Synthesis of New N-Arylpyrimidin-2-amine Derivatives Using a Palladium Catalyst

1
Department of Biomolecular Science, University of Science and Technology, Daejon, Korea
2
Life Sciences Research Division, Korea Institute of Science & Technology, P.O. Box 131, Cheongryang, Seoul 130-650, Korea
*
Author to whom correspondence should be addressed.
Molecules 2008, 13(4), 818-830; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13040818
Submission received: 26 March 2008
/
Revised: 8 April 2008
/
Accepted: 8 April 2007
/
Published: 9 April 2008
{kind=link}
{kind=link}
{kind=link}
Abstract
:New N-aryl-4-(pyridin-3-yl)pyrimidin-2-amine derivatives were synthesized from the corresponding amines, applying optimized Buchwald-Hartwig amination conditions using dichlorobis(triphenylphosphine)Pd(II), xantphos and sodium tert-butoxide in refluxing toluene under a nitrogen atmosphere. The target N-aryl derivatives were obtained in moderate to good yields ranging from 27% to 82%. The procedure described could be widely employed for the preparation of new heterocyclic compounds. The structures of the new compounds were confirmed by FT-NMR, FT-IR and elemental analysis.
Introduction
The 2-aminopyrimidine moiety is a common structural subunit in a large number of both natural products [1] and synthetic compounds with important biological activities. This structural motif, representing a heterocylic guanidine moiety, has been widely used as a drug-like scaffold [1,2,3,4,5,6,7,8,9,10]. 2-Aminopyrimidine derivatives substituted at N- or 4-positions are of particular importance, since they show versatile biological and pharmacological activities [2,3,4,5,6,7,8,9,10]. These activities include antifungal [2] and pesticidal [3] activities and enzyme inhibitory activity against a number of kinases, such as Bcr-Abl kinase [4], rho-associated protein kinase [5] and glycogen synthase kinase (GSK3) [6]. They are active also as inhibitors for N-type Ca-channels [7], endothelin receptors [8], human methionine aminopeptidase [9], and as potential drug candidates for treatment of prion diseases [10]. A representative example of such substituted 2-aminopyrimidines is imatinib, a highly selective Bcr-Abl kinase inhibitor, which has been used successfully for treatment of chronic myeloid leukemia [4]. Furthermore, 4-pyridinylpyrimidines are widely used as ligands for metal complexation [11,12].
In most of the reported literature, the synthesis of N-arylpyrimidin-2-amines was achieved by condensation of substituted guanidines with enones [13,14,15]. This approach however is of restricted use for the preparation of diverse derivatives of N-arylpyrimidin-2-amines because of the limited availability of substituted guanidines. Herein we will describe a detailed synthesis for a number of N-aryl-4-(substituted)pyrimidin-2-amines by applying a Buchwald-Hartwig amination protocol [16].
Results and Discussion
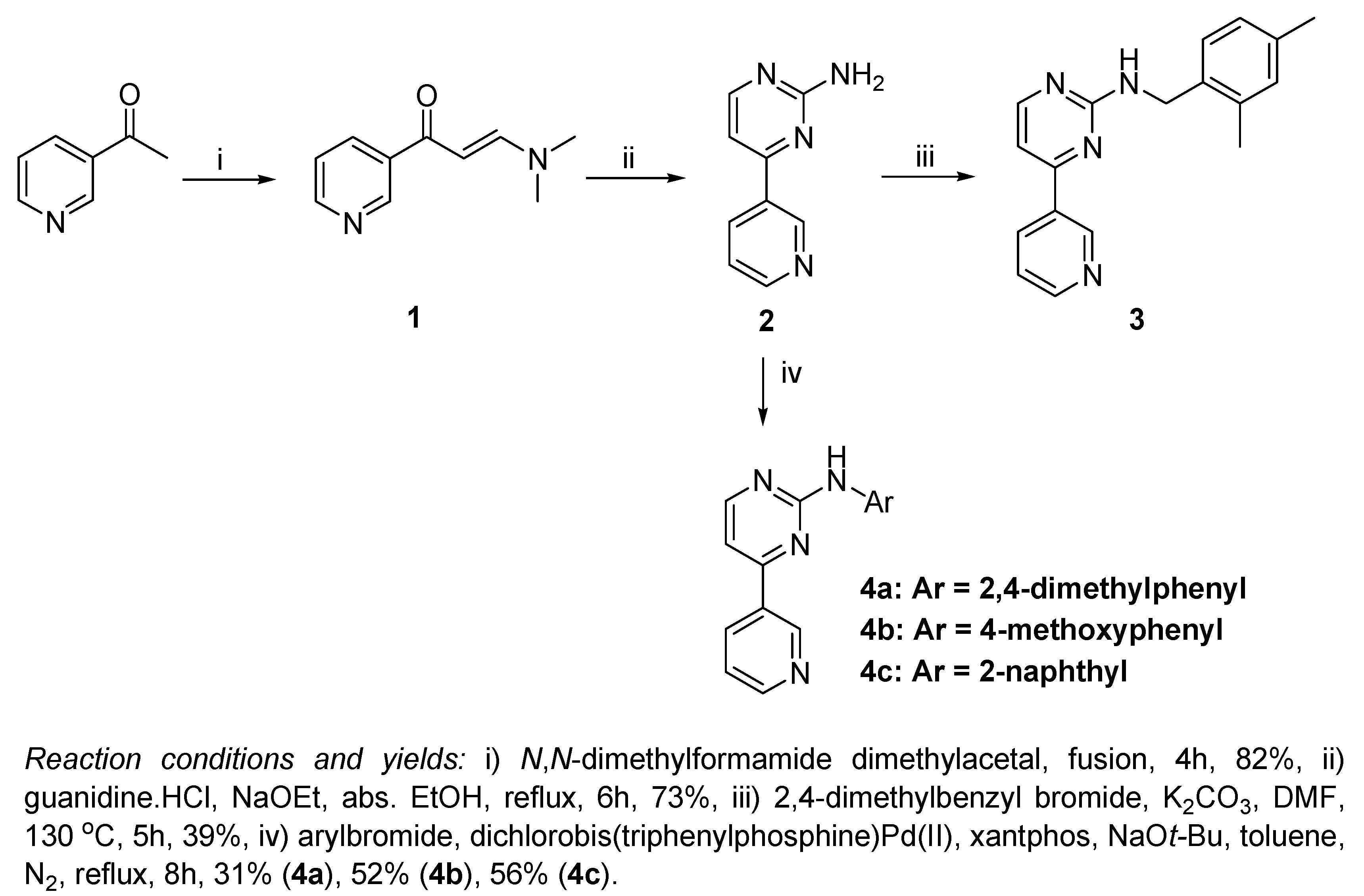
Scheme 1.

