
Dual RNA-Seq Enables Full-Genome Assembly of Measles Virus and Characterization of Host–Pathogen Interactions

 , , ,
, , ,
Abstract
:1. Introduction
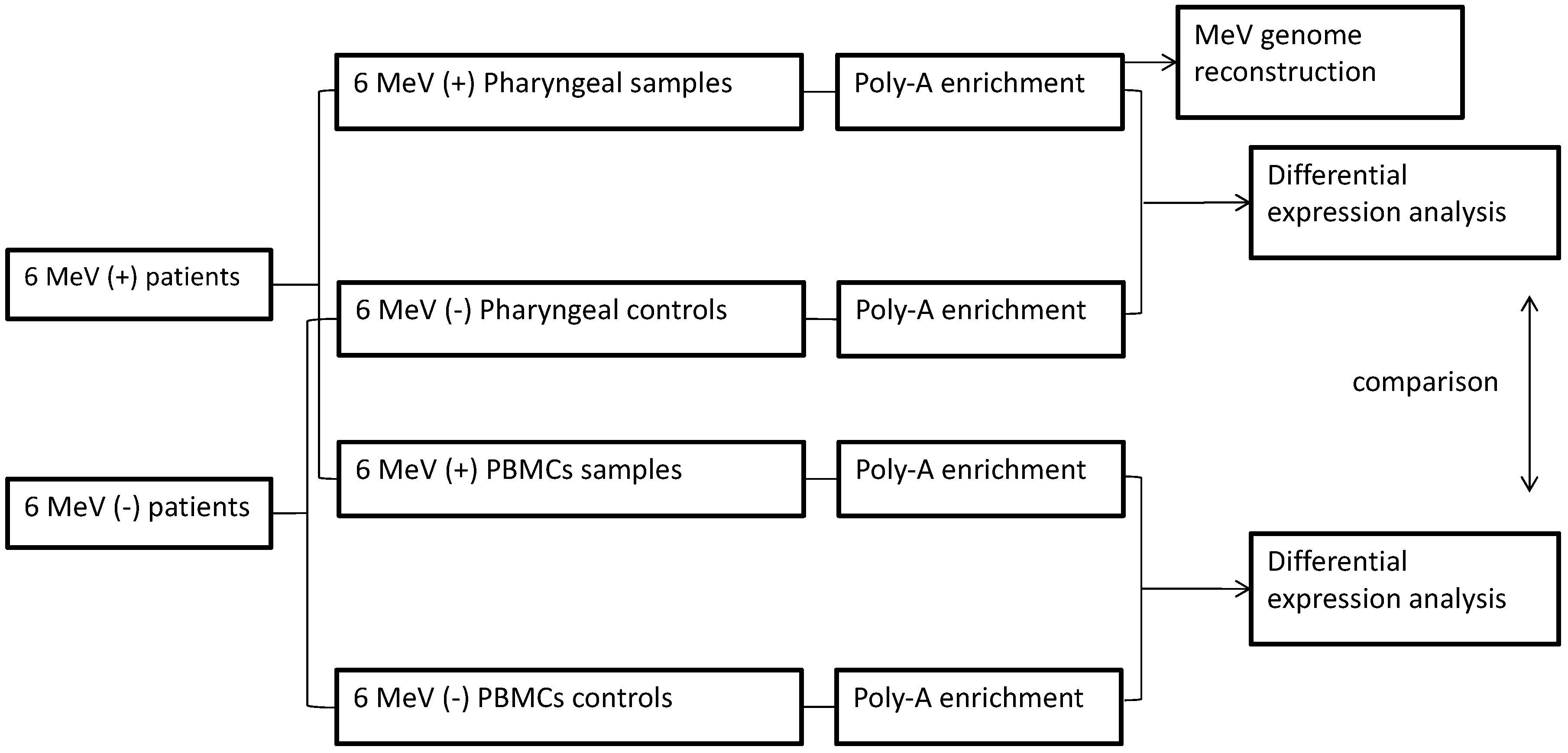
2. Materials and Methods
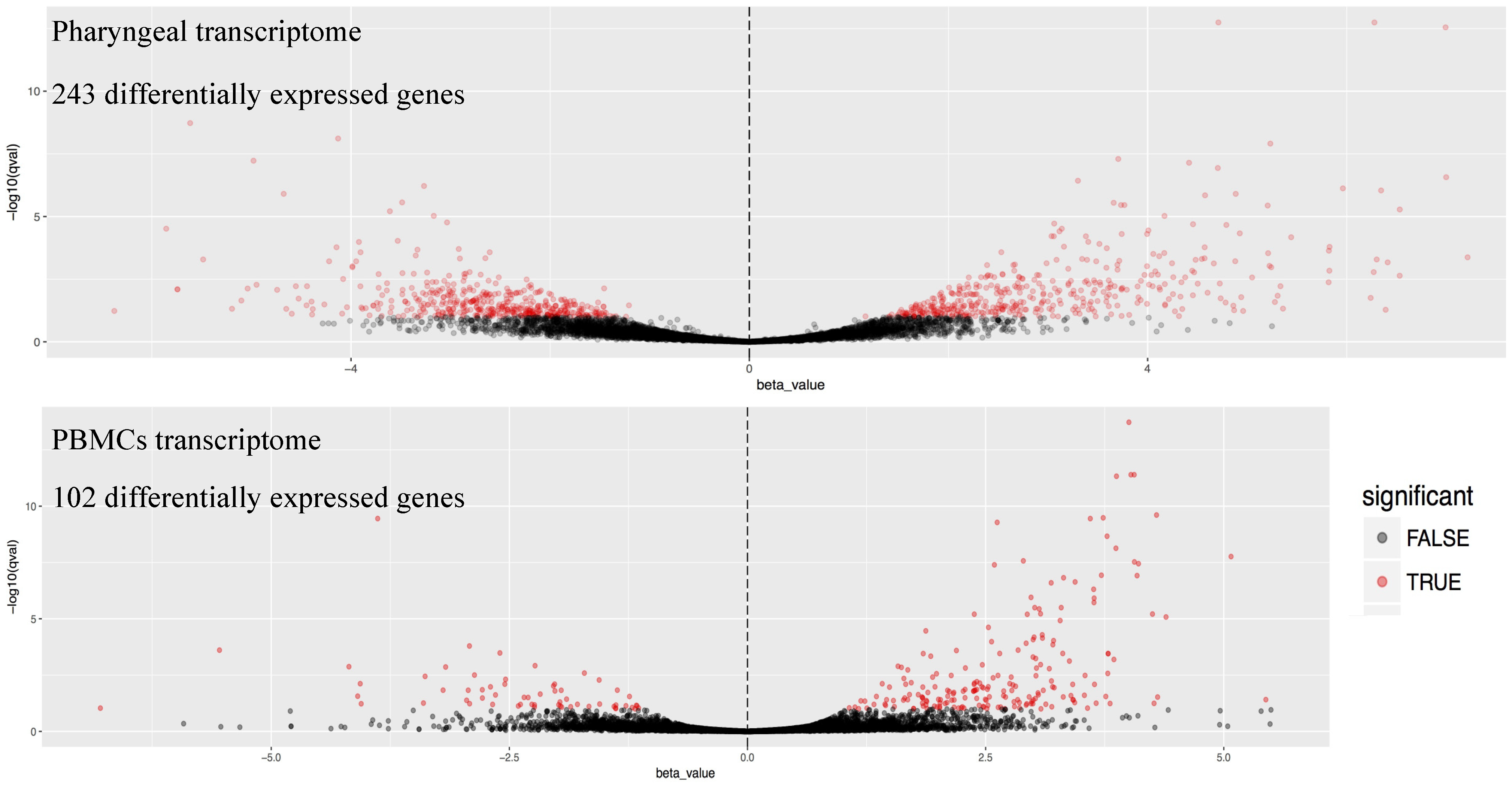
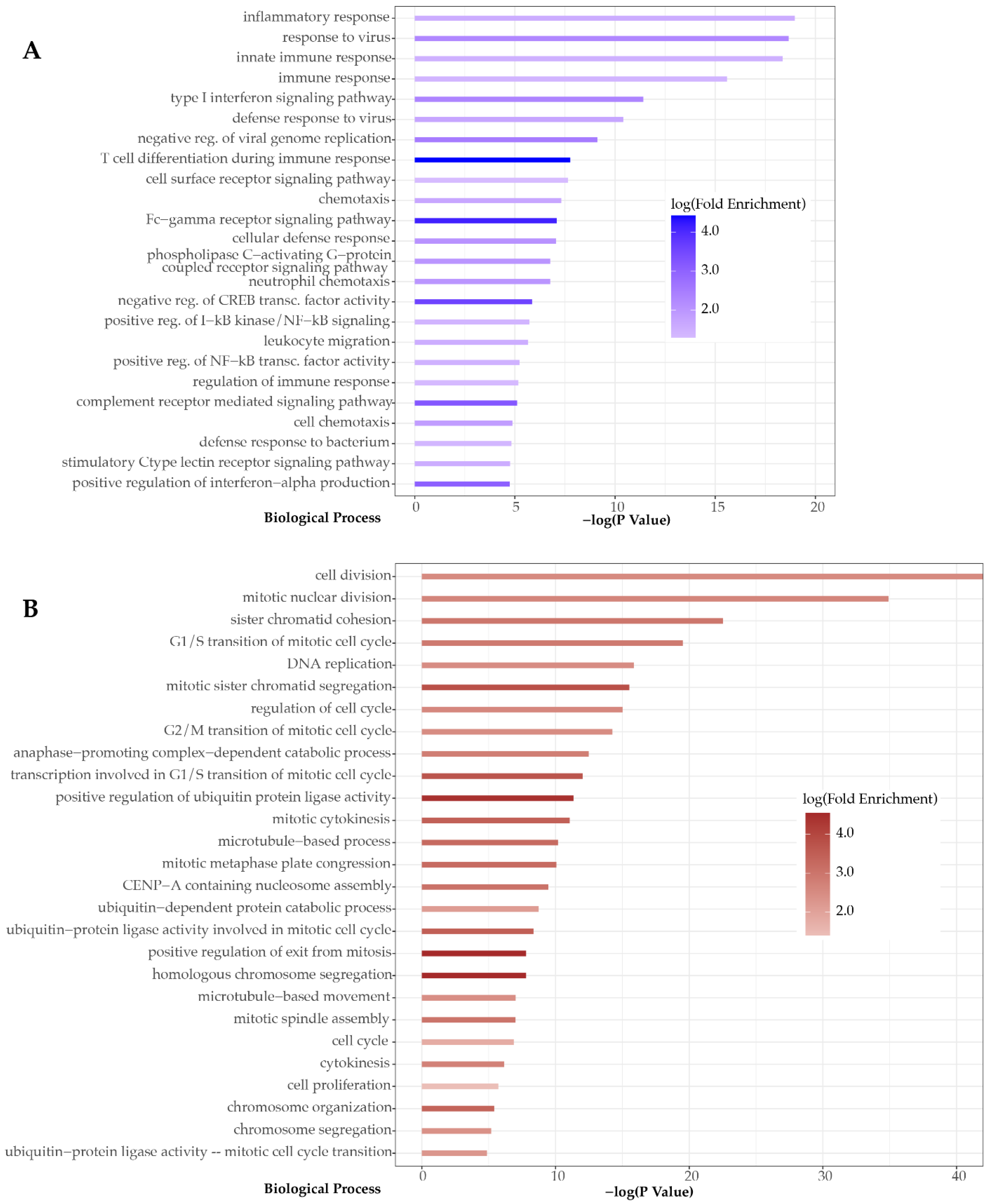
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rota, P.A.; Moss, W.J.; Takeda, M.; De Swart, R.L.; Thompson, K.M.; Goodson, J.L. Measles. Nat. Rev. Dis. Prim. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Wassilak, S.; Emiroglu, N.; Uzicanin, A.; Deshesvoi, S.; Jankovic, D.; Goel, A.; Khetsuriani, N. What will it take to achieve measles elimination in the World Health Organization European Region: Progress from 2003–2009 and essential accelerated actions. J. Infect. Dis. 2011, 204, 325–334. [Google Scholar] [CrossRef]
- Muscat, M.; Bang, H.; Wohlfahrt, J.; Glismann, S.; Mølbak, K. Measles in Europe: An epidemiological assessment. Lancet 2009, 373, 383–389. [Google Scholar] [CrossRef]
- CfDCaP, C. Global Measles and Rubella Laboratory Network, January 2004–June 2005. Morb. Mortal. Wkly. Rep. 2005, 54, 1100–1104. [Google Scholar]
- WHO. Genetic diversity of wildtype measles viruses and the global measles nucleotide surveillance database (MeaNS) = La diversitι gιnιtique des virus rougeoleux de type sauvage et la base de donnιes MeaNS (Measles Nucleotide Surveillance). Wkly. Epidemiol. Rec. Relev. Ιpidιmiologique Hebd. 2015, 90, 373–380. [Google Scholar]
- Harvala, H.; Wiman, A.; Wallensten, A.; Zakikhany, K.; Englund, H.; Brytting, M. Role of sequencing the measles virus hemagglutinin gene and hypervariable region in the measles outbreak investigations in Sweden during 2013–2014. J. Infect. Dis. 2016, 213, 592–599. [Google Scholar] [CrossRef]
- Penedos, A.R.; Myers, R.; Hadef, B.; Aladin, F.; Brown, K.E. Assessment of the utility of whole genome sequencing of measles virus in the characterisation of outbreaks. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Phan, M.V.T.; Schapendonk, C.M.E.; Oude Munnink, B.B.; Koopmans, M.P.G.; de Swart, R.L.; Cotten, M. Complete genome sequences of six measles virus strains. Genome Announc. 2018, 6, 1–2. [Google Scholar] [CrossRef] [Green Version]
- ROBBINS, F.C. Measles: Clinical Features. Am. J. Dis. Child. 1962, 103, 266. [Google Scholar] [CrossRef]
- Simani, O.E.; Adrian, P.V.; Violari, A.; Kuwanda, L.; Otwombe, K.; Nunes, M.C.; Cotton, M.F.; Madhi, S.A. Effect of in-utero HIV exposure and antiretroviral treatment strategies on measles susceptibility and immunogenicity of measles vaccine. Aids 2013, 27, 1583–1591. [Google Scholar] [CrossRef]
