
Carapanolides T–X from Carapa guianensis (Andiroba) Seeds

 ,
,
Abstract
:1. Introduction
2. Results and Discussion



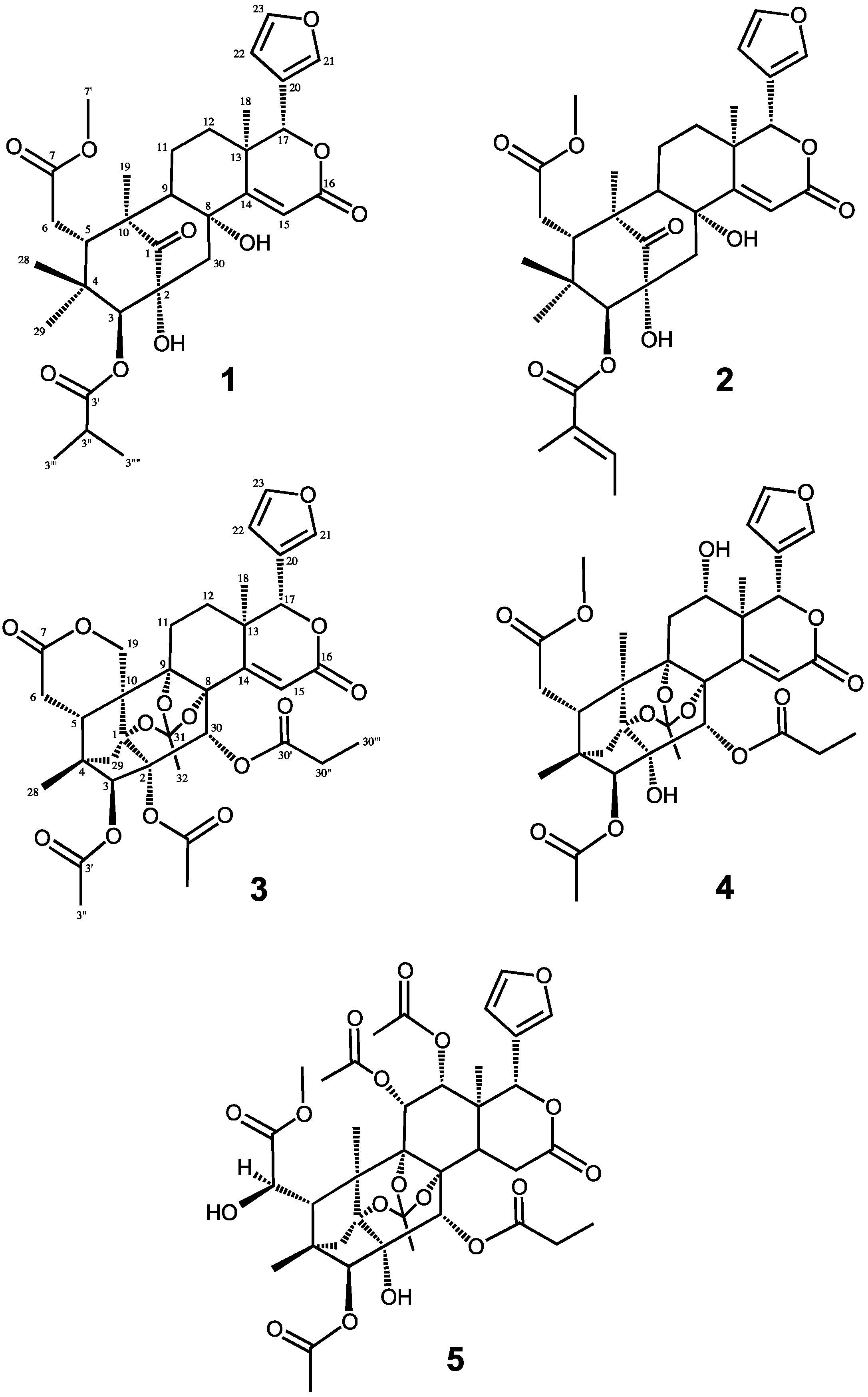{kind=link}
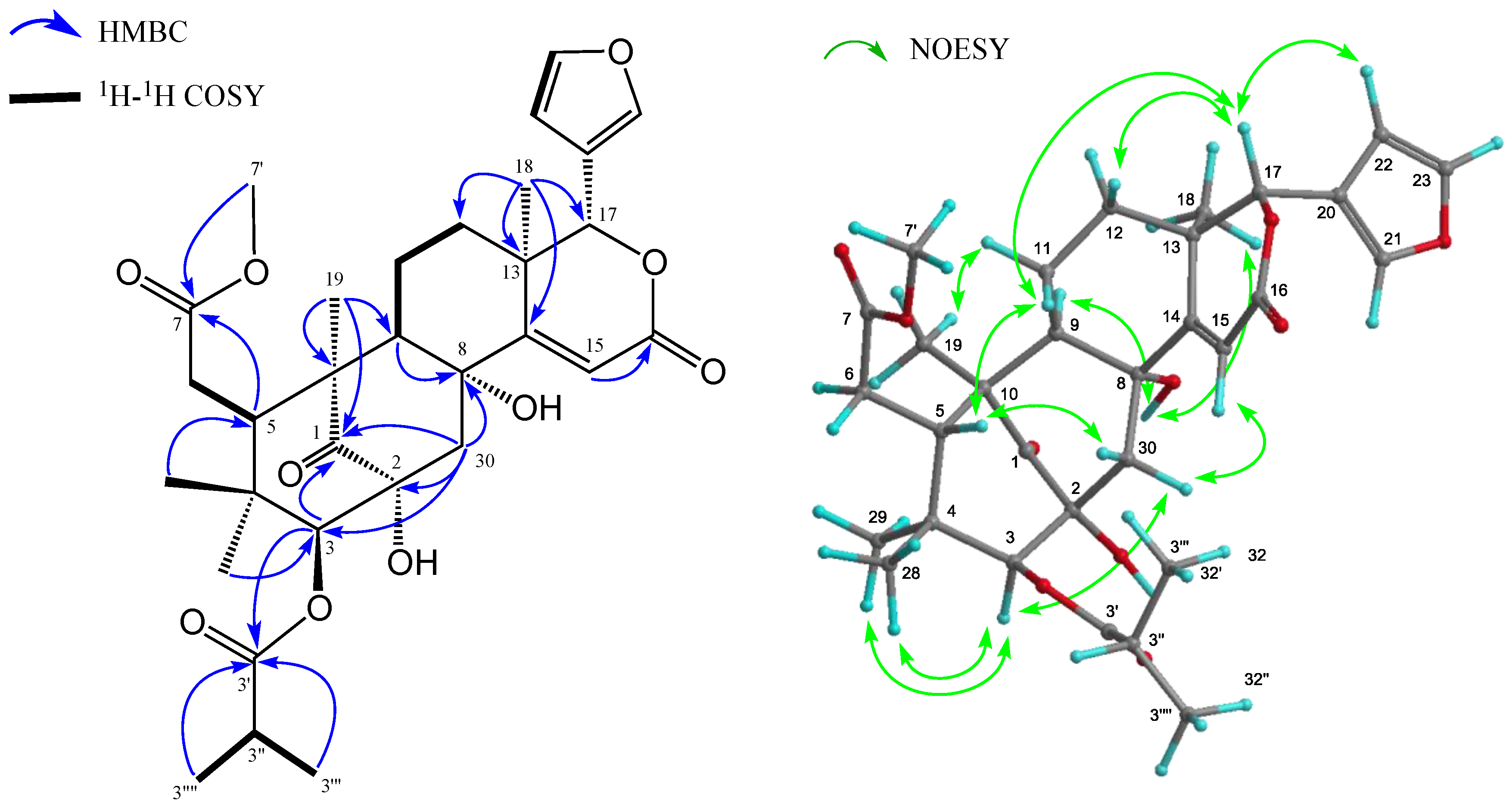{kind=link}
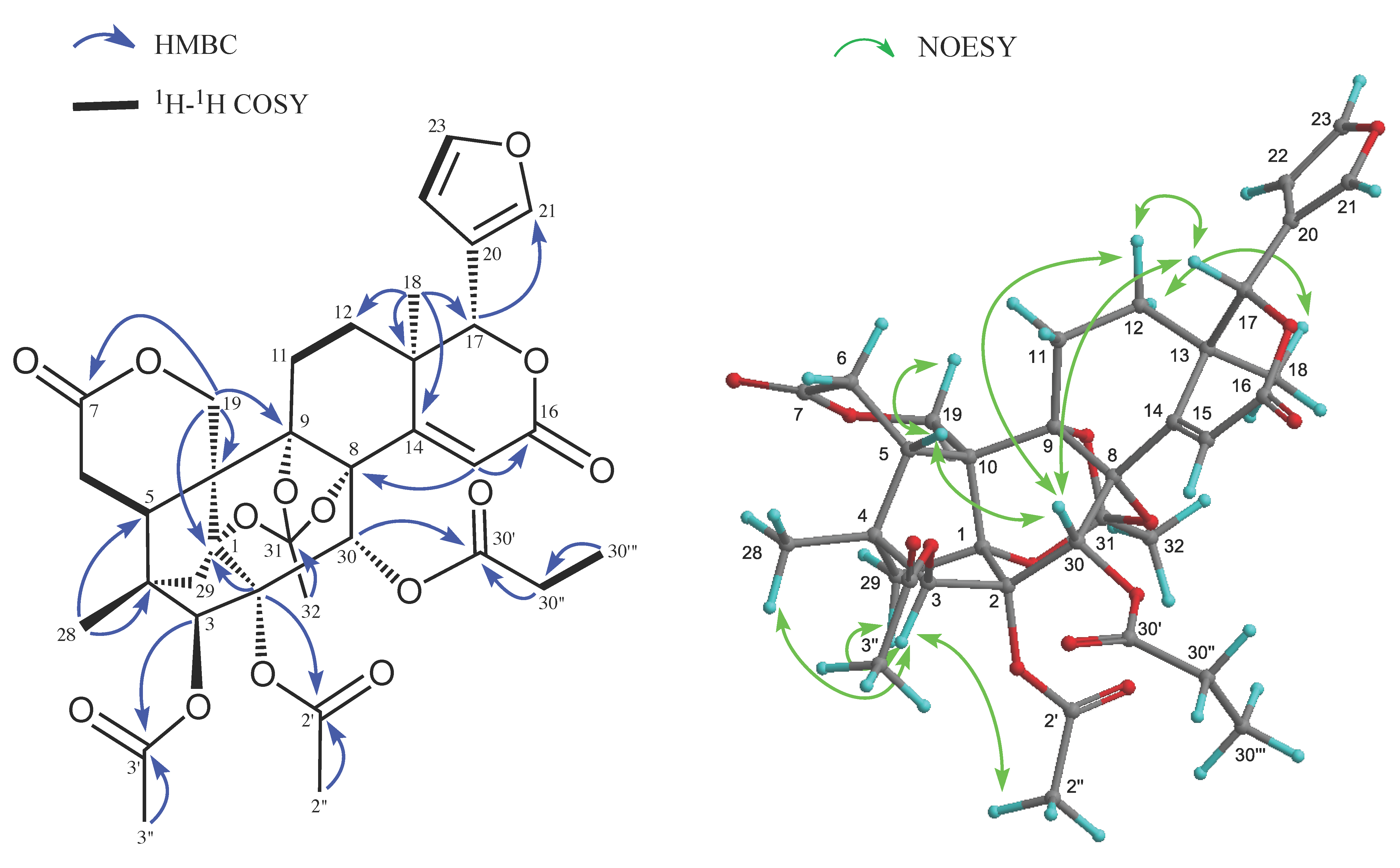{kind=link}
{kind=link}
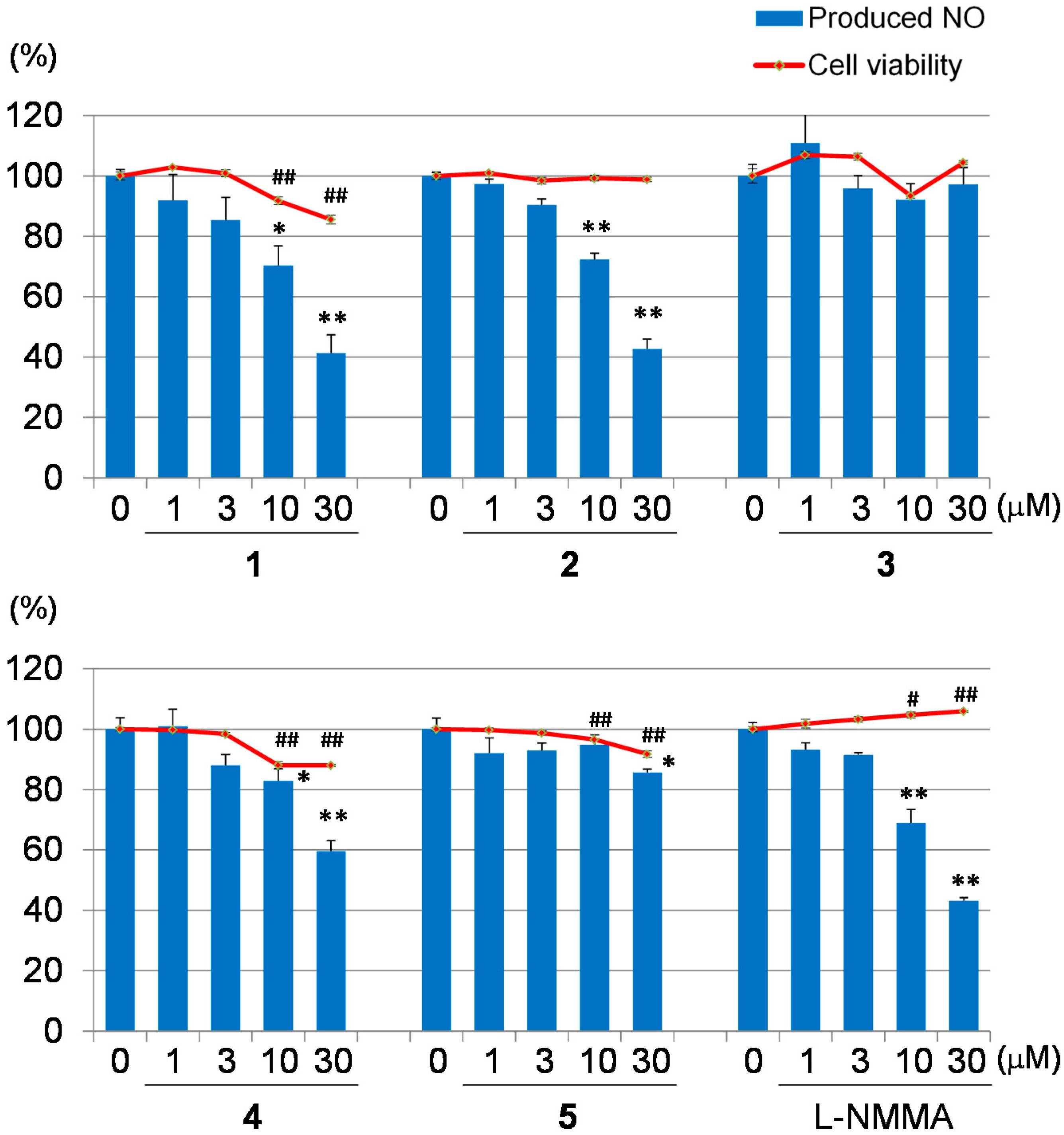{kind=link}
| Position | 1 | 2 | |||||
|---|---|---|---|---|---|---|---|
| 1H a (J, Hz) | 13C b | 1H a (J, Hz) | 13C b | ||||
| 1 | 216.8 | 216.7 | |||||
| 2 | 76.7 | 77.0 | |||||
| 3 | 4.80 | s | 85.5 | 4.88 | s | 85.8 | |
| 4 | 39.7 | 40.0 | |||||
| 5 | 3.36 | dd (9.4, 1.5) | 41.8 | 3.39 | dd (7.9, 1.1) | 41.8 | |
| 6 | α | 2.34 | dd (17.3, 1.5) | 32.9 | 2.37 | m | 32.9 |
| β | 2.38 | dd (17.3, 9.4) | 2.34 | m | |||
| 7 | 173.6 | 173.6 | |||||
| 8 | 72.8 | 72.9 | |||||
| 9 | 1.85 | dd (13.3, 6.0) | 60.2 | 1.87 | t (5.6) | 60.1 | |
| 10 | 47.9 | 47.8 | |||||
| 11 | α | 1.48 | m | 20.7 | 1.67 | m | 20.7 |
| β | 1.52 | m | 1.53 | m | |||
| 12 | α | 1.28 | m | 33.7 | 1.34 | m | 33.8 |
| β | 2.00 | ddd (14.1, 6.8, 3.6) | 2.02 | m | |||
| 13 | 38.5 | 38.5 | |||||
| 14 | 164.7 | 164.6 | |||||
| 15 | 6.18 | s | 116.3 | 6.16 | s | 116.3 | |
| 16 | 167.5 | 167.6 | |||||
| 17 | 5.16 | s | 78.8 | 5.18 | s | 79.8 | |
| 18 | 1.27 | s | 23.2 | 1.28 | s | 23.2 | |
| 19 | 1.23 | s | 18.3 | 1.24 | s | 18.4 | |
| 20 | 119.8 | 119.8 | |||||