The reaction of 3-acetylpyridine with N,N-dimethylformamide dimethylacetal to yield 3-(dimethylamino)-1-(pyridin-3-yl)prop-2-en-1-one (1) is reported to take place in a variety of solvents such as toluene [17] and DMF [18], but it was most conveniently applied without the use of any solvent, by direct fusion of 3-acetylpyridine with 1.4 equivalents of N,N-dimethylformamide dimethylacetal (Scheme 1), which proceeds smoothly in a relatively short time of 4 hours and provides a good yield (82%) [19]. By refluxing compound 1 with guanidine hydrochloride in absolute ethanol and in the presence of sodium ethoxide, 4-(pyridin-3-yl)pyrimidin-2-amine (2) was obtained in a good yield of 73% [15].
The most challenging step in our work was to apply an electrophilic substitution using a benzyl or phenylbromide derivative at the highly electron deficient amino group of the pyrimidin-2-amine. The reaction was easier in case of benzylation of the amino group, since we applied a dropwise addition of 2,4-dimethylbenzyl bromide to a heated solution of 2 and K2CO3 in DMF for the preparation of compound 3 (39% yield). This dropwise addition during 4 hours was essential to avoid N,N-dibenzylation. On the other hand, when K2CO3 was replaced with NaHCO3, no product was obtained at all, which is probably due to the weak basicity of NaHCO3 relative to K2CO3.
The arylation of 2 with different arylbromides to obtain compounds 4a, b and c was much more difficult. The synthesis of compounds 4a and 4b by the condensation of compound 1 with the appropriate substituted guanidine derivatives was reported previously [13]. In order to prepare these compounds using Buchwald-Hartwig amination protocol [16], we first tried to carry out the reaction in refluxing toluene under nitrogen atmosphere, using Pd2(dba)3, 1,3-bis(diphenylphosphino)propane (dppp) as a ligand and sodium tert-butoxide as a base. Under these conditions, the reaction rate was extremely low and the reaction was incomplete, forming the product in only trace amounts that could not be separated from the reaction mixture. Replacing Pd2(dba)3 with dichlorobis(triphenyl-phosphine)Pd(II) improved the reaction slightly, giving a little higher yield of product (~5%) that could be separated, but the reaction conditions still seemed impractical. By replacing the ligand 1,3-bis(diphenylphosphino)propane (dppp) by xantphos, while keeping dichlorobis (triphenylphosphine) Pd(II) as the catalyst, the reaction rate was greatly improved and the yields became more convenient: 31% (4a), 52% (4b), and 56% (4c).
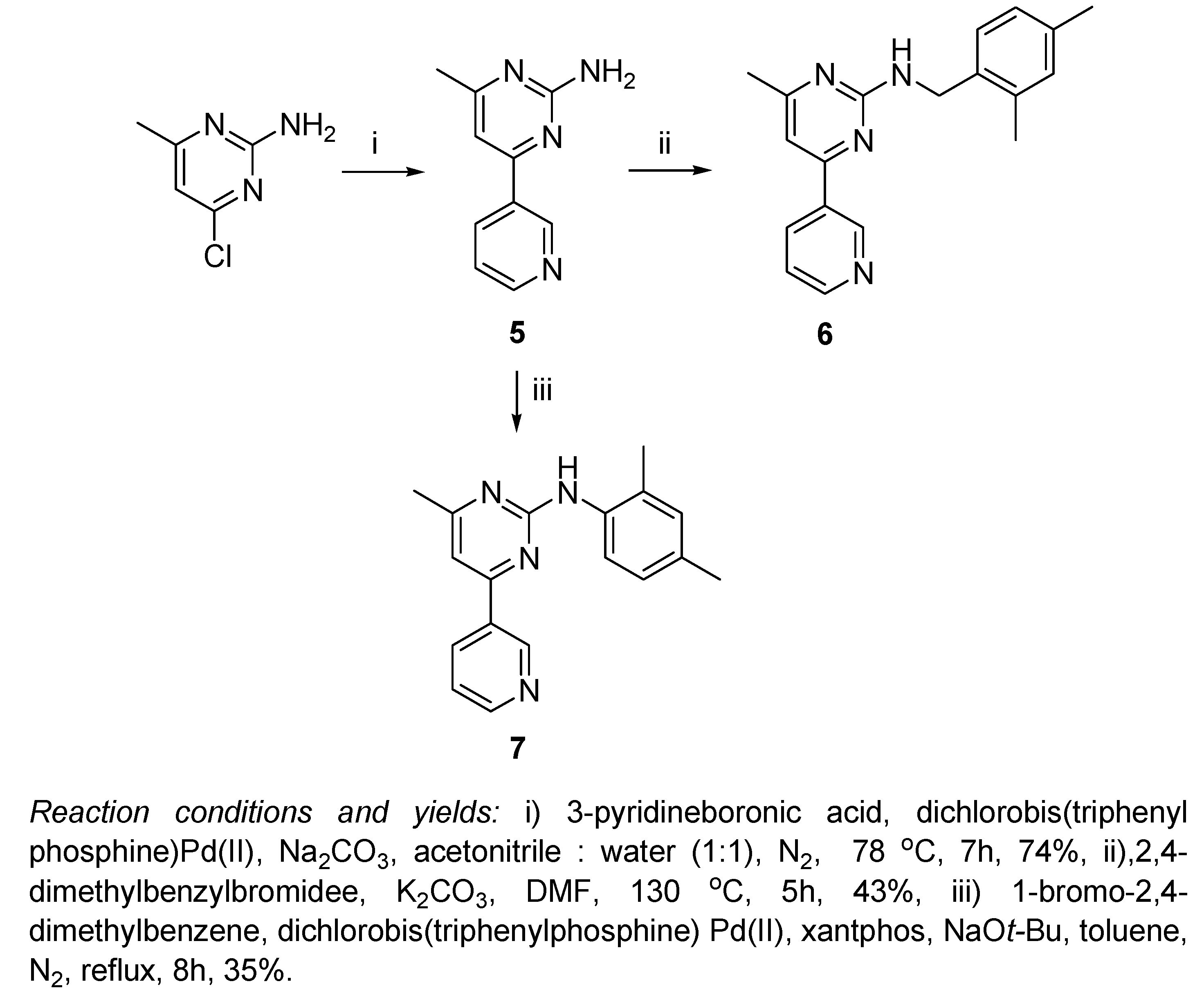
Scheme 2.

As shown in Scheme 2, a completely different procedure was followed for the preparation of the 6-methylpyrimidin-2-amine scaffold, whereby 4-chloro-6-methylpyrimidin-2-amine underwent Suzuki coupling with pyridine-3-boronic acid in a mixed solvent of acetonitrile and water (1:1, v/v) at 78 oC under nitrogen atmosphere, and in the presence of dichlorobis(triphenylphosphine)Pd(II) and Na2CO3 to yield 4-methyl-6-(pyridin-3-yl)pyrimidin-2-amine (5) in 74% yield [20]. The benzylation of compound 5 was achieved by following the same procedure used for the preparation of compound 3, that is by dropwise addition of 2,4-dimethylbenzyl bromide to a heated solution of 5 and K2CO3 in DMF to yield the target compound 6 in 43% yield. Compound 7 was prepared by the reaction of 5 with 1-bromo-2,4-dimethylbenzene following the optimized conditions for Buchwald-Hartwig reaction; in refluxing toluene under nitrogen atmosphere, using dichlorobis(triphenylphosphine)Pd(II), xantphos and sodium tert-butoxide in yield of 35%.
