
Celastrol Induces Cell Apoptosis and Inhibits the Expression of the AML1-ETO/C-KIT Oncoprotein in t(8;21) Leukemia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
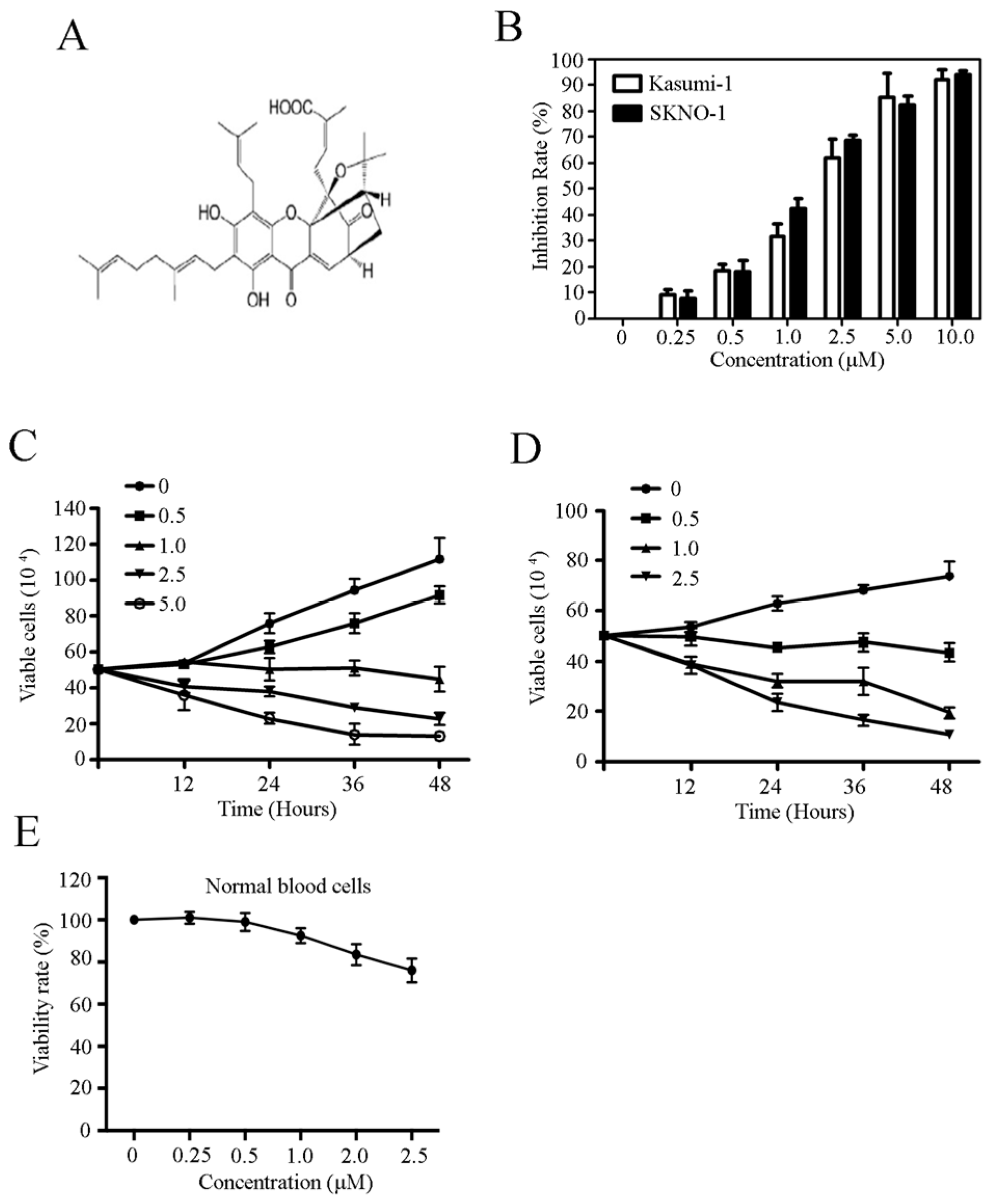
2.1. Celastrol Inhibits Growth and Proliferation in t(8;21) Leukemia Cells
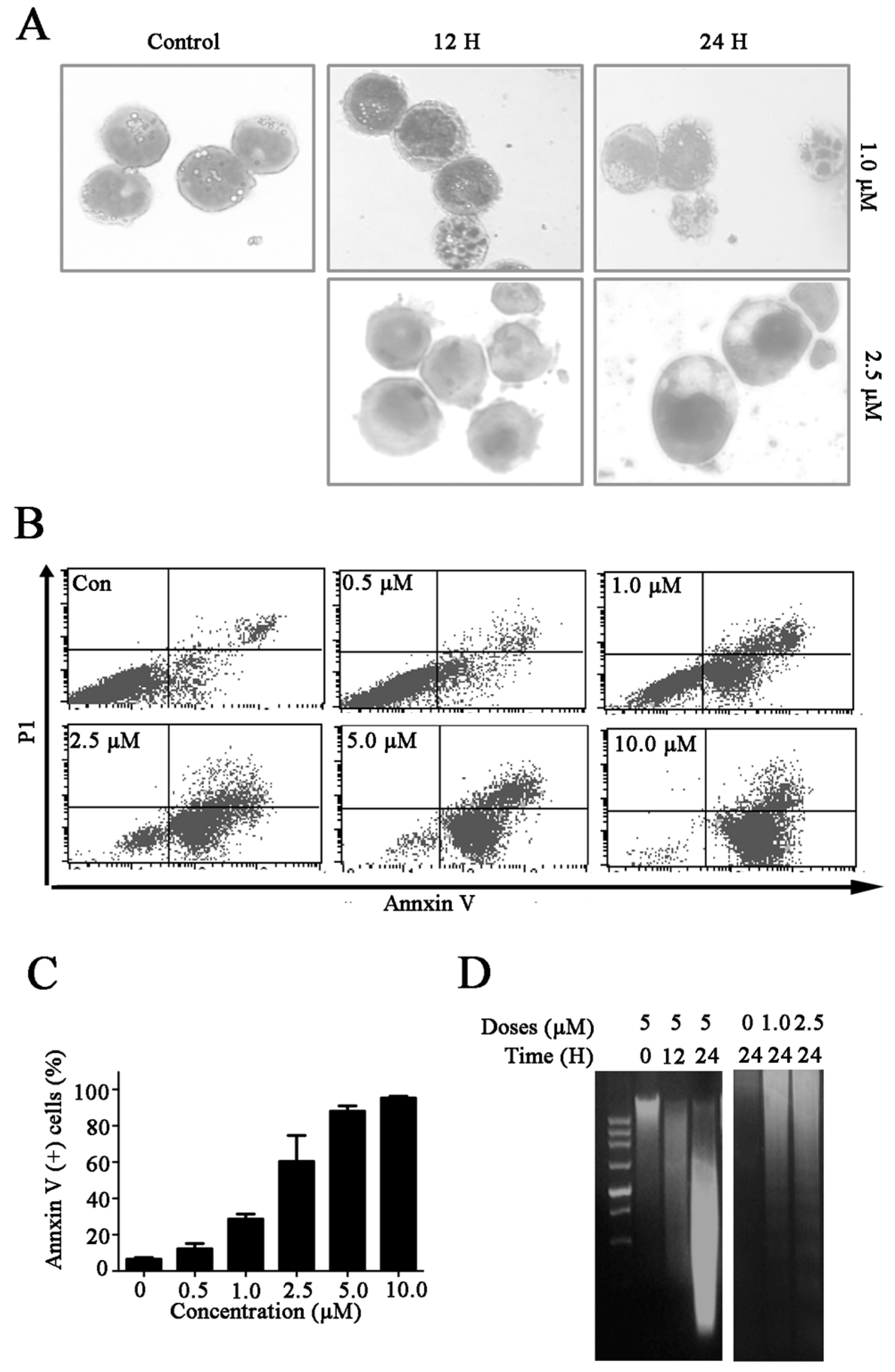
