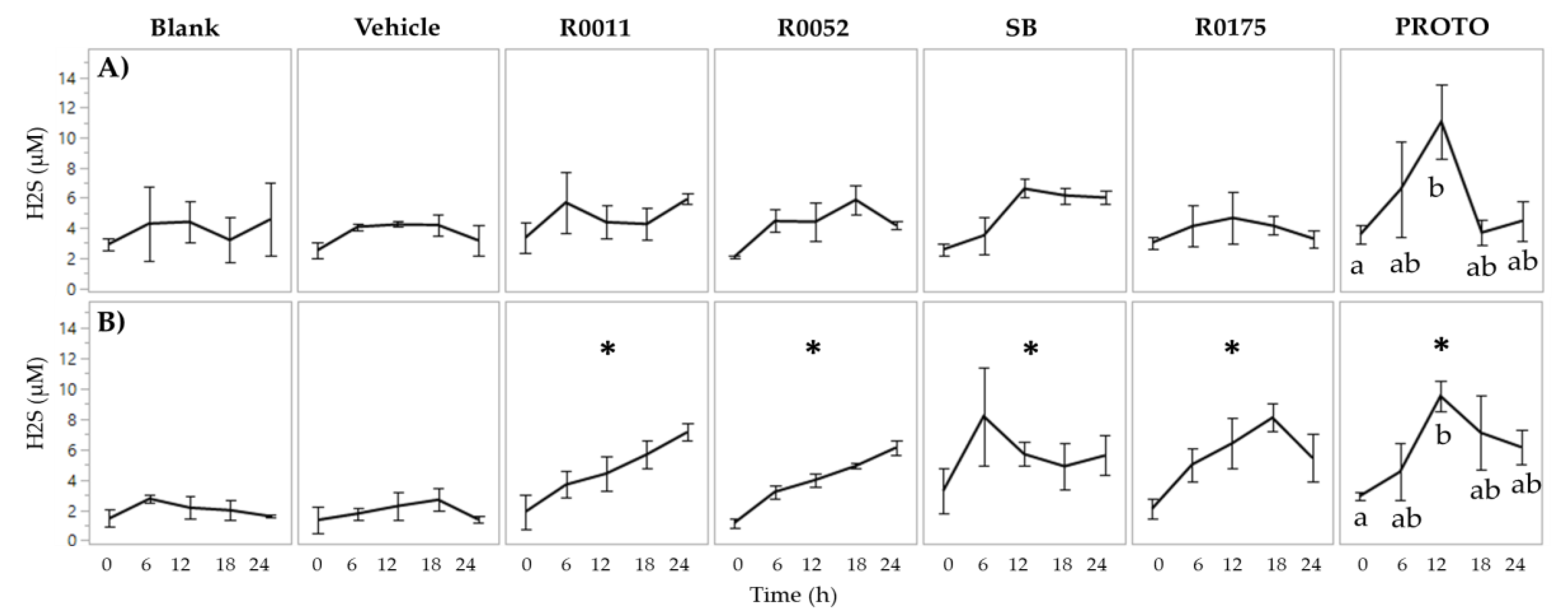
3.2. NH4 and H2S Determination in FW
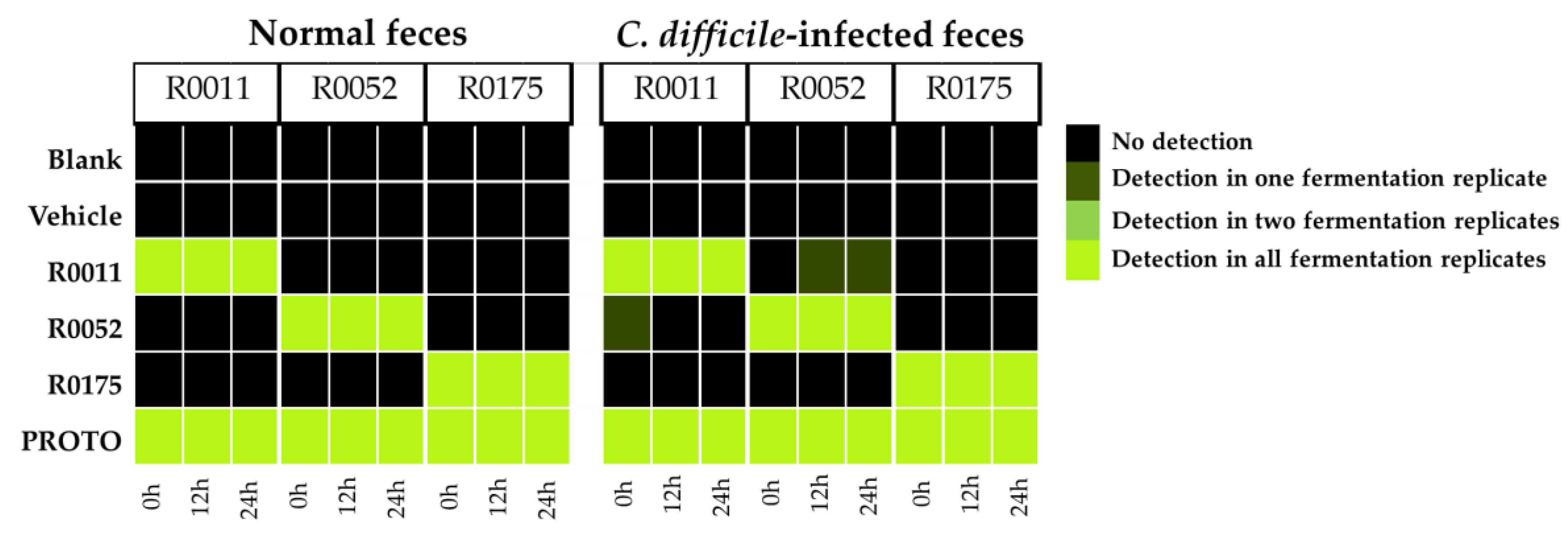
3.3. Detection of Probiotic Strains by qPCR
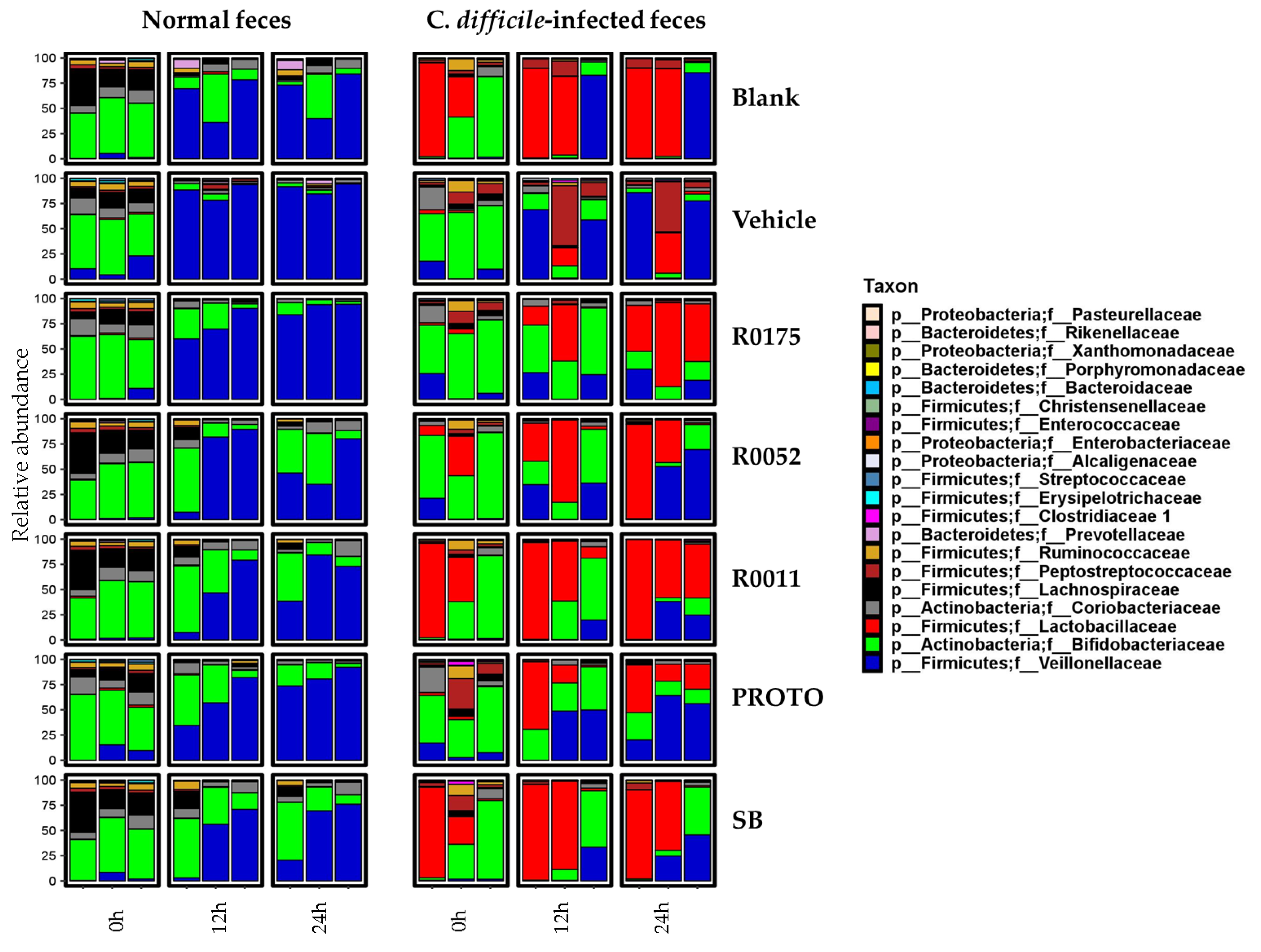
3.4. Microbial Community
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Awad, M.M.; Johanesen, P.A.; Carter, G.P.; Rose, E.; Lyras, D. Clostridium difficile virulence factors: Insights into an anaerobic spore-forming pathogen. Gut Microbes 2014, 5, 579–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupnik, M.; Wilcox, M.H.; Gerding, D.N. Clostridium difficile infection: New developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 526. [Google Scholar] [CrossRef] [PubMed]
- Bibbò, S.; Lopetuso, L.R.; Ianiro, G.; Di Rienzo, T.; Gasbarrini, A.; Cammarota, G. Role of microbiota and innate immunity in recurrent Clostridium difficile infection. J. Immunol. Res. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antharam, V.C.; Li, E.C.; Ishmael, A.; Sharma, A.; Mai, V.; Rand, K.H.; Wang, G.P. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol. 2013, 51, 2884–2892. [Google Scholar] [CrossRef] [Green Version]
- Cook, S.; Sellin, J. Short chain fatty acids in health and disease. Aliment. Pharmacol. Ther. 1998, 12, 499–507. [Google Scholar] [CrossRef]
- Frädrich, C.; Beer, L.-A.; Gerhard, R. Reactive Oxygen Species as Additional Determinants for Cytotoxicity of Clostridium difficile Toxins A and B. Toxins 2016, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Farrow, M.A.; Chumbler, N.M.; Lapierre, L.A.; Franklin, J.L.; Rutherford, S.A.; Goldenring, J.R.; Lacy, D.B. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc. Natl. Acad. Sci. USA 2013, 110, 18674–18679. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Hagen, S.; Pothoulakis, C.; Chen, M.; Medina, N.; Warny, M.; LaMont, J. Clostridium difficile toxin A causes early damage to mitochondria in cultured cells. Gastroenterology 2000, 119, 139–150. [Google Scholar] [CrossRef]
- Surawicz, C.M. Probiotics, antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in humans. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 775–783. [Google Scholar] [CrossRef]
- Goldstein, E.J.C.; Johnson, S.J.; Maziade, P.J.; Evans, C.T.; Sniffen, J.C.; Millette, M.; McFarland, L.V. Probiotics and prevention of Clostridium difficile infection. Anaerobe 2017, 45, 114–119. [Google Scholar] [CrossRef]