Scheme 3.

The target of the reaction sequence shown in Scheme 3 was to obtain new pyrimidine scaffolds substituted with aryl or heteroaryl moieties. In order to achieve this target, 2,5-dibromopyridine was converted into 5-acetyl-2-bromopyridine (8) in 51% yield according to a literature procedure [21], by lithiation of 2,5-dibromopyridine in diethylether at -78 oC under nitrogen atmosphere, followed by acetylation at 5-position by N,N-dimethylacetamide. 1-(6-(Substituted)pyridin-3-yl)ethanones 9a and 9b were obtained by Suzuki coupling of 8 with the appropriate boronic acid derivatives in a mixed solvent of acetonitrile and water (1:1, v/v) at 78 oC under nitrogen atmosphere, and in the presence of dichlorobis(triphenylphosphine)Pd(II) and Na2CO3 in good yields of 67% for 9a and 74% for 9b. The fusion of 9a and 9b with 2 equivalents of N,N-dimethylformamide dimethylacetal yielded the target compounds 10a and 10b in yield of 99% and 88%, respectively.
The reaction of the resulted prop-2-en-1-one derivatives 10a and 10b with guanidine hydrochloride in refluxing absolute ethanol and in the presence of sodium ethoxide afforded the desired amines 11a and 11b in 84% and 90% yields, respectively. The arylation of these amines with a number of aryl bromides using the previously mentioned optimized Buchwald-Hartwig amination conditions in refluxing toluene under nitrogen atmosphere, using dichlorobis(triphenylphosphine)Pd(II), xantphos and sodium tert-butoxide yielded the target compounds 12a, 12b and 12c in 82%, 31% and 27% yield, respectively.
In summary, we have developed a facile and general approach for the synthesis of new N-aryl-4-(pyridine-3-yl)pyrimidin-2-amine derivatives. The functionalities of pyrimidine derivatives proceeded Suzuki coupling and Buchwald-Hartwig type reactions smoothly in moderate to good yield.
Experimental
General
1H-NMR (300 MHz) and 13C-NMR (75 MHz) were recorded on a Bruker Avance 300 spectrometer with TMS as an internal reference. The IR spectra were recorded on Perkin Elmer Spectrum GX spectrometer. Melting points were taken on a Thomas-Hoover capillary melting apparatus and were uncorrected. Chemical analyses were carried out by EA 1108 CHNS-O from Fisons Instruments. Column chromatography was performed on Merck silica gel 60 (230 - 400 mesh). TLC was carried out using glass sheets precoated with silica gel 60 F254 prepared by E. Merck. All the commercially available reagents were obtained from Aldrich and Tokyo Kasei Chemical and generally used without further purification.
3-(Dimethylamino)-1-(pyridin-3-yl)prop-2-en-1-one (1). A mixture of 3-acetylpyridine (10.0 g, 8.3 mmoles) and N,N-dimethylformamide dimethylacetal (13.8 g, 11.6 mmoles) was heated under reflux in an oil bath for 4 hours. The excess unreacted N,N-dimethylformamide dimethylacetal was removed by distillation, followed by removal of the residual part under vacuum. The solid residue was triturated with water (100 mL) and then extracted with methylene chloride (200 mL x 3). The organic layer was separated, dried over anhydrous MgSO4, and then evaporated under vacuum to yield the crude product. Crystallization from the mixed ethanol and chloroform solvent (1:1, v/v) yielded pure 1 as reddish orange crystals: 12.0 g (82%); m.p. 70-71 oC (lit. 62-70 oC [19]); 1H-NMR (CDCl3): δ 2.90 (s, 3H, N-CH3), 3.12 (s, 3H, N-CH3), 5.62 (d, J = 12.2 Hz, 1H, CH=CH-N), 7.30 (dd, J = 3.6, 5.4 Hz, 1H, H5'), 7.79 (d, J = 12.2 Hz, 1H, CH=CH-N), 8.14 (d, J = 7.8 Hz, 1H, H3'), 8.60 (d, J = 4.8 Hz, 1H, H6'), 9.03 (s, 1H, H2'); 13C-NMR (CDCl3): δ 37.35 (N-CH3), 45.18 (N-CH3), 91.79 (CH=CH-N), 123.25 (C5), 135.03 (C2), 135.62 (C3), 148.85 (C2), 151.38 (C6), 154.68 (CH=CH-N), 186.30 (C=O).
4-(Pyridin-3-yl)pyrimidin-2-amine (2). To a solution of NaOEt in absolute ethanol, made by dissolving sodium metal (0.69 g, 30.0 mmoles) in absolute ethanol (100 mL), guanidine hydrochloride (2.86 g, 30.0 mmoles) was added in one portion and stirred at room temperature for one hour. Compound 1 (5.28 g, 30.0 mmoles) was dissolved in absolute ethanol (20 mL) and added to the reaction mixture. The mixture was heated under reflux for 6 hours, and was then left to cool at room temperature, followed by cooling in an ice bath. The formed product was separated by filtration, washed with cold ethanol (20 mL), then with water (50 mL), and left to dry. Crystallization from ethanol yielded pure 2 as pale yellow needle crystals: 3.8 g (73%); m.p. 191-192 oC (lit. 186-188 oC [15]); 1H-NMR (DMSO-d6): δ 6.78 (s, 2H, NH2), 7.20 (d, J = 6.0 Hz, 1H, H5), 7.52 (dd, J = 3.0, 4.8 Hz, 1H, H5'), 8.34–8.40 (m, 2H, H6,4'), 8.67 (d, J = 3.6 Hz, 1H, H6'), 9.23 (s, 1H, H2').
N-(2,4-Dimethylbenzyl)-4-(pyridin-3-yl)pyrimidin-2-amine (3). A solution of 2,4-dimethylbenzyl bromide (0.17 g, 0.87 mmoles) in DMF (5 mL) was added dropwise over a period of 4 hours to a mixture of 4-(pyridin-3-yl)pyrimidin-2-amine (2, 0.15 g, 0.87 mmoles) and K2CO3 (0.12 g, 0.87 mmoles) in DMF (5 mL) with heating (130 oC) and stirring. After complete addition of the 2,4-dimethylbenzylbromide, heating and stirring were maintained for 1 more hour. The reaction mixture was left to cool at room temperature, and then poured over ice water (50 mL). The resulted precipitate was filtered, dried, then purified by column chromatography (silica gel, ethyl acetate-hexane, 1:1) to give 3 as brownish needles: 0.10 g (39%); m.p. 82.5-83.5 oC; IR υ/cm-1: 3231 (N-H), 1597 (aromatic C=C), 1566 (aromatic C=C), 1345 (CH3), 794 (aromatic C-H); 1H-NMR (CDCl3): δ 2.15 (s, 3H, CH3), 2.32 (s, 3H, CH3), 4.66 (d, J = 4.4 Hz, 2H, CH2), 5.86 (s, 1H, NH), 6.98–7.11 (m, 3H, H3",5",6"), 7.26 (d, J = 7.2 Hz, 1H, H5), 7.40 (dd, J = 2.7, 5.1 Hz, 1H, H5'), 8.30–8.33 (m, 2H, H6,4'), 8.69 (d, J = 3.3 Hz, 1H, H6'), 9.23 (s, 1H, H2'); 13C-NMR (CDCl3): δ 19.06 (2"-CH3), 21.03 (4"-CH3), 43.53 (CH2), 106.42, 123.53, 126.70, 128.46, 131.30, 133.05, 133.67, 134.42, 136.31, 137.14, 148.55, 151.24, 158.99, 162.48; Anal. Calcd. for C18H18N4: C, 74.46; H, 6.25; N, 19.30. Found: C, 74.06; H, 6.25; N, 19.20.