| 21 | 7.51 | brs | 141.7 | 7.51 | br s | 141.7 | |
| 22 | 6.49 | dd (2.1, 0.9) | 110.4 | 6.49 | m | 110.4 | |
| 23 | 7.44 | t (2.1) | 143.1 | 7.44 | t (1.4) | 143.1 | |
| 28 | 0.69 | s | 22.3 | 0.70 | s | 22.4 | |
| 29 | 0.86 | s | 22.6 | 0.88 | s | 22.6 | |
| 30 | α | 2.51 | d (16.0) | 45.0 | 2.52 | dd (14.9, 1.2) | 45.0 |
| β | 3.55 | d (16.0) | 3.58 | d (14.9) | |||
| 3′ | 176.2 | 167.2 | |||||
| 3′′ | 2.71 | sept (6.7) | 34.3 | 128.1 | |||
| 3′′′ | 1.25 | d (6.7) | 19.1 | 6.96 | q (7.1) | 138.8 | |
| 3′′′′ | 1.27 | d (6,7) | 19.2 | 1.88 | d (7.1) | 14.7 | |
| 3′′′′′ | 1.92 | s | 12.4 | ||||
| 7' | 3.71 | s | 52.2 | 3.70 | s | 52.1 | |
| 2-OH | 4.05 | s | |||||
| 8-OH | 2.81 | s | |||||

| Position | 3 | 4 | 5 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1H a (J, Hz) | 13C b | 1H a (J, Hz) | 13C b | 1H a (J, Hz) a | δC b | |||||
| 1 | 84.6 | 84.2 | 85.1 | |||||||
| 2 | 84.2 | 83.5 | 79.6 | |||||||
| 3 | 5.27 | s | 81.3 | 5.22 | s | 85.0 | 4.59 | s | 83.7 | |
| 4 | 46.4 | 44.6 | 45.5 | |||||||
| 5 | 2.72 | dd (4.7, 3.2) | 33.8 | 2.11 | m | 39.9 | 3.30 | brs | 40.1 | |
| 6 | α | 2.64 | dd (17.0, 3.2) | 68.6 | 2.34 | m | 33.7 | 5.98 | brs | 71.5 |
| β | 2.59 | dd (17.0, 4.7) | 2.36 | m | ||||||
| 7 | 171.0 | 173.9 | 169.4 | |||||||
| 8 | 82.6 | 83.7 | 84.8 | |||||||
| 9 | 83.9 | 86.1 | 86.0 | |||||||
| 10 | 46.9 | 48.1 | 46.2 | |||||||