- Sanders, M.E. Probiotics: Definition, Sources, Selection, and Uses. Clin. Infect. Dis. 2008, 46, S58–S61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBlanc, J.G.; Chain, F.; Martín, R.; Bermúdez-Humarán, L.G.; Courau, S.; Langella, P. Beneficial effects on host energy metabolism of short-chain fatty acids and vitamins produced by commensal and probiotic bacteria. Microb. Cell Factories 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wullt, M.; Johansson Hagslätt, M.-L.; Odenholt, I.; Berggren, A. Lactobacillus plantarum 299v Enhances the Concentrations of Fecal Short-Chain Fatty Acids in Patients with Recurrent Clostridium difficile-Associated Diarrhea. Dig. Dis. Sci. 2007, 52, 2082. [Google Scholar] [CrossRef] [PubMed]
- Ouwehand, A.C.; Salminen, S.; Isolauri, E. Probiotics: An overview of beneficial effects. In Proceedings of the Lactic Acid Bacteria: Genetics, Metabolism and Applications: Seventh Symposium on Lactic Acid Bacteria: Genetics, Metabolism and Applications, Egmond aan Zee, The Netherlands, 1–5 September 2002; Siezen, R.J., Kok, J., Abee, T., Schasfsma, G., Eds.; Springer: Dordrecht, The Netherlands, 2002; pp. 279–289. [Google Scholar] [CrossRef]
- Ooi, L.-G.; Liong, M.-T. Cholesterol-Lowering Effects of Probiotics and Prebiotics: A Review of in Vivo and in Vitro Findings. Int. J. Mol. Sci. 2010, 11, 2499–2522. [Google Scholar] [CrossRef]
- Marteau, P.; Seksik, P.; Jian, R. Probiotics and intestinal health effects: A clinical perspective. Br. J. Nutr. 2007, 88, s51–s57. [Google Scholar] [CrossRef] [Green Version]
- McFarland, L.V.; Surawicz, C.M.; Greenberg, R.N.; Fekety, R.; Elmer, G.W.; Moyer, K.A.; Melcher, S.A.; Bowen, K.E.; Cox, J.L.; Noorani, Z. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994, 271, 1913–1918. [Google Scholar] [CrossRef]
- Collado, M.C.; Meriluoto, J.; Salminen, S. In vitro analysis of probiotic strain combinations to inhibit pathogen adhesion to human intestinal mucus. Food Res. Int. 2007, 40, 629–636. [Google Scholar] [CrossRef]
- Elmadfa, I.; Klein, P.; Meyer, A.L. Immune-stimulating effects of lactic acid bacteria in vivo and in vitro. Proc. Nutr. Soc. 2010, 69, 416–420. [Google Scholar] [CrossRef] [Green Version]
