
Bacterial Communities in the Embryo of Maize Landraces: Relation with Susceptibility to Fusarium Ear Rot

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Description of Bacterial Embryo-Associated Community
2.2.1. Sterilization of Seeds and Embryo Isolation
2.2.2. Isolation of Cultivable Bacteria
2.2.3. Molecular Characterization of Cultivable Bacteria
2.2.4. Cultivation-Independent Description of Bacterial Community
2.2.5. Validation of Sequencing Data
2.3. In Vitro Characterization of the Antifungal Properties of the Isolated Bacteria
2.3.1. In Vitro Antifungal Assays
2.3.2. In Vivo Biocontrol Assay
2.4. In Field Susceptibility Assay of Selected Maize Accessions
2.5. Statistical Analyses
3. Results
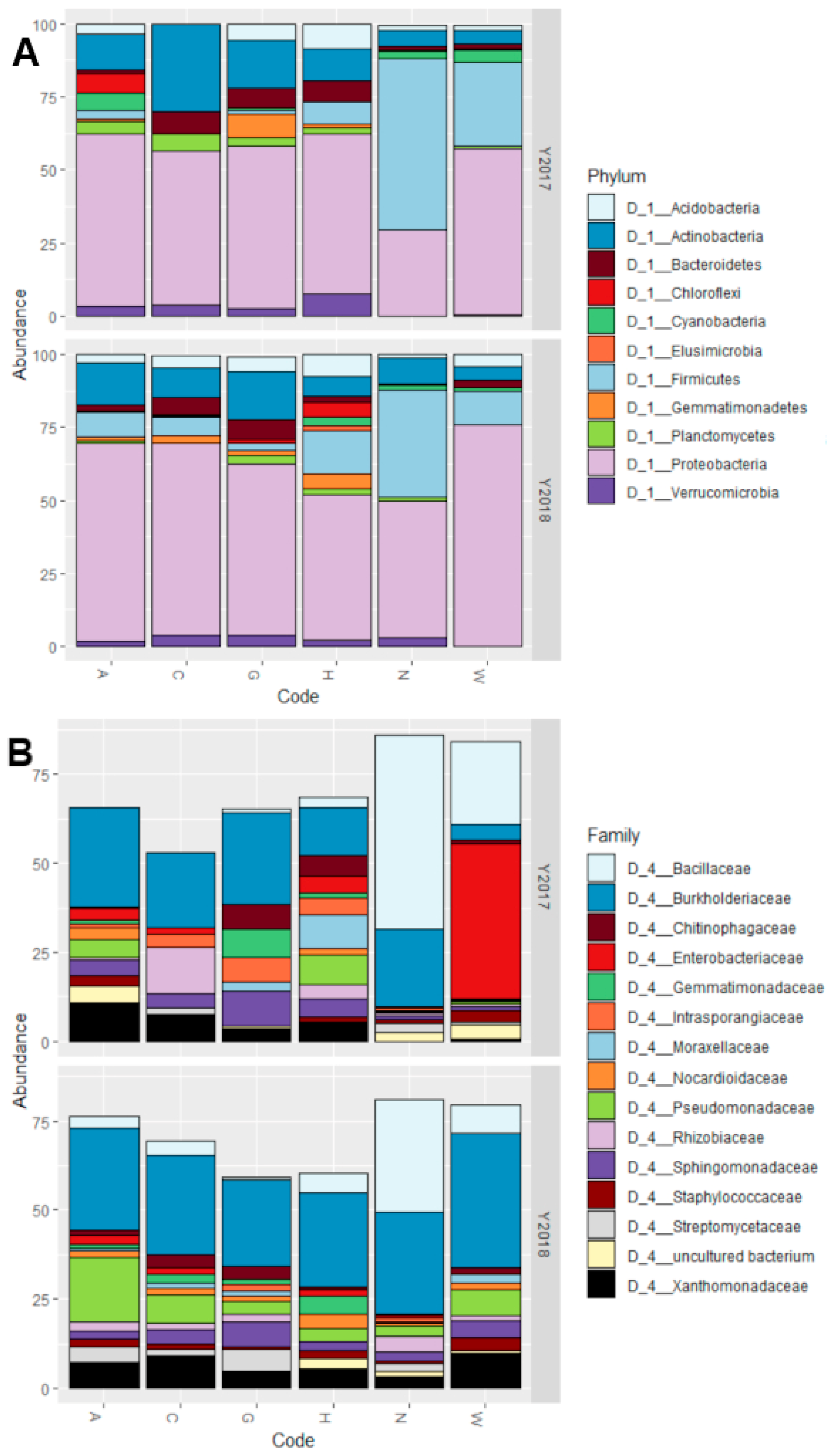
3.1. Gram-Positive Bacteria Dominate the Culturable Fraction of the Maize Embryo Bacterial Microbiota
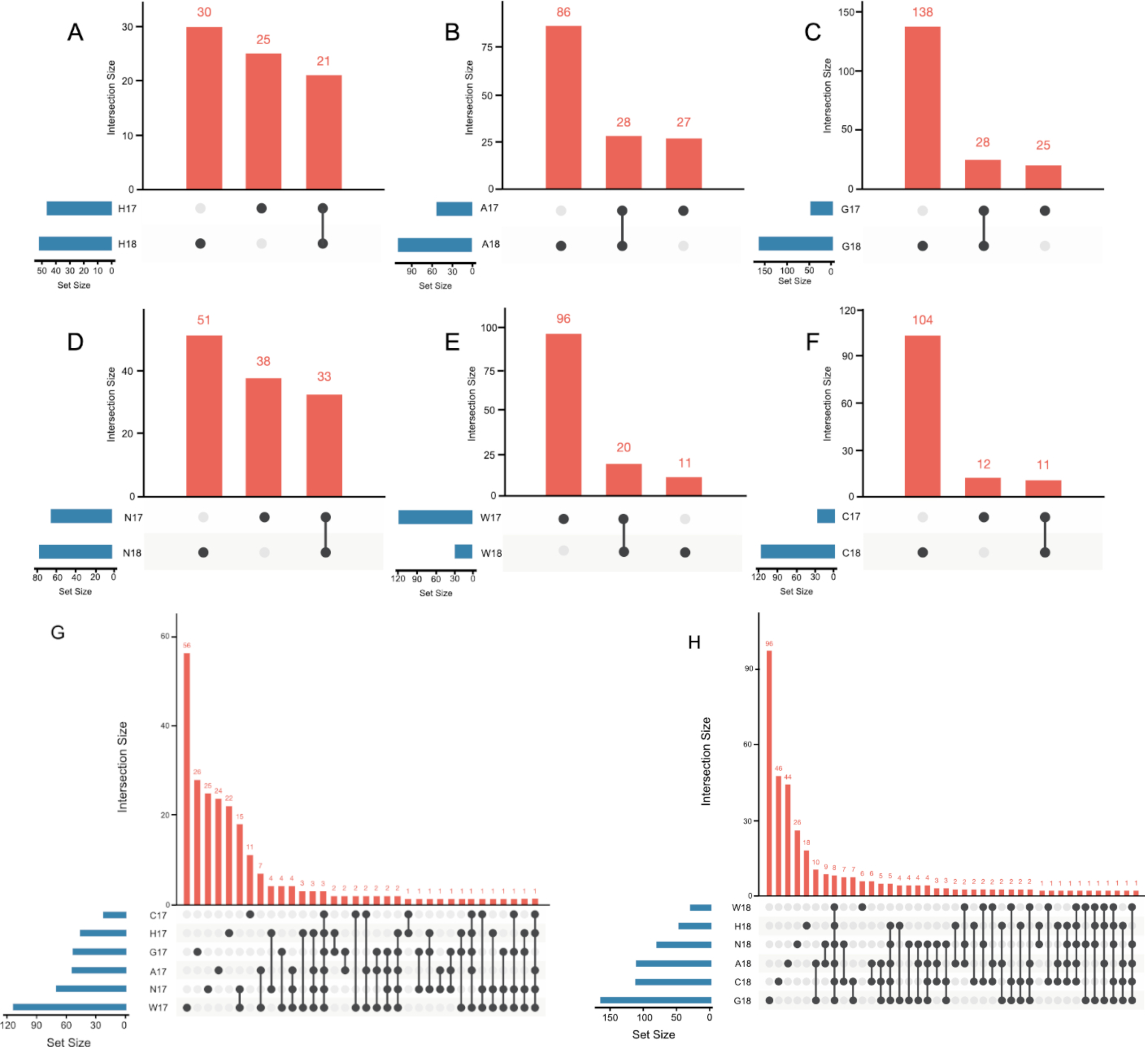
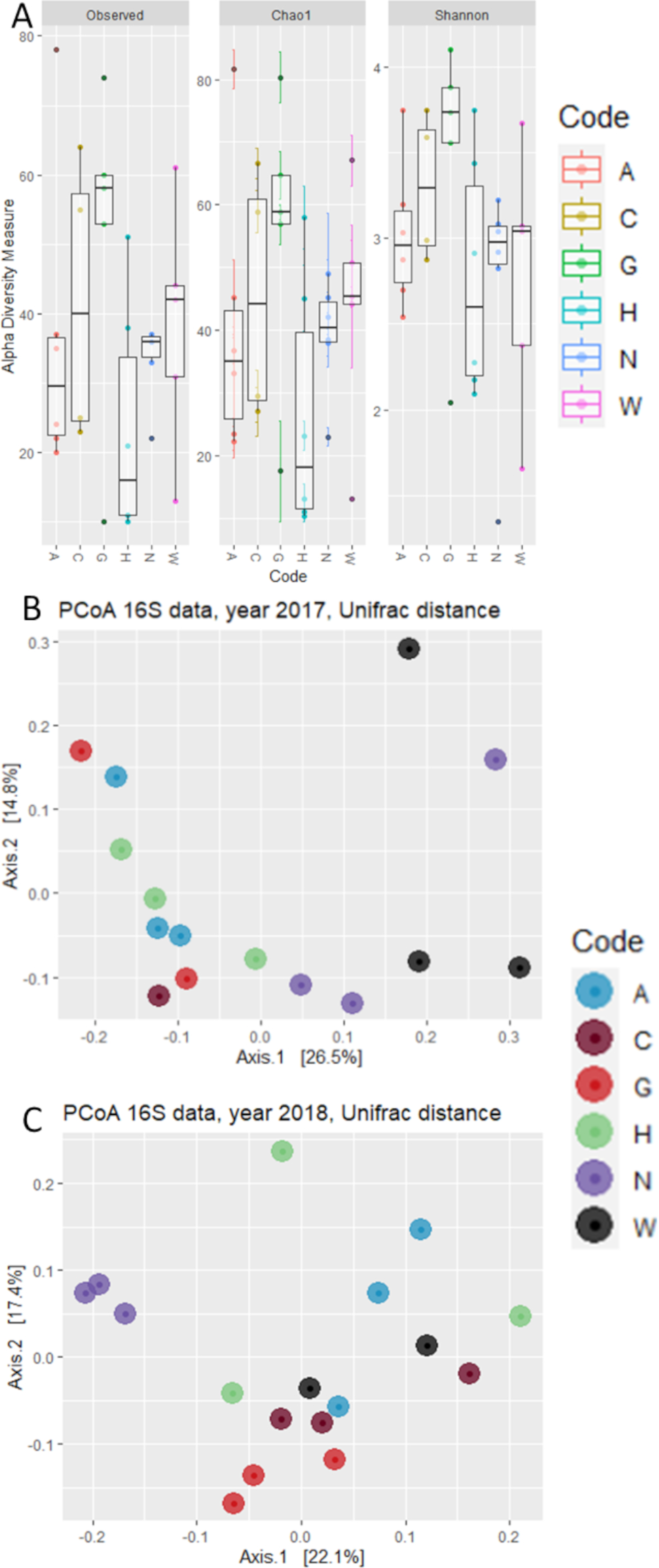
3.2. The Host Genotype as a Driver of the Maize Embryo Bacterial Microbiota
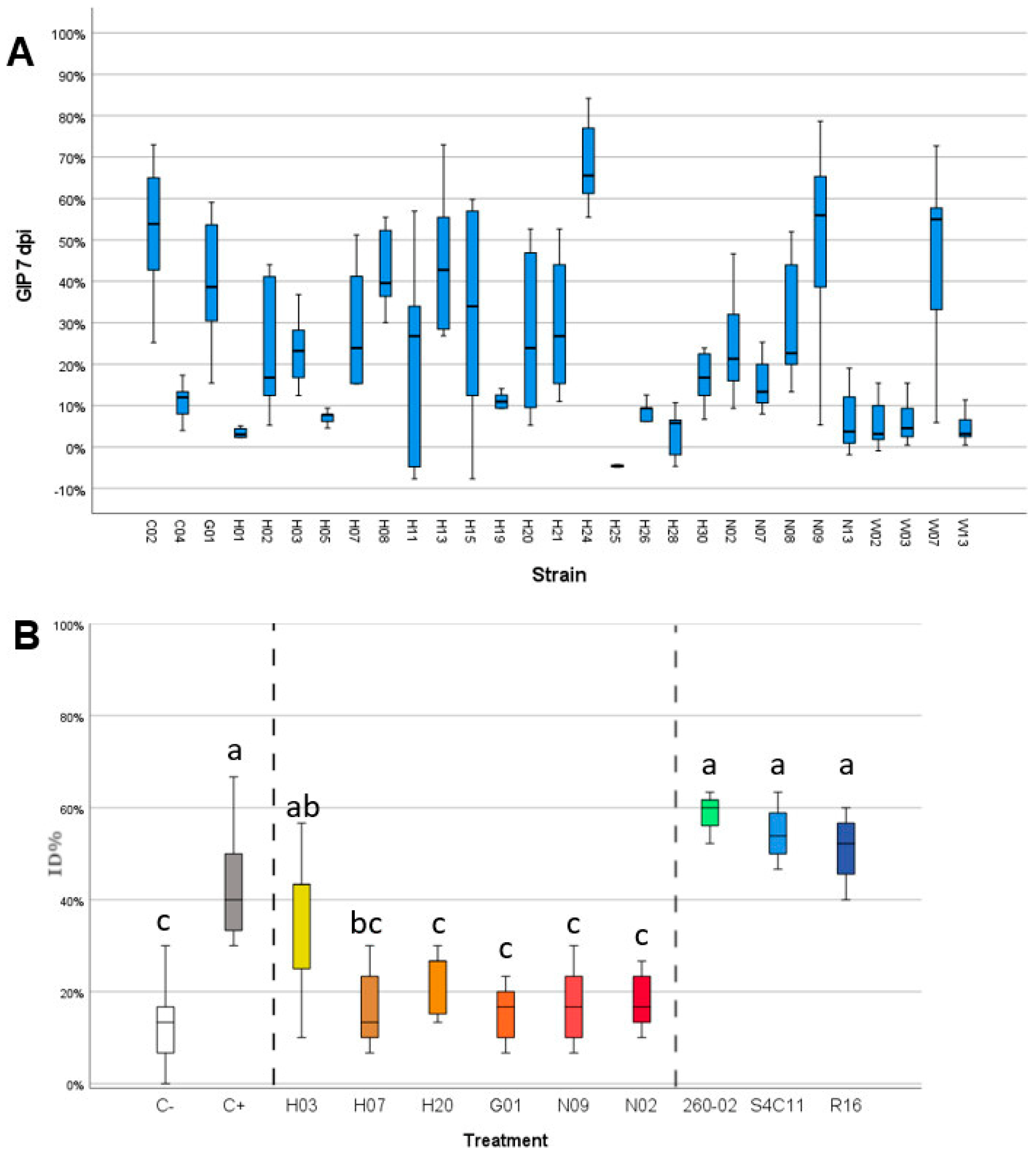
3.3. Gram-Positive Bacteria Isolated from Maize Show Strong Antifungal Ability against F. verticillioides
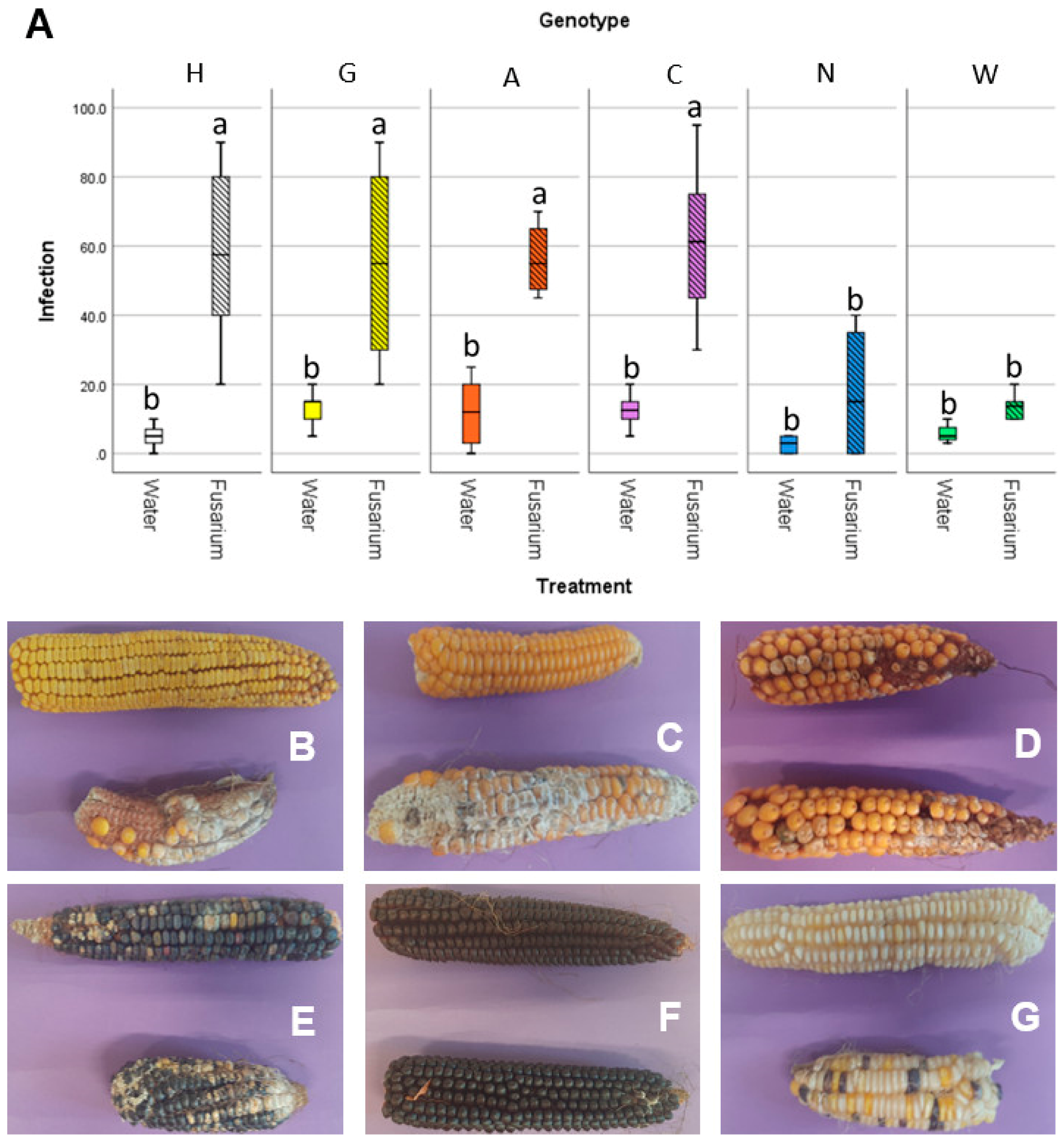
3.4. Accessions Characterized by Similar Embryo Microbiota Show Similar Susceptibility to Fusarium Ear Rot
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haley, C. A cornucopia of maize genes. Nat. Genet. 2011, 43, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Warburton, M.; Crouch, J. Association Mapping for Enhancing Maize (Zea mays L.) Genetic Improvement. Crop. Sci. 2011, 51, 433–449. [Google Scholar] [CrossRef]
- Tester, M.; Langridge, P. Breeding Technologies to Increase Crop Production in a Changing World. Science 2010, 327, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Brandolini, A.; Brandolini, A. Maize introduction, evolution and diffusion in Italy. Maydica 2009, 54, 233–242. [Google Scholar]
- Tafuri, A.; Alfieri, M.; Redaelli, R. Determination of soluble phenolics content in Italian maize varieties and lines. Tec. Mol. Int. 2014, 65, 60–69. [Google Scholar]