| 11 | α | 2.19 | m | 25.6 | 1.97 | m | 34.6 | 4.49 | d (2.3) | 69.4 |
| β | 2.33 | m | 2.21 | dd (14.7, 4.1) | ||||||
| 12 | α | 1.50 | m | 26.5 | 3.86 | dd (13.5, 4.1) | 66.6 | 4.48 | d (2.3) | 71.7 |
| β | 1.64 | m | ||||||||
| 13 | 37.6 | 44.8 | 38.4 | |||||||
| 14 | 159.6 | 153.8 | 2.79 | dd (10.4, 0.6) | 42.3 | |||||
| 15 | 6.05 | s | 121.0 | 6.62 | s | 123.7 | 2.90 | dd (18.7, 10.4) | 26.9 | |
| 3.22 | dd (18.7, 0.6) | |||||||||
| 16 | 163.0 | 163.5 | 170.8 | |||||||
| 17 | 5.10 | s | 80.4 | 5.90 | s | 78.8 | 5.98 | s | 76.9 | |
| 18 | 1.14 | s | 18.7 | 1.48 | s | 13.0 | 1.43 | s | 15.8 | |
| 19 | α | 4.86 | d (14.0) | 31.4 | 1.32 | s | 15.5 | 1.23 | s | 13.8 |
| β | 4.34 | d (14.0) | ||||||||
| 20 | 119.4 | 121.4 | 120.9 | |||||||
| 21 | 7.52 | br s | 141.4 | 7.53 | br s | 144.8 | 7.48 | brs | 141.0 | |
| 22 | 6.44 | dd (1.7, 0.6) | 109.7 | 6.61 | dd (1.7, 0.9) | 109.6 | 6.46 | dd (1.8, 1.5) | 110.9 | |
| 23 | 7.44 | t (1.7) | 143.3 | 7.64 | t (1.7) | 142.4 | 7.00 | t (1.8) | 143.1 | |
| 28 | 1.02 | s | 14.2 | 0.74 | s | 14.5 | 1.10 | s | 15.4 | |
| 29 | pro-R | 1.78 | d (11.6) | 39.2 | 1.72 | d (11.5) | 39.8 | 1.81 | d (10.9)) | 39.9 |
| pro-S | 2.38 | d (11.6) | 1.96 | d (11.5) | 2.06 | d (10.9) | ||||
| 30 | 5.78 | s | 68.1 | 5.35 | s | 74.3 | 6.01 | s | 69.8 | |
| 31 | 120.1 | 119.7 | 119.3 | |||||||
| 32 | 1.68 | s | 20.9 | 1.70 | s | 16.5 | 1.76 | s | 21.1 | |
| 2′ | 170.1 | |||||||||