- Boirivant, M.; Strober, W. The mechanism of action of probiotics. Curr. Opin. Gastroenterol. 2007, 23, 679–692. [Google Scholar] [CrossRef]
- Sartor, R.B. Probiotic therapy of intestinal inflammation and infections. Curr. Opin. Gastroenterol. 2005, 21, 44–50. [Google Scholar]
- Wollowski, I.; Rechkemmer, G.; Pool-Zobel, B.L. Protective role of probiotics and prebiotics in colon cancer. Am. J. Clin. Nutr. 2001, 73, 451s–455s. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, R.D. The role of probiotic cultures in the control of gastrointestinal health. J. Nutr. 2000, 130, 396S–402S. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.A.C.; Balciunas, E.M.; Converti, A.; Cotter, P.D.; de Souza Oliveira, R.P. Bacteriocin production by Bifidobacterium spp. A review. Biotechnol. Adv. 2013, 31, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Marco, M.L.; Pavan, S.; Kleerebezem, M. Towards understanding molecular modes of probiotic action. Curr. Opin. Biotechnol. 2006, 17, 204–210. [Google Scholar] [CrossRef]
- Gibson, G.; McCartney, A.; Rastall, R. Prebiotics and resistance to gastrointestinal infections. Br. J. Nutr. 2005, 93, S31–S34. [Google Scholar] [CrossRef]
- Van Zanten, G.C.; Knudsen, A.; Röytiö, H.; Forssten, S.; Lawther, M.; Blennow, A.; Lahtinen, S.J.; Jakobsen, M.; Svensson, B.; Jespersen, L. The Effect of Selected Synbiotics on Microbial Composition and Short-Chain Fatty Acid Production in a Model System of the Human Colon. PLoS ONE 2012, 7, e47212. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Merkel, G.J.; Bhunia, A.K. Lactobacillus delbrueckii ssp. bulgaricus B-30892 can inhibit cytotoxic effects and adhesion of pathogenic Clostridium difficile to Caco-2 cells. Gut Pathog. 2009, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Trejo, F.M.; Minnaard, J.; Perez, P.F.; De Antoni, G.L. Inhibition of Clostridium difficile growth and adhesion to enterocytes by Bifidobacterium supernatants. Anaerobe 2006, 12, 186–193. [Google Scholar] [CrossRef]
- Castagliuolo, I.; Riegler, M.F.; Valenick, L.; LaMont, J.T.; Pothoulakis, C. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect. Immun. 1999, 67, 302–307. [Google Scholar] [CrossRef]
- Qamar, A.; Aboudola, S.; Warny, M.; Michetti, P.; Pothoulakis, C.; LaMont, J.T.; Kelly, C.P. Microbial immunity and vaccines-Saccharomyces boulardii Stimulates Intestinal Immunoglobulin A Immune Response to Clostridium difficile Toxin A in Mice. Infect. Immun. 2001, 69, 2762–2765. [Google Scholar] [CrossRef] [Green Version]