- Griffin, D.E. The immune response in measles: Virus control, clearance and protective immunity. Viruses 2016, 8, 282. [Google Scholar] [CrossRef]
- Mόhlebach, M.D.; Mateo, M.; Sinn, P.L.; Prόfer, S.; Katharina, M.; Leonard, V.H.J.; Navaratnarajah, C.K.; Frenzke, M.; Xiao, X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 (PVRL4) is the epithelial receptor for measles virus. Nature 2012, 480, 530–533. [Google Scholar] [CrossRef] [Green Version]
- Moss, W.J. Seminar Measles. Lancet 2017, 6736, 12–19. [Google Scholar] [CrossRef]
- Plattet, P.; Alves, L.; Herren, M.; Aguilar, H.C. Measles virus fusion protein: Structure, function and inhibition. Viruses 2016, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Noyce, R.S.; Richardson, C.D. Nectin 4 is the epithelial cell receptor for measles virus. Trends Microbiol. 2012, 20, 429–439. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; Mesman, A.W.; Geijtenbeek, T.B.H.; Duprex, W.P.; De Swart, R.L. The pathogenesis of measles. Curr. Opin. Virol. 2012, 2, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Zilliox, M.J.; Parmigiani, G.; Griffin, D.E. Gene expression patterns in dendritic cells infected with measles virus compared with other pathogens. Proc. Natl. Acad. Sci. USA 2006, 103, 3363–3368. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Cheng, Y.; Shi, B.; Qian, F.; Wang, F.; Liu, X.; Yang, H.; Xu, Q.; Qi, T.; Zha, L.; et al. Measles Virus Infection in Adults Induces Production of IL-10 and Is Associated with Increased CD4+ CD25+ Regulatory T Cells. J. Immunol. 2008, 181, 7356–7366. [Google Scholar] [CrossRef] [Green Version]
- Christ-Crain, M.; Jaccard-Stolz, D.; Bingisser, R.; Gencay, M.M.; Huber, P.R.; Tamm, M.; Mόller, B. Effect of procalcitonin-guided treatment on antibiotic use and outcome in lower respiratory tract infections: Cluster-randomised, single-blinded intervention trial. Lancet 2004, 363, 600–607. [Google Scholar] [CrossRef]
- Bergin, S.P.; Tsalik, E.L. Procalcitonin: The Right Answer but to Which Question? Clin. Infect. Dis. 2017, 65, 191–193. [Google Scholar] [CrossRef] [Green Version]