- Pilu, R.; Cassani, E.; Sirizzotti, A.; Petroni, K.; Tonelli, C. Effect of flavonoid pigments on the accumulation of fumonisin B1 in the maize kernel. J. Appl. Genet. 2010, 52, 145–152. [Google Scholar] [CrossRef]
- Balconi, C.; Berardo, N.; Locatelli, S.; Lanzanova, C.; Torri, A.; Redaelli, R. Evaluation of ear rot (Fusarium verticillioides) resistance and fumonisin accumulation in Italian maize inbred lines. Phytopathol. Mediterr. 2014, 53, 14–26. [Google Scholar] [CrossRef]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [Green Version]
- Venturini, G.; Babazadeh, L.; Casati, P.; Pilu, R.; Salomoni, D.; Toffolatti, S.L. Assessing pigmented pericarp of maize kernels as possible source of resistance to fusarium ear rot, Fusarium spp. infection and fumonisin accumulation. Int. J. Food Microbiol. 2016, 227, 56–62. [Google Scholar] [CrossRef]
- Bacon, C.W.; Glenn, A.E.; Yates, I.E. Fusarium Verticillioides: Managing the Endophytic Association with Maize for Reduced Fumonisins Accumulation. Toxin Rev. 2008, 27, 411–446. [Google Scholar] [CrossRef]
- Quattrini, M.; Bernardi, C.; Stuknytė, M.; Masotti, F.; Passera, A.; Ricci, G.; Vallone, L.; De Noni, I.; Brasca, M.; Fortina, M.G. Functional characterization of Lactobacillus plantarum ITEM 17215: A potential biocontrol agent of fungi with plant growth promoting traits, able to enhance the nutritional value of cereal products. Food Res. Int. 2018, 106, 936–944. [Google Scholar] [CrossRef]
- Edwards, S.G.; Godley, N. Reduction of Fusariumhead blight and deoxynivalenol in wheat with early fungicide applications of prothioconazole. Food Addit. Contam. Part A 2010, 27, 629–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa-López, A.M.; Cordero-Ramírez, J.D.; Martínez-Álvarez, J.C.; López-Meyer, M.; Lizárraga-Sánchez, G.J.; Félix-Gastélum, R.; Castro-Martínez, C.; Maldonado-Mendoza, I.E. Rhizospheric bacteria of maize with potential for biocontrol of Fusarium verticillioides. SpringerPlus 2016, 5, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasanna, B.M. Diversity in global maize germplasm: Characterization and utilization. J. Biosci. 2012, 37, 843–855. [Google Scholar] [CrossRef]