- Castagliuolo, I.; LaMont, J.T.; Nikulasson, S.T.; Pothoulakis, C. Saccharomyces boulardii protease inhibits Clostridium difficile toxin A effects in the rat ileum. Infect. Immun. 1996, 64, 5225–5232. [Google Scholar] [PubMed]
- Goldenberg, J.Z.; Yap, C.; Lytvyn, L.; Lo, C.K.F.; Beardsley, J.; Mertz, D.; Johnston, B.C. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V. Meta-analysis of probiotics for the prevention of antibiotic associated diarrhea and the treatment of Clostridium difficile disease. Am. J. Gastroenterol. 2006, 101, 812. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.; Salewski, R.P.; Christman, M.C.; Girard, S.A.; Tompkins, T.A. Effectiveness of Lactobacillus helveticus and Lactobacillus rhamnosus for the management of antibiotic-associated diarrhoea in healthy adults: A randomised, double-blind, placebo-controlled trial. Br. J. Nutr. 2016, 116, 94–103. [Google Scholar] [CrossRef]
- Molly, K.; Woestyne, M.V.; Verstraete, W. Development of a 5-step multi-chamber reactor as a simulation of the human intestinal microbial ecosystem. Appl. Microbiol. Biotechnol. 1993, 39, 254–258. [Google Scholar] [CrossRef]
- Payne, A.N.; Zihler, A.; Chassard, C.; Lacroix, C. Advances and perspectives in in vitro human gut fermentation modeling. Trends Biotechnol. 2012, 30, 17–25. [Google Scholar] [CrossRef]
- Theriot, C.M.; Young, V.B. Interactions between the Gastrointestinal Microbiome and Clostridium difficile. Ann. Rev. Microbiol. 2015, 69, 445–461. [Google Scholar] [CrossRef] [Green Version]
- Nyangale, E.P.; Mottram, D.S.; Gibson, G.R. Gut Microbial Activity, Implications for Health and Disease: The Potential Role of Metabolite Analysis. J. Proteome Res. 2012, 11, 5573–5585. [Google Scholar] [CrossRef]
- Medani, M.; Collins, D.; Docherty, N.G.; Baird, A.W.; O’Connell, P.R.; Winter, D.C. Emerging role of hydrogen sulfide in colonic physiology and pathophysiology. Inflamm. Bowel Dis. 2010, 17, 1620–1625. [Google Scholar] [CrossRef]
- Schneider, S.-M.; Girard-Pipau, F.; Filippi, J.; Hebuterne, X.; Moyse, D.; Hinojosa, G.-C.; Pompei, A.; Rampal, P. Effects of Saccharomyces boulardii on fecal short-chain fatty acids and microflora in patients on long-term total enteral nutrition. World J. Gastroenterol. 2005, 11, 6165–6169. [Google Scholar] [CrossRef]