- Holcomb, Z.E.; Tsalik, E.L.; Woods, C.W.; McChain, M.T. Host-Based Peripheral Blood Gene Expression Analysis for Diagnosis of Infectious Diseases. J. Clin. Microbiol. 2017, 55, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Yu, J.; Crosby, S.D.; Storch, G.A. Gene expression profiles in febrile children with defined viral and bacterial infection. Proc. Natl. Acad. Sci. USA 2013, 110, 12792–12797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsalik, E.L.; Henao, R.; Nichols, M.; Burke, T.; Ko, E.R.; McClain, M.T.; Hudson, L.L.; Mazur, A.; Freeman, D.H.; Veldman, T.; et al. Host gene expression classifiers diagnose acute respiratory illness etiology. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, N.M.; Bunsow, E.; Falsey, A.R.; Walsh, E.E.; Mejias, A.; Ramilo, O. Superiority of transcriptional profiling over procalcitonin for distinguishing bacterial from viral lower respiratory tract infections in hospitalized adults. J. Infect. Dis. 2015, 212, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef]
- Wesolowska-Andersen, A.; Everman, J.L.; Davidson, R.; Rios, C.; Herrin, R.; Eng, C.; Janssen, W.J.; Liu, A.H.; Oh, S.S.; Kumar, R.; et al. Dual RNA-seq reveals viral infections in asthmatic children without respiratory illness which are associated with changes in the airway transcriptome. Genome Biol. 2017, 18, 12. [Google Scholar] [CrossRef] [Green Version]
- Poole, A.; Urbanek, C.; Eng, C. Dissecting Childhood Asthma with Nasal Transcriptomics Distinguishes Subphenotypes of Disease. Bone 2008, 23, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S. Bowtie2. Nat. Methods 2013, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdσttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Church, D.M.; Schneider, V.A.; Steinberg, K.M.; Schatz, M.C.; Quinlan, A.R.; Chin, C.S.; Kitts, P.A.; Aken, B.; Marth, G.T.; Hoffman, M.M.; et al. Extending reference assembly models. Genome Biol. 2015, 16, 13. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Melchjorsen, J.; Kristiansen, H.; Christiansen, R.; Rintahaka, J.; Matikainen, S.; Paludan, S.R.; Hartmann, R. Differential regulation of the OASL and OAS1 genes in response to viral infections. J. Interf. Cytokine Res. 2009, 29, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Boucas, A.P.; Oliveira, F. dos S. de; Canani, L.H.; Crispim, D. The role of interferon induced with helicase C domain 1 (IFIH1) in the development of type 1 diabetes mellitus. Arq. Bras. Endocrinol. Metabol. 2013, 57, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Durmus, S.; Cakir, T.; Ozgur, A.; Guthke, R. A review on computational systems biology of pathogen-host interactions. Front. Microbiol. 2015, 6, 1–19. [Google Scholar] [CrossRef]