- Lago, C.; Landoni, M.; Cassani, E.; Cantaluppi, E.; Doria, E.; Nielsen, E.; Giorgi, A.; Pilu, R. Study and Characterization of an Ancient European Flint White Maize Rich in Anthocyanins: Millo Corvo from Galicia. PLoS ONE 2015, 10, e0126521. [Google Scholar] [CrossRef] [Green Version]
- Cassani, E.; Puglisi, D.; Cantaluppi, E.; Landoni, M.; Giupponi, L.; Giorgi, A.; Pilu, R. Genetic studies regarding the control of seed pigmentation of an ancient European pointed maize (Zea mays L.) rich in phlobaphenes: The “Nero Spinoso” from the Camonica valley. Genet. Resour. Crop. Evol. 2016, 64, 761–773. [Google Scholar] [CrossRef]
- Landoni, M.; Puglisi, D.; Cassani, E.; Borlini, G.; Brunoldi, G.; Comaschi, C.; Pilu, R. Phlobaphenes modify pericarp thickness in maize and accumulation of the fumonisin mycotoxins. Sci. Rep. 2020, 10, 1417. [Google Scholar] [CrossRef] [Green Version]
- Springer, N.M. Epigenetics and crop improvement. Trends Genet. 2013, 29, 241–247. [Google Scholar] [CrossRef]
- Links, M.; Demeke, T.; Gräfenhan, T.; Hill, J.; Hemmingsen, S.M.; Dumonceaux, T.J. Simultaneous profiling of seed-associated bacteria and fungi reveals antagonistic interactions between microorganisms within a shared epiphytic microbiome on T riticum and B rassica seeds. New Phytol. 2014, 202, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Johnston-Monje, D.; Gutiérrez, J.P.; Lopez-Lavalle, L.A.B. Seed-Transmitted Bacteria and Fungi Dominate Juvenile Plant Microbiomes. Front. Microbiol. 2021, 12, 2945. [Google Scholar] [CrossRef]
- Sheibani-Tezerji, R.; Rattei, T.; Sessitsch, A.; Trognitz, F.; Mitter, B. Transcriptome Profiling of the Endophyte Burkholderia phytofirmans PsJN Indicates Sensing of the Plant Environment and Drought Stress. mBio 2015, 6, e00621-15. [Google Scholar] [CrossRef] [Green Version]