General procedure for the synthesis of compounds 4a-4c
A mixture of 4-(pyridin-3-yl)pyrimidin-2-amine (2, 0.15 g, 0.87 mmoles), the appropriate arylbromide (0.87 mmoles), dichlorobis(triphenylphosphine)Pd(II) (61 mg, 0.087 mmoles), xantphos (0.05 g, 0.087 mmoles) and sodium tert-butoxide (0.25 g, 2.61 mmoles) was refluxed in toluene (15 mL) under nitrogen atmosphere for 8 hours. Toluene was removed under vacuum, and the residue was triturated with water (50 mL) to give compounds 4a-4c.
N-(2,4-Dimethylphenyl)-4-(pyridin-3-yl)pyrimidin-2-amine (4a). The aqueous mixture was extracted with ethyl acetate (100 mL x 2). The organic layer was separated, dried over anhydrous MgSO4, and then evaporated under vacuum. The crude product was purified by column chromatography (silica gel, ethyl acetate-hexane, 1:1) to give 4a as a yellow powder: 74 mg (31%); m.p. 122-123 oC (lit. 113-115 oC [13]); IR υ/cm-1: 3210 (N-H), 2922 (CH3), 1595 (aromatic C=C), 1571 (aromatic C=C), 1451 (aromatic C=C), 1290 (CH3), 797 (aromatic C-H); 1H-NMR (CDCl3): δ 2.32 (s, 3H, CH3), 2.34 (s, 3H, CH3), 6.94 (s, 1H, NH), 7.08–7.14 (m, 3H, H5,3",6"), 7.42 (dd, J = 3.0, 4.8 Hz, H5'), 7.82 (d, J = 8.1 Hz, 1H, H5"), 8.34 (d, J = 8.1 Hz, 1H, H6), 8.47 (d, J = 5.1 Hz, 1H, H4'), 8.71 (d, J = 3.3 Hz, 1H, H6'), 9.25 (S, 1H, H2'); 13C-NMR (CDCl3): δ 18.12, 20.89, 107.77, 122.91, 123.62, 127.14, 129.84, 131.30, 132.79, 133.94, 134.50, 148.56, 151.40, 159.16, 162.56.
N-(4-Methoxyphenyl)-4-(pyridin-3-yl)pyrimidin-2-amine (4b). After trituration with water, the formed solid was filtered, washed with cold water, dried, and then crystallized from ethanol to give 4b as yellow crystals: 0.13 g (52%); m.p. 118-119 oC (lit. 121-122 oC [13]); IR υ/cm-1: 3435 (N-H), 1575 (aromatic C=C), 1509 (aromatic C=C), 1423 (aromatic C=C), 1242 (Ar-OCH3), 801 (aromatic C-H); 1H-NMR (DMSO-d6): δ 3.74 (s, 3H, OCH3), 6.92 (d, J = 9.0 Hz, 2H, H2",6"), 7.42 (d, J = 5.2 Hz, 1H, H5), 7.59 (dd, J = 4.0, 4.5 Hz, 1H, H5'), 7.68 (d, J = 8.7 Hz, 2H, H3",5"), 8.49 (d, J = 7.8 Hz, 1H, H6), 8.55 (d, J = 4.7 Hz, 1H, H4'), 8.73 (d, J = 4.0 Hz, 1H, H6'), 9.34 (s, 1H, NH), 9.58 (s, 1H, H2'); 13C-NMR (DMSO-d6): δ 55.64 (OCH3), 108.07, 114.13, 121.31, 124.37, 132.78, 133.93, 134.79, 18.58, 151.90, 154.82, 159.91, 160.80, 161.99.
N-(Naphthalen-2-yl)-4-(pyridin-3-yl)pyrimidin-2-amine (4c). After trituration with water, the formed solid was filtered, washed with cold water, dried, and then crystallized from ethanol to give 4c as yellow plate-like crystals: 0.14 g (56%); m.p. 199-200 oC; IR υ/cm-1: 3247 (N-H), 1571 (aromatic C=C), 1548 (aromatic C=C), 1447 (aromatic C=C), 1434 (aromatic C=C), 798 (aromatic C-H); 1H-NMR (DMSO-d6): δ 7.36 (t, J = 7.8 Hz, 1H, Ar-H), 7.46 (t, J = 7.3 Hz, 1H, Ar-H), 7.56 (d, J = 5.1 Hz, 1H, H5), 7.63 (dd, J = 2.9, 4.8 Hz, 1H, H5'), 7.78–7.88 (m, 4 H, Ar-H), 8.54–8.57 (m, 2H, Ar-H), 8.67 (d, J = 5.1 Hz, 1H, H4'), 8.76 (d, J = 4.5 Hz, 1H, H6'), 9.40 (s, 1H, H2'), 10.03 (s, 1H, NH); 13C-NMR (DMSO): δ 108.98, 114.43, 121.19, 124.49, 126.77, 127.46, 127.89, 128.50, 129.46, 134.18, 134.93, 138.55, 148.68, 152.03, 160.69; Anal. Calcd. for C19H14N4: C, 76.49; H, 4.73; N, 18.78. Found: C, 76.60; H, 4.70; N, 18.38.
4-Methyl-6-(pyridin-3-yl)pyrimidin-2-amine (5). A mixture of 2-amino-4-chloro-6-methyl-pyridine (2.70 g, 18.76 mmoles), 3-pyridineboronic acid (2.54 g, 20.66 mmoles), dichlorobis(triphenyl- phosphine)Pd(II) (0.36 g, 0.512 mmoles) and Na2CO3 (1.40 g, 13.2 mmoles) was placed in mixed solvent of acetonitrile and water (1:1, 150 mL). N2 gas was bubbled into this mixture for 10 minutes, and then the mixture was heated at 78 oC while stirring under N2 atmosphere for 7 hours. The reaction mixture was left to cool at room temperature, poured into ice water (100 mL), and then extracted with ethylacetate (100 ml x 3). The organic layer was separated, washed with water (100 mL x 3), dried over anhydrous MgSO4, and then evaporated under vacuum to yield the crude product which was then crystallized from ethanol to yield the pure yellow crystals of 5: 2.58 g, (74%); m.p. 191-192 oC (lit. 192-194 oC [20]); 1H-NMR (CD3OD): δ 2.41 (s, 3H, CH3), 7.13 (s, 1H, H5), 7.56 (dd, J = 2.9, 4.9 Hz, 1H, H5'), 8.49 (d, J = 8.0 Hz, 1H, H4'), 8.64 (d, J = 5.0 Hz, 1H, H6'), 9.21 (s, 1H, H2').