- Sumegi, J.; Barnes, M.G.; Nestheide, S.V.; Molleran-Lee, S.; Villanueva, J.; Zhang, K.; Risma, K.A.; Grom, A.A.; Filipovich, A.H. Gene expression profiling of peripheral blood mononuclear cells from children with active hemophagocytic lymphohistiocytosis. Blood 2011, 117, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Cilloniz, C.; Ebihara, H.; Ni, C.; Neumann, G.; Korth, M.J.; Kelly, S.M.; Kawaoka, Y.; Feldmann, H.; Katze, M.G. Functional Genomics Reveals the Induction of Inflammatory Response and Metalloproteinase Gene Expression during Lethal Ebola Virus Infection. J. Virol. 2011, 85, 9060–9068. [Google Scholar] [CrossRef] [Green Version]
- Chiffoleau, E. C-type lectin-like receptors as emerging orchestrators of sterile inflammation represent potential therapeutic targets. Front. Immunol. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Boro, M.; Balaji, K.N. CXCL1 and CXCL2 Regulate NLRP3 Inflammasome Activation via G-Protein–Coupled Receptor CXCR2. J. Immunol. 2017, 199, 1660–1671. [Google Scholar] [CrossRef] [PubMed]
- Komune, N.; Ichinohe, T.; Ito, M.; Yanagi, Y. Measles Virus V Protein Inhibits NLRP3 Inflammasome-Mediated Interleukin-1 Secretion. J. Virol. 2011, 85, 13019–13026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigola, J.; He, J.; Kinkelin, K.; Pye, V.E.; Renault, L.; Douglas, M.E.; Remus, D.; Cherepanov, P.; Costa, A.; Diffley, J.F.X. Cdt1 stabilizes an open MCM ring for helicase loading. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Shen, T.; Huang, S. The Role of Cdc25A in the Regulation of Cell Proliferation and Apoptosis. Anticancer. Agents Med. Chem. 2012, 12, 631–639. [Google Scholar] [CrossRef]
- Hégarat, N.; Rata, S.; Hochegger, H. Bistability of mitotic entry and exit switches during open mitosis in mammalian cells. BioEssays 2016, 38, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Laksono, B.M.; de Vries, R.D.; Verburgh, R.J.; Visser, E.G.; de Jong, A.; Fraaij, P.L.A.; Ruijs, W.L.M.; Nieuwenhuijse, D.F.; van den Ham, H.J.; Koopmans, M.P.G.; et al. Studies into the mechanism of measles-associated immune suppression during a measles outbreak in the Netherlands. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-Gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol. Cell 2018, 71, 117–128.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakachi, I.; Helfrich, B.A.; Spillman, M.A.; Mickler, E.A.; Olson, C.J.; Rice, J.L.; Coldren, C.D.; Heasley, L.E.; Geraci, M.W.; Stearman, R.S. PTTG1 Levels Are Predictive of Saracatinib Sensitivity in Ovarian Cancer Cell Lines. Clin. Transl. Sci. 2016, 9, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, C.; Iankov, I.D.; Galanis, E. A key anti-viral protein, RSAD2/VIPERIN, restricts the release of Measles virus in infected cells. Physiol. Behav. 2016, 176, 100–106. [Google Scholar] [CrossRef]