- Barac, T.; Taghavi, S.; Borremans, B.; Provoost, A.; Oeyen, L.; Colpaert, J.V.; Vangronsveld, J.; Van Der Lelie, D. Engineered endophytic bacteria improve phytoremediation of water-soluble, volatile, organic pollutants. Nat. Biotechnol. 2004, 22, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Rijavec, T.; Lapanje, A.; Dermastia, M.; Rupnik, M. Isolation of bacterial endophytes from germinated maize kernels. Can. J. Microbiol. 2007, 53, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Johnston-Monje, D.; Mousa, W.K.; Lazarovits, G.; Raizada, M.N. Impact of swapping soils on the endophytic bacterial com-munities of pre-domesticated, ancient and modern maize. BMC Plant. Biol. 2014, 14, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puglisi, D.; Landoni, M.; Cassani, E.; Toschi, I.; Lucchini, G.; Cesari, V.; Borlini, G.; Scapin, A.; Pilu, R. Traditional farmers’ varieties: A valuable source of genetic variability for biofortification programs. Maydica 2018, 63, M11. [Google Scholar]
- Cantaluppi, E.; Manzi, S.; Egal, A.A.; Puglisi, D.; Cassani, E.; Toschi, I.; Cesari, V.T.; Landoni, M.; Scapin, A.; Pilu, R. Nutritional and phenotypical characterization of two South African maize (Zea mays L.) varieties sampled in the Qwa-Qwa region. Maydica 2018, 62, M7. [Google Scholar]
- Wilson, K. Preparation of Genomic DNA from Bacteria. Curr. Protoc. Mol. Biol. 2001, 56, 2–4. [Google Scholar] [CrossRef]
- Morandi, S.; Cremonesi, P.; Capra, E.; Silvetti, T.; Decimo, M.; Bianchini, V.; Alves, A.C.; de Vargas, A.C.; Costa, G.M.; Ribeiro, M.G.; et al. Molecular typing and differences in biofilm formation and antibiotic susceptibilities among Prototheca strains isolated in Italy and Brazil. J. Dairy Sci. 2016, 99, 6436–6445. [Google Scholar] [CrossRef]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley and Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Angelini, E.; Clair, D.; Borgo, M.; Bertaccini, A.; Boudon-Padieu, E. Flavescence dorée in France and Italy—Occurrence of closely related phytoplasma isolates and their near relationships to Palatinate grapevineyellows and an alder yellows phyto-plasma. Vitis 2001, 40, 79–86. [Google Scholar] [CrossRef]