| 2′′ | 2.17 | s | 21.8 | |||||||
| 3′ | 169.1 | 169.1 | 169.6 | |||||||
| 3′′ | 2.04 | s | 20.8 | 2.09 | s | 21.7 | 2.18 | s | 21.2 | |
| 7′ | 3.71 | s | 52.3 | 3.69 | s | 53.1 | ||||
| 11′ | 169.7 | |||||||||
| 11′′ | 2.22 | s | 21.4 | |||||||
| 12′ | 169.6 | |||||||||
| 12′′ | 1.70 | s | 20.1 | |||||||
| 30′ | 173.3 | 173.8 | 172.5 | |||||||
| 30′′ | A | 2.25 | m | 27.4 | 2.45 | m | 28.1 | 2.38 | dq (11.2, 7.3) | 27.9 |
| B | 2.29 | m | 2.38 | dq (11.2, 7.3) | ||||||
| 30′′′ | 1.07 | t (7.6) | 8.5 | 1.16 | t (7.7) | 8.9 | 1.09 | t (7.3) | 8.6 | |


3. Experimental Section
3.1. General Procedures
3.2. Plant Material
3.3. Isolation of Compounds 1–5
3.4. Analytical Data
3.5. Determination of RAW264.7 Cell Proliferation
3.6. Inhibitory Assay of NO Production
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tan, Q.G.; Luo, X.D. Meliaceous limonoids: Chemistry and biological activities. Chem. Rev. 2011, 111, 7437–7522. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Di, Y.T.; Hao, X.J. The advaces in the limonoid chemistry of the Meliaceae family. Curr. Org. Chem. 2011, 15, 1363–1391. [Google Scholar] [CrossRef]
- Liao, S.G.; Chen, H.D.; Yue, J.M. Plant orthoesters. Chem. Rev. 2009, 109, 1092–1140. [Google Scholar] [CrossRef] [PubMed]