- Le Barz, M.; Anhê, F.F.; Varin, T.V.; Desjardins, Y.; Levy, E.; Roy, D.; Urdaci, M.C.; Marette, A. Probiotics as complementary treatment for metabolic disorders. Diabetes Metab. J. 2015, 39, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi Ekbatan, S.; Sleno, L.; Sabally, K.; Khairallah, J.; Azadi, B.; Rodes, L.; Prakash, S.; Donnelly, D.J.; Kubow, S. Biotransformation of polyphenols in a dynamic multistage gastrointestinal model. Food Chem. 2016, 204, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Kubow, S.; Iskandar, M.M.; Sabally, K.; Azadi, B.; Sadeghi Ekbatan, S.; Kumarathasan, P.; Das, D.D.; Prakash, S.; Burgos, G.; zum Felde, T. Biotransformation of anthocyanins from two purple-fleshed sweet potato accessions in a dynamic gastrointestinal system. Food Chem. 2016, 192, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.S.; Wilcox, M.H.; Chilton, C.H. An In Vitro Model of the Human Colon: Studies of Intestinal Biofilms and Clostridium difficile Infection. In Clostridium Difficile: Methods and Protocols; Roberts, A.P., Mullany, P., Eds.; Springer: New York, NY, USA, 2016; pp. 223–234. [Google Scholar] [CrossRef]
- Ursell, L.K.; Haiser, H.J.; Van Treuren, W.; Garg, N.; Reddivari, L.; Vanamala, J.; Dorrestein, P.C.; Turnbaugh, P.J.; Knight, R. The Intestinal Metabolome: An Intersection between Microbiota and Host. Gastroenterology 2014, 146, 1470–1476. [Google Scholar] [CrossRef] [Green Version]
- Gaisawat, M.B.; Iskandar, M.M.; MacPherson, C.W.; Tompkins, T.A.; Kubow, S. Probiotic Supplementation is Associated with Increased Antioxidant Capacity and Copper Chelation in C. difficile-Infected Fecal Water. Nutrients 2019, 11, 2007. [Google Scholar] [CrossRef] [Green Version]
- Tzounis, X.; Vulevic, J.; Kuhnle, G.G.; George, T.; Leonczak, J.; Gibson, G.R.; Kwik-Uribe, C.; Spencer, J.P. Flavanol monomer-induced changes to the human faecal microflora. Br. J. Nutr. 2008, 99, 782–792. [Google Scholar] [CrossRef] [Green Version]
- Molly, K.; Vande Woestyne, M.; de Smet, J.; Verstraete, W. Validation of the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) reactor using microorganism-associated activities. Microb. Ecol. Health Dis. 1994, 7. [Google Scholar] [CrossRef]