- Moronta-Barrios, F.; Gionechetti, F.; Pallavicini, A.; Marys, E.; Venturi, V. Bacterial Microbiota of Rice Roots: 16S-Based Taxonomic Profiling of Endophytic and Rhizospheric Diversity, Endophytes Isolation and Simplified Endophytic Community. Microorganisms 2018, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Robertson-Albertyn, S.; Alegria Terrazas, R.; Balbirnie, K.; Blank, M.; Janiak, A.; Szarejko, I.; Chmielewska, B.; Karcz, J.; Morris, J.; Hedley, P.E.; et al. Root Hair Mutations Displace the Barley Rhizosphere Microbiota. Front. Plant. Sci. 2017, 8, 1094. [Google Scholar] [CrossRef] [PubMed]
- Caradonia, F.; Ronga, D.; Catellani, M.; Giaretta Azevedo, C.V.; Alegria Terrazas, R.; Robertson-Albertyn, S.; Francia, E.; Bulgarelli, D. Nitrogen Fertilizers Shape the Composition and Predicted Functions of the Microbiota of Field-Grown Tomato Plants. Phytobiomes J. 2019, 3, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Pietrangelo, L.; Bucci, A.; Maiuro, L.; Bulgarelli, D.; Naclerio, G. Unraveling the Composition of the Root-Associated Bacterial Microbiota of Phragmites australis and Typha latifolia. Front. Microbiol. 2018, 9, 1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yorza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids. Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, B.; Simpson, G.; Solymos, P.; Stevens, H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.2-1. 2. 1-2. 2015. Available online: https://github.com/vegandevs/vegan (accessed on 16 November 2021).
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 16 November 2021).
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gregoris, T.B.; Aldred, N.; Clare, A.S.; Burgess, J.C. Improvement of phylum- and class-specific primers for real-time PCR quantification of bacterial taxa. J. Microbiol. Methods 2011, 86, 351–356. [Google Scholar] [CrossRef]