N-(2,4-Dimethylbenzyl)-4-methyl-6-(pyridin-3-yl) pyrimidin-2-amine (6). The procedure used for the synthesis of compound 3 was adapted for the preparation of this compound. After pouring the reaction mixture over ice water, the aqueous solution was extracted with ethyl acetate (100 mL x 2). The organic layer was separated, dried over anhydrous MgSO4, then evaporated under vacuum to yield the crude product, which was then purified by column chromatography (silica gel, ethyl acetate-hexane, 1:1) to give 6 as brown needle-like crystals: 0.11 g (43%); m.p. 125-126 oC; IR υ/cm-1: 3254 (N-H), 2920 (CH3), 1604 (aromatic C=C), 1557 (aromatic C=C), 1341 (CH3), 808 (aromatic C-H); 1H-NMR (CDCl3): 2.32 (s, 3H, 4"-CH3), 2.37 (s, 3H, 2"-CH3), 2.43 (s, 3H, 4-CH3), 4.68 (d, J = 7.5 Hz, 2H, CH2), 5.41 (s, 1H, NH), 6.90 (s, 1H, H5), 6.98–7.02 (m, 2H, H3",5"), 7.25 (d, J = 7.8 Hz, 1H, H6"), 7.33 (dd, J = 3.5, 4.8 Hz, 1H, H5'), 8.31 (d, J = 7.8 Hz, 1H, H4'), 8.67 (d, J = 4.8 Hz, 1H, H6'), δ 9.22 (s, 1H, H2'); 13C-NMR (CDCl3): δ 19.08 (4"-CH3), 21.01(2"-CH3), 24.38(4-CH3), 43.44(CH2), 106.23, 123.48, 126.66, 128.34, 131.24, 133.34, 133.94, 134.45, 136.21, 137.03, 148.55, 151.01, 162.13, 162.48, 168.95; Anal. Calcd. for C19H20N4: C, 74.97; H, 6.62; N, 18.41. Found: C, 74.99; H, 6.65; N, 18.90.
4-Methyl-N-(2,4-dimethylphenyl)-6-(pyridin-3-yl)pyrimidin-2-amine (7). The procedure used for the synthesis of compound 4a was adapted for the synthesis of this compound to give 7 as an off-white powder: 88 mg (35%); m.p. 95-96 oC; IR υ/cm-1: 3215 (N-H), 2921 (CH3), 1597 (aromatic C=C), 1581 (aromatic C=C), 1552 (aromatic C=C), 1448 (aromatic C=C), 1343 (CH3), 804 (aromatic C-H); 1H-NMR (CDCl3): δ 2.34 (s, 6H, 2"-, 4"-CH3), 2.49 (s, 3H, 4-CH3), 6.90 (s, 1H, NH), 7.04–7.10 (m, 3H, H5,3",6"), 7.41 (dd, J = 3.0, 5.0 Hz, 1H, H5') 7.97 (d, J = 8.1 Hz, 1H, H5"), 8.32 (d, J = 8.1 Hz, 1H, H4'), 8.69 (d, J = 3.9 Hz, 1H, H6'), 9.24 (s, 1H, H2'); 13C-NMR (CDCl3): δ 18.16 (2"-CH3), 20.83 (4"-CH3), 24.42 (4-CH3), 107.49, 121.97, 123.55, 127.02, 128.68, 131.16, 133.04, 133.08, 134.49, 134.90, 148.58, 151.19, 160.79, 162.18, 169.15; Anal. Calcd. for C18H18N4: C, 74.46; H, 6.25; N, 19.30. Found: C, 74.46; H, 6.50; N, 19.00.
5-Acetyl-2-bromopyridine (8). Under a N2 atmosphere, n-BuLi (8.1 mL, 1.6 M in hexane, 13 mmoles) was added to a mixture of 2,5-dibromopyridine (3.1 g, 13.0 mmoles) and anhydrous Et2O (50 mL) at ‑78 oC. The reaction mixture was stirred for 30 minutes and then N,N-dimethylacetamide (1.4 mL, 15.0 mmoles) was added and stirred at -78 oC to ambient temperature within 1 hour. The whole mixture was poured into saturated NH4Cl and then extracted with Et2O (150 mL x 3). The combined organic layer was washed with brine, dried, and concentrated. The crude product was purified by column chromatography (silica gel, ethyl acetate-hexane (1:7.5, v/v) to give 8 as a yellowish white solid: 1.30 g (51%), m.p. 127-129 oC (lit. 124-128 oC [21]); 1H-NMR (CDCl3): δ 2.62 (s, 3H, CH3), 7.61 (d, J = 8.2 Hz, 1H, H3), 8.07 (d, J = 7.6 Hz, 1H, H4), 8.89 (s, 1H, H6); 13C-NMR (CDCl3): δ 26.79 (CH3), 128.44 (C3), 131.41 (C5), 137.70 (C4), 146.96 (C2), 150.42 (C6), 195.65 (C=O).
General procedure for the synthesis of compounds 9a-9b
A mixture of 5-acetyl-2-bromopyridine (8, 2.0 g, 10 mmoles), the appropriate aryl boronic acid (11.0 mmoles), dichlorobis(triphenylphosphine)Pd(II) (0.19 g, 0.27 mmoles) and Na2CO3 (0.75 g, 7.0 mmoles) was placed in mixed solvent of acetonitrile and water (1:1, 80 mL). N2 gas was bubbled into this mixture for 10 minutes, and then the mixture was heated at 78 oC while stirring under N2 atmosphere for 4 hours. The reaction mixture was left to cool at room temperature, poured into ice water (100 mL), and then extracted with methylene chloride (150 mL x 3). The organic layer was separated, dried over anhydrous MgSO4, and then evaporated under vacuum to yield the crude product which was then crystallized from ethanol to yield the pure compounds 9a and 9b.
1-(6-Phenylpyridin-3-yl)ethanone (9a). It was obtained as silvery needle-like crystals: 1.32 g (67%); m.p. 119-120 oC (lit. 118 oC [22]); IR υ/cm-1: 1677 (C=O), 1588 (aromatic C=C), 1262 (CH3), 740 (aromatic C-H); 1H-NMR (CDCl3): δ 2.68 (s, 3H, CH3), 7.49–7.55 (m, 3H, H3",4",5"), 7.86 (d, J = 8.4 Hz, 1H, H5'), 8.08 (d, J = 7.2 Hz, 2H, H2",6"), 8.31 (dd, J = 2.2, 6.2 Hz, 1H, H4'), 9.25 (s, 1H); 13C-NMR (CDCl3): δ 26.76 (CH3), 120.18, 127.39, 128.96, 130.11, 136.43, 138.13, 150.12, 160.60 (C=O).