- Prophiro, J.S.; da Silva Mario, A.N.; Kanis, L.A.; da Rocha, L.C.B.P.; Duque-Luna, J.E.; da Silva, O.S. First report on susceptibility of wild Aedes aegypty (Diptera: Culicidae) using Carapa guianensis (Meliaceae) and Copaifera sp. (Leguminosae). Parasitol. Res. 2012, 110, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Penido, C.; Costa, K.A.; Pennaforte, R.J.; Costa, M.F.S.; Pereira, J.F.G.; Siani, A.C.; Henriques, M.G.M.O. Anti-allergic effects of natural tetranortriterpenoids isolated from Carapa guianensis Aublet onallergen-induced vascular permeability and hyperalgesia. Inflamm. Res. 2005, 54, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Nayak, B.S.; Kanhai, J.; Milne, D.M.; Pereira, L.P.; Swanston, W.H. Experimental Evaluation of Ethanolic Extract of Carapa guianensis L. Leaf for Its Wound Healing Activity Using Three Wound Models. Evid. Based Complement. Altern. Med. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Penido, C.; Conte, F.P.; Chagas, M.S.S.; Rodrigue, C.A.B.; Pereira, J.F.G.; Henriques, M.G.M.O. Antiinflammatory effects of natural tetranortriterpenoids isolated from Carapa guianensis Aublet on zymosan-induced arthritis in mice. Inflamm. Res. 2006, 55, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Nayak, B.S.; Kanhai, J.; Milne, D.M.; Swanston, W.H.; Mayers, S.; Eversley, M.; Rao, A.V. Investigation of the wound healing activity of Carapa guianensis L. (Meliaceae) bark extract in rats using excision, incision, and dead space wound