- Passera, A.; Venturini, G.; Battelli, G.; Casati, P.; Penaca, F.; Quaglino, F.; Bianco, P.A. Competition assays revealed Paenibacillus pasadenensis strain R16 as a novel antifungal agent. Microbiol. Res. 2017, 198, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Passera, A.; Compant, S.; Casati, P.; Maturo, M.G.; Battelli, G.; Quaglino, F.; Antonielli, L.; Salerno, D.; Brasca, M.; Toffolatti, S.L.; et al. Not Just a Pathogen? Description of a Plant-Beneficial Pseudomonas syringae Strain. Front. Microbiol. 2019, 10, 1409. [Google Scholar] [CrossRef] [Green Version]
- Passera, A.; Rossato, M.; Oliver, J.S.; Battelli, G.; Shahzad, G.-I.; Cosentino, E.; Sage, J.M.; Toffolatti, S.L.; Lopatriello, G.; Davis, J.R.; et al. Characterization of Lysinibacillus fusiformis strain S4C11: In vitro, in planta, and in silico analyses reveal a plant-beneficial microbe. Microbiol. Res. 2020, 244, 126665. [Google Scholar] [CrossRef]
- Townsend, G.R.; Heuberger, J.W. Methods for estimating losses caused by diseases in fungicide experiments. Plant. Dis. Rep. 1943, 27, 340–343. [Google Scholar]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.R.; Tang, C.; Salvato, F.; Clouse, K.M.; Bartlett, A.; Vintila, S.; Phillips, L.; Sermons, S.; Hoffmann, M.; Balint-Kurti, P.J.; et al. Microbe-dependent heterosis in maize. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Mousa, W.K.; Shearer, C.; Limay-Rios, V.; Ettinger, C.L.; Eisen, J.A.; Raizada, M.N. Root-hair endophyte stacking in finger millet creates a physicochemical barrier to trap the fungal pathogen Fusarium graminearum. Nat. Microbiol. 2016, 1, 16167. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, H.; Fan, X.; Wang, Y.; Kusstatscher, P.; Duan, J.; Wu, S.; Chen, S.; Qiao, K.; Wang, Y.; Ma, B.; et al. Bacterial seed endophyte shapes disease resistance in rice. Nat. Plants 2021, 7, 60–72. [Google Scholar] [CrossRef]