- Landry, M.L.; Foxman, E.F. Antiviral Response in the Nasopharynx Identifies Patients with Respiratory Virus Infection. J. Infect. Dis. 2018, 217, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-Y.; Yaneva, R.; Cresswell, P. Viperin: A multifunctional, interferon-inducible protein that regulates virus replication. Bone 2012, 23, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Weidner, J.M.; Qing, M.; Pan, X.-B.; Guo, H.; Xu, C.; Zhang, X.; Birk, A.; Chang, J.; Shi, P.-Y.; et al. Identification of Five Interferon-Induced Cellular Proteins That Inhibit West Nile Virus and Dengue Virus Infections. J. Virol. 2010, 84, 8332–8341. [Google Scholar] [CrossRef] [Green Version]
- John, W.; Schoggins, C.M.R. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2012, 1, 519–525. [Google Scholar] [CrossRef]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Bone 2012, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Okamoto, M.; Morioka, Y.; Okabe, M.; Matsumoto, M.; Seya, T. DDX60 Is Involved in RIG-I-Dependent and Independent Antiviral Responses, and Its Function Is Attenuated by Virus-Induced EGFR Activation. Cell Rep. 2015, 11, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Childs, K.; Randall, R.; Goodbourn, S. Paramyxovirus V proteins interact with the RNA Helicase LGP2 to inhibit RIG-I-dependent interferon induction. J. Virol. 2012, 86, 3411–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizaki, Y.; Takemoto, M.; Kira, R.; Kusuhara, K.; Torisu, H.; Sakai, Y.; Sanefuji, M.; Yukaya, N.; Hara, T. Association of toll-like receptor 3 gene polymorphism with subacute sclerosing panencephalitis. J. Neurovirol. 2008, 14, 486–491. [Google Scholar] [CrossRef]
- Sato, H.; Honma, R.; Yoneda, M.; Miura, R.; Tsukiyama-Kohara, K.; Ikeda, F.; Seki, T.; Watanabe, S.; Kai, C. Measles virus induces cell-type specific changes in gene expression. Virology 2008, 375, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Donohue, R.C.; Pfaller, C.K.; Cattaneo, R. Cyclical adaptation of measles virus quasispecies to epithelial and lymphocytic cells: To V, or not to V. PLoS Pathog. 2019, 15, e1007605. [Google Scholar] [CrossRef]