models. J. Med. Food. 2010, 13, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Bickii, J.; Njifutie, N.; Foyere, J.A.; Basco, L.K.; Ringwald, P.J. In vitro antimalarial activity of limonoids from Khaya grandifoliola C.D.C. (Meliaceae). J. Ethnopharmacol. 2000, 69, 27–33. [Google Scholar] [CrossRef]
- Ferraris, F.K.; Rodrigues, R.; da Silva, V.P.; Figueiredo, R.; Penido, C.; Henriques, M.G.M.O. Modulation of T lymphocyte and eosinophil functions in vitro by natural tetranortriterpenoids isolated from Carapa guianensis Aublet. Int. Immunopharmacol. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Miranda, J.R.N.C.; Dolabela, M.F.; da Silva, M.N.; Povoa, M.M.; Maia, J.G.S. Antiplasmoidal activity of the andiroba (Carapa guianensis Aublet., Meliaceae) oil and its limonoid-rich fraction. J. Ethnopharmacol. 2012, 142, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, H.; Lima, C.R.; Silva, E.J.R.; Araujo, A.V.; Fraga, M.C.C.R.; Ribeiro, E.; Ribwiro, A.; Arruda, A.C.; Lafayette, S.S.L.; Wanderley, J. Acute and subacute toxicity of the Carapa guianensis Aublet (Meliaceae) seed oil. J. Ethnopharmacol. 2008, 116, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Nagai, Y.; Mitooka, A.; Ujike, R.; Muraoka, O.; Yamada, T.; Tanaka, R. Carapanolides A and B: Unusual 9,10-seco-mexicanolides having a 2R,9S-oxygen bridge From the seeds of Carapa guianensis. Tetrahedron Lett. 2012, 53, 6685–6688. [Google Scholar] [CrossRef]