- Thompson, M.; Raizada, M.N. Fungal Pathogens of Maize Gaining Free Passage Along the Silk Road. Pathogens 2018, 7, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousa, W.K.; Shearer, C.R.; Limay-Rios, V.; Zhou, T.; Raizada, M.N. Bacterial endophytes from wild maize suppress Fusarium graminearum in modern maize and inhibit mycotoxin accumulation. Front. Plant. Sci. 2015, 6, 805. [Google Scholar] [CrossRef] [Green Version]
- Kuźniar, A.; Włodarczyk, K.; Grządziel, J.; Goraj, W.; Gałązka, A.; Wolińska, A. Culture-independent analysis of an endophytic core microbiome in two species of wheat: Triticum aestivum L. (cv. ‘Hondia’) and the first report of microbiota in Triticum spelta L. (cv. ‘Rokosz’). Syst. Appl. Microbiol. 2019, 43, 126025. [Google Scholar] [CrossRef]
- Guimarães, R.A.; Pherez-Perrony, P.E.; Müller, H.; Berg, G.; Medeiros, F.H.V.; Cernava, T. Microbiome-guided evaluation of Bacillus subtilis BIOUFLA2 application to reduce mycotoxins in maize kernels. Biol. Control. 2020, 150, 104370. [Google Scholar] [CrossRef]
- Johnston-Monje, D.; Raizada, M.N. Conservation and Diversity of Seed Associated Endophytes in Zea across Boundaries of Evolution, Ethnography and Ecology. PLoS ONE 2011, 6, e20396. [Google Scholar] [CrossRef] [Green Version]
- Mitter, B.; Pfaffenbichler, N.; Flavell, R.; Compant, S.; Antonielli, L.; Petric, A.; Berninger, T.; Naveed, M.; Sheibani-Tezerji, R.; von Maltzahn, G.; et al. A New Approach to Modify Plant Microbiomes and Traits by Introducing Beneficial Bacteria at Flowering into Progeny Seeds. Front. Microbiol. 2017, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, F.; Kostic, T.; Sessitsch, A.; Mitter, B. The potential of plant microbiota in reducing postharvest food loss. Microb. Biotechnol. 2018, 11, 971–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Code | Genetic Constitution | Origin | FAO Classification | Pigmentation | Reference |
|---|---|---|---|---|---|---|
| B73 × Mo17 | H | Hybrid | USA | 700 | Yellow | [25] |
| Ottofile Basia | A | opv | North Italy | 300 | Orange (Phlobaphenes) | Unpublished results of our group |