- Marazzi, I.; Ho, J.S.Y.; Kim, J.; Manicassamy, B.; Dewell, S.; Albrecht, R.A.; Seibert, C.W.; Schaefer, U.; Jeffrey, K.L.; Prinjha, R.K.; et al. Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483, 428–433. [Google Scholar] [CrossRef]
- Law, G.L.; Korth, M.J.; Benecke, A.G.; Katze, M.G. Systems virology: Host-directed approaches to viral pathogenesis and drug targeting. Systems 2014, 11, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Runge, S.; Sparrer, K.M.J.; Lδssig, C.; Hembach, K.; Baum, A.; Garcνa-Sastre, A.; Sφding, J.; Conzelmann, K.K.; Hopfner, K.P. In Vivo Ligands of MDA5 and RIG-I in Measles Virus-Infected Cells. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Yuan, Y. Tumor necrosis factor alpha-induced proteins in malignant tumors: Progress and prospects. Onco. Targets. Ther. 2020, 13, 3303–3318. [Google Scholar] [CrossRef] [Green Version]
- Wenzl, K.; Hofer, S.; Troppan, K.; Lassnig, M.; Steinbauer, E.; Wiltgen, M.; Zulus, B.; Renner, W.; Beham-Schmid, C.; Neumeister, P.; et al. Higher incidence of the SNP Met 788 Ile in the coding region of A20 in diffuse large B cell lymphomas. Tumor Biol. 2016, 37, 4785–4789. [Google Scholar] [CrossRef] [PubMed]
- Mcelroy, R.; Ennis, M.; Schock, B.C. TNFAIP3 (Tumor Necrosis Factor, Alpha-Induced Protein 3). Encycl. Signal. Mol. 2017, 3. [Google Scholar] [CrossRef]
- Dyer, D.P.; Salanga, C.L.; Johns, S.C.; Valdambrini, E.; Fuster, M.M.; Milner, C.M.; Day, A.J.; Handel, T.M. The anti-inflammatory protein TSG-6 regulates chemokine function by inhibiting chemokine/glycosaminoglycan interactions. J. Biol. Chem. 2016, 291, 12627–12640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capp, E.; Milner, C.M.; Williams, J.; Hauck, L.; Jauckus, J.; Strowitzki, T.; Germeyer, A. Modulation of tumor necrosis factor-stimulated gene-6 (TSG-6) expression in human endometrium. Arch. Gynecol. Obstet. 2014, 289, 893–901. [Google Scholar] [CrossRef]
- Huysamen, C.; Brown, G.D. The fungal pattern recognition receptor, Dectin-1, and the associated cluster of C-type lectin-like receptors. FEMS Microbiol. Lett. 2009, 290, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Patin, E.C.; Willcocks, S.; Orr, S.; Ward, T.H.; Lang, R.; Schaible, U.E. Mincle-mediated anti-inflammatory IL-10 response counter-regulates IL-12 in vitro. Innate Immun. 2016, 22, 181–185. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Samperio, P. Expression and regulation of chemokines in mycobacterial infection. J. Infect. 2008, 57, 374–384. [Google Scholar] [CrossRef]