1-(6-(Pyridin-3-yl)pyridin-3-yl)ethanone (9b). It was obtained as buff plates: 1.46 g (74%); m.p. 103-104 oC; IR υ/cm-1: 1680 (C=O), 1586 (aromatic C=C), 1269 (CH3), 818 (aromatic C-H); 1H-NMR (CD3OD): δ 2.68 (s, 3H, CH3), 7.63 (dd, J = 3.0, 4.8 Hz, 1H, H5"), 8.10 (d, J = 8.1 Hz, 1H, H5'),8.44 (dd, J = 2.1, 6.0 Hz, 1H, H4"), 8.58 (d, J = 8.1 Hz, 1H, H4'), 8.66 (d, J = 4.5 Hz, 1H, H6"), 9.23 (s, 1H, H2"), 9.28 (s, 1H, H2); 13C-NMR (CDCl3): δ 25.49 (CH3), 120.51, 124.25, 131.48, 134.36, 135.80, 137.00, 147.29, 149.29, 149.78, 157.27, 197.02 (C=O); Anal. Calcd. for C12H10N2O: C, 72.71; H, 5.08; N, 14.13. Found: C, 72.96; H, 5.25; N, 14.30.
General procedure for the synthesis of compounds 10a-10b
The appropriate 1-(6-(substituted)pyridin-3-yl)ethanone 9a or 9b (2.5 mmoles) and N,N-dimethyl-formamide dimethylacetal (0.6 g, 5.0 mmoles) were refluxed together in an oil bath for 3 hours. The excess unreacted N,N-dimethylformamide dimethylacetal was removed by distillation, followed by removal of the residual part under vacuum. The solid residue was triturated with water (30 mL) and then extracted with methylene chloride (100 mL x 2). The organic layer was separated, dried over anhydrous MgSO4, and then evaporated under vacuum to yield the crude product which was used for the next step without further purification.
3-(Dimethylamino)-1-(6-phenylpyridin-3-yl)prop-2-en-1-one (10a). It was obtained as a yellow powder: 0.62 g (99%); m.p. 151-152 oC; IR υ/cm-1: 1644 (C=O), 1589 (aromatic C=C), 1567 (aromatic C=C), 1537 (aromatic C=C), 1285 (N-CH3), 1242 (N-CH3), 769 (aromatic C-H), 745 (aromatic C-H); 1H-NMR (CDCl3): δ 2.97 (s, 3H, N-CH3), 3.19 (s, 3H, N-CH3), 5.74 (d, J = 12.3 Hz, 1H, CH=CH-N), 7.42–7.53 (m, 3H, H3",4",5"), 7.81 (d, J = 8.3 Hz, 1H, H5'), 7.87 (d, J = 12.2 Hz, 1H, CH=CH-N), 8.05 (d, J = 7.4 Hz, 2H, H2",6"), 8.28 (dd, J = 1.6, 6.6 Hz, 1H, H4'), 9.18 (s, 1H, H2'); 13C-NMR (CDCl3): δ 37.46 (N-CH3), 45.30 (N-CH3), 91.71 (CH=CH-N), 120.46, 127.32, 128.95, 129.84, 134.14, 136.96, 137.79, 148.10, 154.74, 158.37, 185.40 (C=O); Anal. Calcd. for C16H16N2O: C, 76.16; H, 6.39; N, 11.10. Found: C, 76.86; H, 6.25; N, 11.50.
3-(Dimethylamino)-1-(6-(pyridin-3-yl)pyridin-3-yl)prop-2-en-1-one (10b). It was obtained as a brown powder: 0.56 g (88%); m.p. 163-164 oC; IR υ/cm-1: 1639 (C=O), 1590 (aromatic C=C), 1565 (aromatic C=C), 1538 (aromatic C=C), 1274 (N-CH3), 1260 (N-CH3), 780 (aromatic C-H); 1H-NMR (CDCl3): δ 2.95 (s, 3H, N-CH3), 3.17 (s, 3H, N-CH3), 5.70 (d, J = 12.2 Hz, 1H, CH=CH-N), 7.40 (dd, J = 2.8, 5.0 Hz, 1H, H5"), 7.79 (d, J = 8.2 Hz, 1H, H5'), 7.85 (d, J = 12.2 Hz, 1H, CH=CH-N), 8.27 (d, J = 8.2 Hz, 1H, H4"), 8.33 (d, J = 7.8 Hz, 1H, H4'), 8.64 (d, J = 4.5 Hz, 1H, H6'), 9.17 (s, 1H, H2"), 9.22 (s, 1H, H2'); 13C-NMR (CDCl3): δ 37.42 (N-CH3), 45.29 (N-CH3), 91.74 (CH=CH-N), 120.02, 123.63, 134.30, 134.52, 136.20, 148.43, 149.29, 150.25, 154.71, 156.26, 185.70 (C=O); Anal. Calcd. for C15H15N3O: C, 71.13; H, 5.97; N, 16.59. Found: C, 71.16; H, 5.99; N, 16.70.
General procedure for the synthesis of compounds 11a-11b
To a solution of NaOEt in absolute ethanol, made by dissolving sodium metal (27 mg, 1.19 mmoles) in absolute ethanol (20 mL), guanidine hydrochloride (0.11 g, 1.19 mmoles) was added in one portion and stirred at room temperature for one hour. The appropriate 3-(dimethylamino)-1-(6-(substituted)pyridin-3-yl)prop-2-en-1-one 10a or 10b (1.19 mmoles) was dissolved in absolute ethanol (10 mL) and added to the reaction mixture. The mixture was heated under reflux for 5 hours, and was then left to cool at room temperature, followed by cooling in an ice bath. The formed product was separated by filtration, washed with cold ethanol (20 mL), then with water (50 mL), and left to dry.
4-(6-Phenylpyridin-3-yl)pyrimidin-2-amine (11a). It was obtained as silver crystals: 0.25 g (84%); m.p. 211-212 oC; IR υ/cm-1: 3317 (N-H), 3156 (N-H), 1648 (aromatic C=N), 1590 (aromatic C=C), 1571 (aromatic C=C), 1542 (aromatic C=C), 1478 (aromatic C=C), 743 (aromatic C-H); 1H-NMR (DMSO-d6): δ 6.80 (s, 2H, NH2), 7.26 (d, J = 3.0 Hz, 1H, H5), 7.48–7.55 (m, 3H, H3", 4",5"), 8.11 (d, J = 8.7 Hz, 1H, H5'), 8.17 (d, J = 7.1 Hz, 2H, H2",6"), 8.36 (d, J = 2.7 Hz, 1H, H6), 8.50 (dd, J = 2.1, 6.3 Hz, 1H, H4'), 9.32 (s, 1H, H2'); 13C-NMR (CDCl3): δ 106.38, 120.48, 127.21, 129.34, 130.05, 131.54, 135.69, 138.40, 148.49, 157.84, 159.83, 161.73, 164.28; Anal. Calcd. for C15H12N4: C, 72.56; H, 4.87; N, 22.57. Found: C, 72.99; H, 4.90; N, 22.80.