- Inoue, T.; Matsui, Y.; Kikuchi, T.; In, Y.; Yamada, T.; Muraoka, O.; Matsunaga, S.; Tanaka, R. Guianolides A and B, New Carbon Skeletal Limonoids from the seeds of Carapa guianensis. Org. Lett. 2013, 15, 3018–3021. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Matsui, Y.; Kikuchi, T.; In, Y.; Muraoka, O.; Yamada, T.; Tanaka, R. Carapanolides C–I from the seeds of andiroba (Carapa guianensis, Meliaceae). Fitoterapia 2014, 96, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Kikuchi, T.; Inoue, T.; Muraoka, O.; Yamada, T.; Tanaka, R. Carapanolides J–L from the Seeds of Carapa guianensis (Andiroba) and Their Effects on LPS-Activated NO Production. Molecules 2014, 19, 17130–17140. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Matsui, Y.; Kikuchi, T.; Yamada, T.; In, Y.; Muraoka, O.; Sakai, C.; Ninomiya, K.; Morikawa, T.; Tanaka, R. Carapanolides M–S from seeds of andiroba (Carapa guianensis, Meliaceae) and triglyceride metabolism-promoting activity in high glucose-pretreated HepG2 cells. Tetrahedron 2015, 71, 2753–2760. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Dong, B.; Ma, X.; Hou, L.; Cao, X.; Wang, C. Anti-inflammatory Activity and Mechanism of Surfactin in Lipopolysaccharide-Activated Macrophages. Inflammation 2015, 38, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Tanaka, Y.; Yamada, T.; Kikuchi, T.; Muraoka, O.; Ninomiya, K.; Morikawa, T.; Tanaka, R. Andirolides W-Y from the Flower Oil of Andiroba (Carapa guianensis, Meliaceae). Fitoterapia 2015, 100, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Muroga, Y.; Jinno, M.; Kajimoto, T.; Usami, Y.; Numata, A.; Tanaka, R. New class azaphilone produced by a marine fish-derived Chaetomium globosum. The stereochemistry and biological activities. Bioorg. Med. Chem. 2011, 19, 4106–4113. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyake, T.; Ishimoto, S.; Ishimatsu, N.; Higuchi, K.; Minoura, K.; Kikuchi, T.; Yamada, T.; Muraoka, O.; Tanaka, R. Carapanolides T–X from Carapa guianensis (Andiroba) Seeds. Molecules 2015, 20, 20955-20966. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119737
Miyake T, Ishimoto S, Ishimatsu N, Higuchi K, Minoura K, Kikuchi T, Yamada T, Muraoka O, Tanaka R. Carapanolides T–X from Carapa guianensis (Andiroba) Seeds. Molecules. 2015; 20(11):20955-20966. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119737
Chicago/Turabian StyleMiyake, Teppei, Sari Ishimoto, Naoko Ishimatsu, Keiichiro Higuchi, Katsuhiko Minoura, Takashi Kikuchi, Takeshi Yamada, Osamu Muraoka, and Reiko Tanaka. 2015. "Carapanolides T–X from Carapa guianensis (Andiroba) Seeds" Molecules 20, no. 11: 20955-20966. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119737