- Zilliox, M.J.; Moss, W.J.; Griffin, D.E. Gene expression changes in peripheral blood mononuclear cells during measles virus infection. Clin. Vaccine Immunol. 2007, 14, 918–923. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Diffley, J.F.X. Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase. Nat. Cell Biol. 2002, 4, 198–207. [Google Scholar] [CrossRef]
- Zou, L.; Stillman, B. Assembly of a Complex Containing Cdc45p, Replication Protein A, and Mcm2p at Replication Origins Controlled by S-Phase Cyclin-Dependent Kinases and Cdc7p-Dbf4p Kinase. Mol. Cell. Biol. 2000, 20, 3086–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Ilves, I.; Tamberg, N.; Petojevic, T.; Nogales, E.; Botchan, M.R.; Berger, J.M. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 2011, 18, 471–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, R.; Ramadurai, S.; Tóth, K.; Togashi, D.M.; Ryder, A.G.; Langowski, J.; Nasheuer, H.P. Cell cycle-dependent mobility of Cdc45 determined in vivo by Fluorescence Correlation Spectroscopy. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Köhler, C.; Koalick, D.; Fabricius, A.; Parplys, A.C.; Borgmann, K.; Pospiech, H.; Grosse, F. Cdc45 is limiting for replication initiation in humans. Cell Cycle 2016, 15, 974–985. [Google Scholar] [CrossRef] [Green Version]
- Suryadinata, R.; Sadowski, M.; Sarcevic, B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci. Rep. 2010, 30, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Blais, A.; Dynlacht, B.D. E2F-associated chromatin modifiers and cell cycle control. Bone 2013, 23, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohe, A.; Erdmann, F.; Babler, C.; Wichapong, K.; Sippl, W.; Schmidt, M. In vitro and in silico studies on substrate recognition and acceptance of human PKMYT1, a Cdk1 inhibitory kinase. Bioorg. Med. Chem. Lett. 2012, 22, 1219–1223. [Google Scholar] [CrossRef]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhao, X.; Zhang, C.; Wang, W.; Li, F.; Liu, D.; Wu, K.; Zhu, D.; Liu, S.; Shen, C.; et al. Overexpressed PKMYT1 promotes tumor progression and associates with poor survival in esophageal squamous cell carcinoma. Cancer Manag. Res. 2019, 11, 7813–7824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, V.N.; Sawatsky, B.; Han, A.X.; Laksono, B.M.; Walz, L.; Parker, E.; Pieper, K.; Anderson, C.A.; De Vries, R.D.; Lanzavecchia, A.; et al. Incomplete genetic reconstitution of B cell pools contributes to prolonged immunosuppression after measles. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Mina, M.J.; Kula, T.; Leng, Y.; Li, M.; Vries, R.D.; De Knip, M.; Siljander, H.; Rewers, M.; Choy, D.F.; Wilson, M.S.; et al. other pathogens. Science 2019, 606, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Wesemann, D.R. Game of clones: How measles remodels the B cell landscape. Sci. Immunol. 2019, 4, 2–5. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D.; McQuaid, S.; van Amerongen, G.; Yόksel, S.; Verburgh, R.J.; Osterhaus, A.D.M.E.; Duprex, W.P.; de Swart, R.L. Measles Immune Suppression: Lessons from the Macaque Model. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Moussallem, T.M.; Guedes, F.; Fernandes, E.R.; Pagliari, C.; Lancellotti, C.L.P.; de Andrade, H.F.; Duarte, M.I.S. Lung involvement in childhood measles: Severe immune dysfunction revealed by quantitative immunohistochemistry. Hum. Pathol. 2007, 38, 1239–1247. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample | Enrichment Method | Total Reads | Total MeV Genome Reads | MeV Reads (%) | Average Depth (×) | MeV Genome Coverage (%) |
|---|---|---|---|---|---|---|
| Pharg_MeV1 | poly-A | 34,370,942 | 405,851 | 1.18 | 1953.95 | 100.00 |
| Pharg_MeV2 | poly-A | 34,817,617 | 282,092 | 0.81 | 1357.61 | 99.25 |
| Pharg_MeV3 | poly-A | 28,188,497 | 90,601 | 0.32 | 436.01 | 100.00 |
| Pharg_MeV4 | poly-A | 29,716,507 | 59,901 | 0.20 | 288.41 | 100.00 |
| Pharg_MeV5 | poly-A | 12,783,974 | 620,934 | 4.86 | 2990.41 | 100.00 |
| Pharg_MeV6 | poly-A | 3,126,258 | 198,309 | 6.34 | 955.13 | 100.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karamitros, T.; Pogka, V.; Papadopoulou, G.; Tsitsilonis, O.; Evangelidou, M.; Sympardi, S.; Mentis, A. Dual RNA-Seq Enables Full-Genome Assembly of Measles Virus and Characterization of Host–Pathogen Interactions. Microorganisms 2021, 9, 1538. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071538
Karamitros T, Pogka V, Papadopoulou G, Tsitsilonis O, Evangelidou M, Sympardi S, Mentis A. Dual RNA-Seq Enables Full-Genome Assembly of Measles Virus and Characterization of Host–Pathogen Interactions. Microorganisms. 2021; 9(7):1538. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071538
Chicago/Turabian StyleKaramitros, Timokratis, Vasiliki Pogka, Gethsimani Papadopoulou, Ourania Tsitsilonis, Maria Evangelidou, Styliani Sympardi, and Andreas Mentis. 2021. "Dual RNA-Seq Enables Full-Genome Assembly of Measles Virus and Characterization of Host–Pathogen Interactions" Microorganisms 9, no. 7: 1538. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071538