- Gilboa-Garber, N. Direct spectrophotometric determination of inorganic sulfide in biological materials and in other complex mixtures. Anal. Biochem. 1971, 43, 129–133. [Google Scholar] [CrossRef]
- Koroleff, F. Methods of Sea Waters Analysis; Grasshoff, K., Ed.; Verlag Chemie: New York, NY, USA, 1976; pp. 126–133. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]
- MacPherson, C.W.; Mathieu, O.; Tremblay, J.; Champagne, J.; Nantel, A.; Girard, S.-A.; Tompkins, T.A. Gut Bacterial Microbiota and its Resistome Rapidly Recover to Basal State Levels after Short-term Amoxicillin-Clavulanic Acid Treatment in Healthy Adults. Sci. Rep. 2018, 8, 11192. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.; Yergeau, E. Systematic processing of ribosomal RNA gene amplicon sequencing data. GigaScience 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; Gonzalez, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. 2011, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haarman, M.; Knol, J. Quantitative real-time PCR assays to identify and quantify fecal Bifidobacterium species in infants receiving a prebiotic infant formula. Appl. Environ. Microbiol. 2005, 71, 2318–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. Chapter Three—The Role of Short-Chain Fatty Acids in Health and Disease. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 121, pp. 91–119. [Google Scholar]
- Shetty, S.A.; Marathe, N.P.; Lanjekar, V.; Ranade, D.; Shouche, Y.S. Comparative Genome Analysis of Megasphaera sp. Reveals Niche Specialization and Its Potential Role in the Human Gut. PLoS ONE 2013, 8, e79353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhu, R.; Altman, E.; Eiteman, M.A. Lactate and acrylate metabolism by Megasphaera elsdenii under batch and steady-state conditions. Appl. Environ. Microbiol. 2012, 78, 8564–8570. [Google Scholar] [CrossRef] [Green Version]
- Theriot, C.M.; Young, V.B. Microbial and metabolic interactions between the gastrointestinal tract and Clostridium difficile infection. Gut Microbes 2014, 5, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Pessione, E. Lactic acid bacteria contribution to gut microbiota complexity: Lights and shadows. Front. Cell. Infect. Microbiol. 2012, 2, 86. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef]