4-(6-(Pyridin-3-yl)pyridin-3-yl)pyrimidin-2-amine (11b). It was obtained as yellow plates: 0.27 g (90%); m.p. 250 oC <; IR υ/cm-1: 3438 (N-H), 3324 (N-H), 1621 (aromatic C=N), 1586 (aromatic C=C), 1566 (aromatic C=C), 1545 (aromatic C=C), 1477 (aromatic C=C), 803 (aromatic C-H); 1H-NMR (DMSO-d6): δ 6.82 (s, 2H, NH2), 7.29 (d, J = 5.4 Hz, 1H, H5), 7.55 (dd, J = 3.2 , 4.8 Hz, 1H, H5"), 8.21 (d, J = 8.1 Hz, 1H, H5'), 8.37 (d, J = 5.1 Hz, 1H, H6), 8.50–8.55 (m, 2H, H4',4"), 8.67 (d, J = 3.6 Hz, 1H, H6"), 9.34 (d, J = 1.5Hz, 1H, H2"), 9.36 (d, J = 1.5 Hz, 1H, H2'); 13C-NMR (DMSO-d6): 106.52, 121.01, 124.41, 132.14, 133.87, 134.67, 135.91, 148.35, 148.70, 150.74, 155.63, 159.92, 161.55, 164.23; Anal. Calcd. for C14H11N5: C, 67.46; H, 4.45; N, 28.10. Found: C, 67.15; H, 4.55; N, 28.30.
General procedure for the synthesis of compounds 12a-12c
A mixture of the appropriate 4-(6-(substituted)pyridin-3-yl)pyrimidin-2-amine 11a or 11b (0.29 mmoles), the appropriate arylbromide (0.29 mmoles), dichlorobis(triphenylphosphine)Pd(II) (41 mg, 0.058 mmoles), xantphos (34 mg, 0.058 mmoles) and sodium tert-butoxide (84 mg, 0.87 mmoles) was refluxed in toluene (7 mL) under nitrogen atmosphere for 8 hours. The reaction mixture was left to cool at room temperature, and then cooled in an ice bath. The formed solid was filtered, washed with cold toluene (10 mL), then with water (50 mL).
N-(4-Methoxyphenyl)-4-(6-phenylpyridin-3-yl)pyrimidin-2-amine (12a). It was obtained as a yellow crystalline powder: 61 mg (82%); m.p. 250 oC <; IR υ/cm-1: 3462 (N-H), 1579 (aromatic C=C), 1511 (aromatic C=C), 1456 (aromatic C=C), 1430 (aromatic C=C), 1262 (CH3), 1240 (CH3), 1097 (C-O), 1039 (C-O), 804 (aromatic C-H); 1H-NMR (DMSO-d6): δ 3.74 (s, 3H, OCH3), 6.92 (d, J = 9.0 Hz, 2H, H2"',6'''), 7.48–7.56 (m, 4H, H5,3",4",5''), 7.71 (d, J = 8.7 Hz, 2H, H3''',5'''), (m, 3H, H5',2",6"), 8.55 – 8.58 (m, 2H, H6,4'), 9.41 (s, 1H, H2'), 9.57 (s, 1H, NH); 13C-NMR (DMSO-d6): δ 55.65 (OCH3), 107.96, 114.24, 120.66, 121.38, 127.25, 129.39, 131.32, 133.90, 135.90, 138.31, 148.59, 154.83, 158.12, 159.79, 160.78; Anal. Calcd. for C22H18N4O: C, 74.56; H, 5.12; N, 15.81. Found: C, 74.30; H, 5.60; N, 15.90.
N1,N1-Diphenyl-N4-(4-(6-phenylpyridin-3-yl)pyrimidin-2-yl)benzene-1,4-diamine (12b). It was obtained as a yellow powder: 44 mg (31%); m.p. 210-211 oC; IR υ/cm-1: 3460 (N-H), 1575 (aromatic C=C), 1527 (aromatic C=C), 1507 (aromatic C=C), 1493 (aromatic C=C), 1422 (aromatic C=C), 1277 (C-N), 744 (aromatic C-H), 695 (aromatic C-H); 1H-NMR (CDCl3): δ 6.98–7.72 (m, 18H, Ar-H), 7.89 (d, J = 8.1 Hz, 1H, Ar-H), 8.10 (d, J = 6.9 Hz, 2H, Ar-H), 8.45 (dd, J = 2.5, 6.1 Hz, 1H, Ar-H), 8.51 (d, J = 5.1 Hz, 1H, Ar-H), 9.40 (s, 1H, NH); 13C-NMR (CDCl3): δ 107.92, 108.12, 119.44, 120.31, 120.46, 122.30, 122.74, 123.60, 125.62, 127.13, 128.90, 129.00, 129.10, 129.60, 130.93, 134.90, 135.26, 138.58, 142.75, 147.98, 148.53, 158.92, 159.15, 160.33, 162.48; Anal. Calcd. for C33H25N5: C, 80.63; H, 5.13; N, 14.25. Found: C, 80.46; H, 5.55; N, 14.20.
N1,N1-Diphenyl-N4-(4-(6-(pyridin-3-yl)pyridin-3-yl)pyrimidin-2-yl)benzene-1,4-diamine (12c). It was obtained as a yellow powder: 39 mg (27%); m.p. 230-231 oC; IR υ/cm-1: 3429 (N-H), 1582 (aromatic C=C), 1530 (aromatic C=C), 1507 (aromatic C=C), 1493 (aromatic C=C), 1422 (aromatic C=C), 1281 (C-N), 751 (aromatic C-H), 695 (aromatic C-H); 1H-NMR (CDCl3): δ 6.96 (d, J = 6.9 Hz, 6H, Ar-H); 7.05 (d, J = 8.8 Hz, 2H, Ar-H), 7.24–7.30 (m, 4H, Ar-H), 7.56 (d, J = 5.1 Hz, 2H, Ar-H), 7.81 (d, J = 8.7 Hz, 2H, Ar-H), 8.25 (d, J = 8.1 Hz, 1H, Ar-H), 8.52 (d, J = 8.1 Hz, 1H, Ar-H), 8.60–8.68 (m, 3H, Ar-H), 9.35 (s, 1H, Ar-H), 9.45 (s, 1H, Ar-H), 9.82 (s, 1H, NH); 13C-NMR (DMSO-d6): 100.37, 108.54, 120.84, 121.17, 122.59, 123.10, 124.43, 126.18, 129.86, 131.87, 133.82, 134.72, 136.15, 136.96, 141.37, 148.00, 148.40, 148.88, 159.92, 161.57; Anal. Calcd. for C32H24N6: C, 78.03; H, 4.91; N, 17.06. Found: C, 78.06; H, 4.99; N, 17.20.
Acknowledgements
This study was supported by Korea Institute of Science and Technology.