| Ottofile Tortonese | G | opv | North Italy | 300 | Yellow | [25] |
| Spinoso Nero della Val Camonica | N | opv | North Italy | 400 | Black (Phlobaphenes) | [16] |
| Mais Bianco Qwa-Qwa | W | opv | South Africa | 800 | White | [26] |
| Millo Corvo | C | opv | Spain | 400 | Black (Anthocyanins) | [15] |
| Accession Code | Isolated Colonies | After Morphology Screening | After RAPD | N° of Genera |
|---|---|---|---|---|
| H | 50 | 31 | 30 | 7 |
| A | 28 | 6 | 5 | 5 |
| G | 82 | 3 | 2 | 2 |
| N | 18 | 17 | 13 | 4 |
| W | 62 | 21 | 16 | 2 |
| C | 63 | 4 | 4 | 2 |
| Accession Code | Year | Reads | OTUs |
|---|---|---|---|
| H | 2017 | 548 | 46 |
| 2018 | 560 | 51 | |
| A | 2017 | 312 | 55 |
| 2018 | 674 | 114 | |
| G | 2017 | 162 | 53 |
| 2018 | 694 | 166 | |
| N | 2017 | 836 | 71 |
| 2018 | 371 | 84 | |
| W | 2017 | 1163 | 116 |
| 2018 | 112 | 31 | |
| C | 2017 | 53 | 23 |
| 2018 | 522 | 115 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passera, A.; Follador, A.; Morandi, S.; Miotti, N.; Ghidoli, M.; Venturini, G.; Quaglino, F.; Brasca, M.; Casati, P.; Pilu, R.; et al. Bacterial Communities in the Embryo of Maize Landraces: Relation with Susceptibility to Fusarium Ear Rot. Microorganisms 2021, 9, 2388. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9112388
Passera A, Follador A, Morandi S, Miotti N, Ghidoli M, Venturini G, Quaglino F, Brasca M, Casati P, Pilu R, et al. Bacterial Communities in the Embryo of Maize Landraces: Relation with Susceptibility to Fusarium Ear Rot. Microorganisms. 2021; 9(11):2388. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9112388
Chicago/Turabian StylePassera, Alessandro, Alessia Follador, Stefano Morandi, Niccolò Miotti, Martina Ghidoli, Giovanni Venturini, Fabio Quaglino, Milena Brasca, Paola Casati, Roberto Pilu, and et al. 2021. "Bacterial Communities in the Embryo of Maize Landraces: Relation with Susceptibility to Fusarium Ear Rot" Microorganisms 9, no. 11: 2388. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9112388