- Sivieri, K.; Morales, M.L.V.; Adorno, M.A.T.; Sakamoto, I.K.; Saad, S.M.I.; Rossi, E.A. Lactobacillus acidophilus CRL 1014 improved “gut health” in the SHIME®reactor. BMC Gastroenterol. 2013, 13, 100. [Google Scholar] [CrossRef]
- Girard-pipau, F.; Pompei, A.; Nano, J.L.; Boquet, X.; Rampal, P. Intestinal Microflora, Short Chain and Cellular Fatty Acids, Influence of a Probiotic Saccharomyces boulardii. Microb. Ecol. Health Dis. 2002, 14, 221–228. [Google Scholar] [CrossRef] [Green Version]
- De Vuyst, L.; Leroy, F. Cross-feeding between bifidobacteria and butyrate-producing colon bacteria explains bifdobacterial competitiveness, butyrate production, and gas production. Int. J. Food Microbiol. 2011, 149, 73–80. [Google Scholar] [CrossRef]
- Falony, G.; Vlachou, A.; Verbrugghe, K.; Vuyst, L.D. Cross-Feeding between Bifidobacterium longum BB536 and Acetate-Converting, Butyrate-Producing Colon Bacteria during Growth on Oligofructose. Appl. Environ. Microbiol. 2006, 72, 7835–7841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windey, K.; De Preter, V.; Verbeke, K. Relevance of protein fermentation to gut health. Mol. Nutr. Food Res. 2012, 56, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Magee, E.; Bingham, S. Protein degradation in the large intestine: Relevance to colorectal cancer. Curr. Issues Intest. Microbiol. 2000, 1, 51–58. [Google Scholar] [PubMed]
- Wrong, O.M.; Vince, A. Urea and ammonia metabolism in the human large intestine. Proc. Nutr. Soc. 2007, 43, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, D.R.; Levitt, M.D.; Farrugia, G.; Szurszewski, J.H. Endogenous production of H2S in the gastrointestinal tract: Still in search of a physiologic function. Antioxid. Redox Signal. 2010, 12, 1135–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasova, L.; Konopelski, P.; Ufnal, M. Gut Bacteria and Hydrogen Sulfide: The New Old Players in Circulatory System Homeostasis. Molecules 2016, 21, 1558. [Google Scholar] [CrossRef]
- Britton, R.A.; Young, V.B. Role of the Intestinal Microbiota in Resistance to Colonization by Clostridium difficile. Gastroenterology 2014, 146, 1547–1553. [Google Scholar] [CrossRef] [Green Version]