References
- Lagoja, I. M. Pyridine as constituent of natural biologically active compounds. Chem. Biodivers. 2005, 2, 1–50. [Google Scholar] [CrossRef]
- Ackermann, P.; Stierli, D.; Jung, P. M. J.; Maienfisch, P.; Cederbaum, F. E. M.; Wenger, J. F. Microbiocidal N-phenyl-N-[4-(4-pyridyl)-2-pyrimidin-2-yl]-amine derivatives. Int. Pat. Appl. WO 03/047347 A1, 2003. [Google Scholar]
- Bretschneider, T.; Es-Sayed, M.; Fischer, R.; Maurer, F.; Erdelen, C.; Lősel, P. Pyridyl pyrimidines for use as pesticides. Int. Pat. Appl. WO 02/067684 A1, 2002. [Google Scholar]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nature Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef]
- Sehon, C. A.; Lee, D.; Goodman, K. B.; Wang, G. Z.; Viet, A. Q. Novel inhibitors of rho-kinases. Int. Pat. Appl. WO 2006/009889 A1, 2006. [Google Scholar]
- Goff, D. A.; Harrison, S. D.; Nuss, J. M.; Ring, D. B.; Zhou, X. A. Inhibitors of glycogen synthase kinase 3. U.S. Pat. 6,417,185 B1, 2002. [Google Scholar]
- Ohno, S.; Otani, K.; Niwa, S.; Iwayama, S.; Takahara, A.; Koganei, H.; Ono, Y.; Fujita, S.; Takeda, T.; Hagihara, M.; Okajima, A. Novel pyrimidine derivative and novel pyridine derivative. Int. Pat. Appl. WO 02/22588 A1, 2002. [Google Scholar]
- Breu, V.; Coassolo, P.; Neidhart, W.; Roux, S.; Weiss, P. 4-Heterocyclysulfonamidyl-6-methoxy-5-(2-methoxy-phenoxy)-2-pyridyl-pyrimidine derivatives, their preparation and use as endothelin receptor antagonists. Int. Pat. Appl. 2000. [Google Scholar]
- Hu, X.; Addlagatta, A.; Matthews, B. W.; Liu, J. O. Identification of pyridinylpyrimidines as inhibitors of human methionine aminopeptidases. Angew. Chem. Int. Ed. 2006, 45, 3772–3775. [Google Scholar] [CrossRef]
- Stein-Gerlach, M.; Salassidis, K.; Bacher, G.; Mueller, S. Pyridylpyrimidine derivatives as effective compounds against prion diseases. Int. Pat. Appl. WO 02/093164 A3, 2002. [Google Scholar]
- Beauchamp, D. A.; Loeb, S. J. Molecular squares, rectangles and infinite helical chains utilizing the simple corner ligand 4-(2-pyridyl)-pyrimidine. Chem. Commun. 2002, 21, 2484–2485. [Google Scholar] [CrossRef]
- Polson, M. I. J.; Lotoski, J. A.; Johansson, K. O.; Taylor, N. J.; Hanan, G. S.; Hasenknopf, B.; Thouvenot, R.; Loiseau, F.; Passalaqua, R.; Campagna, S. Symmetric and asymmetric coupling of pyridylpyrimidines for the synthesis of polynucleating ligands. Eur. J. Inorg. Chem. 2002, 10, 2549–2552. [Google Scholar]
- Paul, R.; Hallett, W. A.; Hanifin, J. W.; Reich, M. F.; Johnson, B. D.; Lenhard, R. H.; Dusza, J. P.; Kerwar, S. S.; Lin, Y.-I.; Pickett, W. C.; Seifert, C. M.; Torley, L. W. Preparation of substituted N-phenyl-4-aryl-2-pyrimidinamides as mediator release inhibitors. J. Med. Chem. 1993, 36, 2716–2725. [Google Scholar] [CrossRef]
- Zimmermann, J.; Buchdunger, E.; Mett, H.; Meyer, T.; Lydon, N. B. Potent and selective inhibitors of the ABL-kinase: phenylaminopyrimidine (PAP) derivatives. Bioorg. Med. Chem. Lett. 1997, 7, 187–192. [Google Scholar] [CrossRef]
- Huang, W.-S.; Shakespeare, W. C. An efficient synthesis of Nilotinib (AMN107). Synthesis 2007, 14, 2121–2124. [Google Scholar] [CrossRef]
- Shekhar, S.; Ryberg, P.; Hartwig, J. F.; Mathew, J. S.; Blackmond, D. G.; Strieter, E. R.; Buchwald, S. L. A reevaluation of the mechanism of the amination of aryl halides catalyzed by BINAP-ligated palladium complexes. J. Am. Chem. Soc. 2006, 128, 3584–3591. [Google Scholar] [CrossRef]
- Kil, K.-E.; Ding, Y.-S.; Lin, K.-S.; Alexoff, D.; Kim, S. W.; Shea, C.; Xu, Y.; Muench, L.; Fowler, J. S. Synthesis and positron emission tomography studies of carbon-11-labeled imatinib (Gleevec). Nucl. Med. Biol. 2007, 34, 153–163. [Google Scholar] [CrossRef]
- Plate, R.; Plaum, M. J. M.; de Boer, T.; Andrews, J. S.; Rae, D. R.; Gibson, S. Synthesis and muscarinic activities of 3-(pyrazolyl)-1,2,5,6-tetrahydropyridine derivatives. Bioorg. Med. Chem. 1996, 4, 227–237. [Google Scholar]
- Adams, H.; Batten, S. R.; Davies, G. M.; Duriska, M. B.; Jeffery, J. C.; Jensen, P.; Lu, J.; Motson, G. R.; Coles, S. J.; Hursthouse, M. B.; Ward, M. D. New bis-, tris- and tetrakis(pyrazolyl)borate ligands with 3-pyridyl and 4-pyridyl substituents: synthesis and coordination chemistry. Dalton Trans. 2005, 11, 1910–1923. [Google Scholar]
- Aakeröy, C. B.; Schultheiss, N.; Desper, J. Directed supramolecular assembly of infinite 1-D M(II)-containing chains (M = Cu, Co, Ni) using structurally bifunctional ligands. Inorg. Chem. 2005, 44, 4983–4991. [Google Scholar] [CrossRef]
- Hatanaka, M.; Takahashi, K.; Nakamura, S.; Mashino, T. Preparation and antioxidant activity of α-pyridoin and its derivatives. Bioorg. Med. Chem. 2005, 13, 6763–6770. [Google Scholar] [CrossRef]
- Giam, C. S.; Knaus, E. E.; Pasutto, F. M. Carbon vs. nitrogen acylation in reactions of organolithium-pyridine adducts with acid chlorides and esters. J. Org. Chem. 1974, 39, 3565–3568. [Google Scholar] [CrossRef]
- Sample Availability: Milligram quantities of compounds 1-12 are available from the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
El-Deeb, I.M.; Ryu, J.C.; Lee, S.H. Synthesis of New N-Arylpyrimidin-2-amine Derivatives Using a Palladium Catalyst. Molecules 2008, 13, 818-830. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13040818
AMA Style
El-Deeb IM, Ryu JC, Lee SH. Synthesis of New N-Arylpyrimidin-2-amine Derivatives Using a Palladium Catalyst. Molecules. 2008; 13(4):818-830. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13040818
Chicago/Turabian StyleEl-Deeb, Ibrahim Mustafa, Jae Chun Ryu, and So Ha Lee. 2008. "Synthesis of New N-Arylpyrimidin-2-amine Derivatives Using a Palladium Catalyst" Molecules 13, no. 4: 818-830. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13040818