- Lahtinen, S.J.; Forssten, S.; Aakko, J.; Granlund, L.; Rautonen, N.; Salminen, S.; Viitanen, M.; Ouwehand, A.C. Probiotic cheese containing Lactobacillus rhamnosus HN001 and Lactobacillus acidophilus NCFM® modifies subpopulations of fecal lactobacilli and Clostridium difficile in the elderly. Age 2012, 34, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Forssten, S.D.; Röytiö, H.; Hibberd, A.A.; Ouwehand, A.C. The effect of polydextrose and probiotic lactobacilli in a Clostridium difficile–infected human colonic model. Microb. Ecol. Health Dis. 2015, 26, 27988. [Google Scholar] [CrossRef]
- Lee, Y.K.; Ho, P.S.; Low, C.S.; Arvilommi, H.; Salminen, S. Permanent colonization by Lactobacillus casei is hindered by the low rate of cell division in mouse gut. Appl. Environ. Microbiol. 2004, 70, 670–674. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strain | Primer Name | Targeted Sequence | Gene Target | Amplicon Size (bp) |
|---|---|---|---|---|
| B. longum R0175 | R175_AP_HP10_F | GTC GCC ACA TTT CAT CGC AA | Hypothetical protein | 99 |
| R175_AP_HP10_R | GAG AGC TTC GAT TGG CGA AC | |||
| L. helveticus R0052 | pIR52-1-orf5 F1 | AGA ATC AAG CAG AGA CTG GCT ACG | An ORF in a plasmid specific to R0052 | 150 |
| pIR52-1-orf5 R1 | GGA CCG GAT TTG AGT AGA GGT A | |||
| L. rhamnosus R0011 | 113A29_293FL | ACT CCA AAG AGC ATT ACC TCC G | 113A29 phage head protein | 71 |
| 113A29_321RU | TGA ATA TGC CGG ATC TAA GTC CA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaisawat, M.B.; MacPherson, C.W.; Tremblay, J.; Piano, A.; Iskandar, M.M.; Tompkins, T.A.; Kubow, S. Probiotic Supplementation in a Clostridium difficile-Infected Gastrointestinal Model Is Associated with Restoring Metabolic Function of Microbiota. Microorganisms 2020, 8, 60. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8010060
Gaisawat MB, MacPherson CW, Tremblay J, Piano A, Iskandar MM, Tompkins TA, Kubow S. Probiotic Supplementation in a Clostridium difficile-Infected Gastrointestinal Model Is Associated with Restoring Metabolic Function of Microbiota. Microorganisms. 2020; 8(1):60. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8010060
Chicago/Turabian StyleGaisawat, Mohd Baasir, Chad W. MacPherson, Julien Tremblay, Amanda Piano, Michèle M. Iskandar, Thomas A. Tompkins, and Stan Kubow. 2020. "Probiotic Supplementation in a Clostridium difficile-Infected Gastrointestinal Model Is Associated with Restoring Metabolic Function of Microbiota" Microorganisms 8, no. 1: 60. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8010060