2.2. Celastrol Induces Cell Death in Kasumi-1 Cells
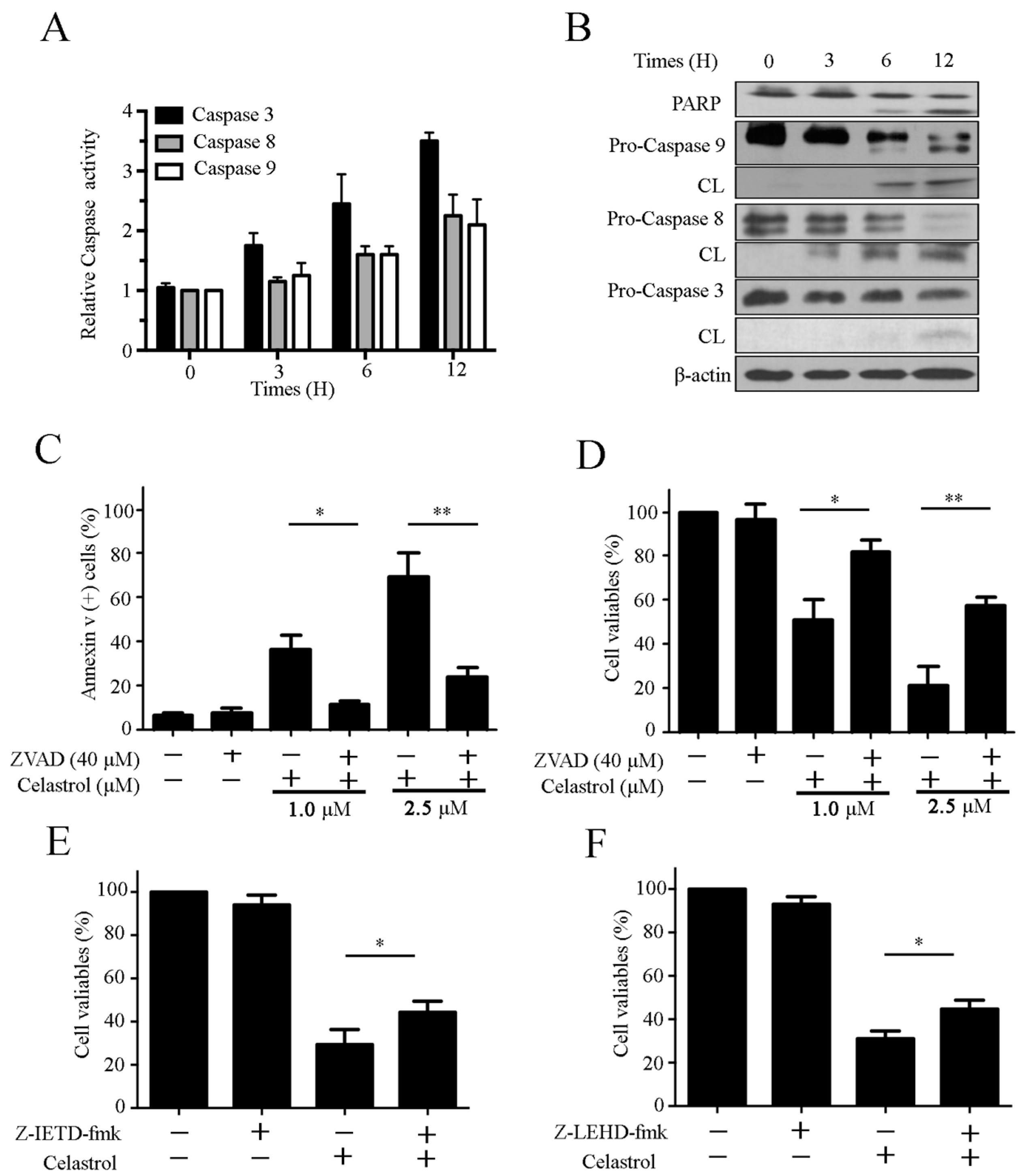
2.3. Celastrol Triggers Caspase Activation in Kasumi-1 Cells
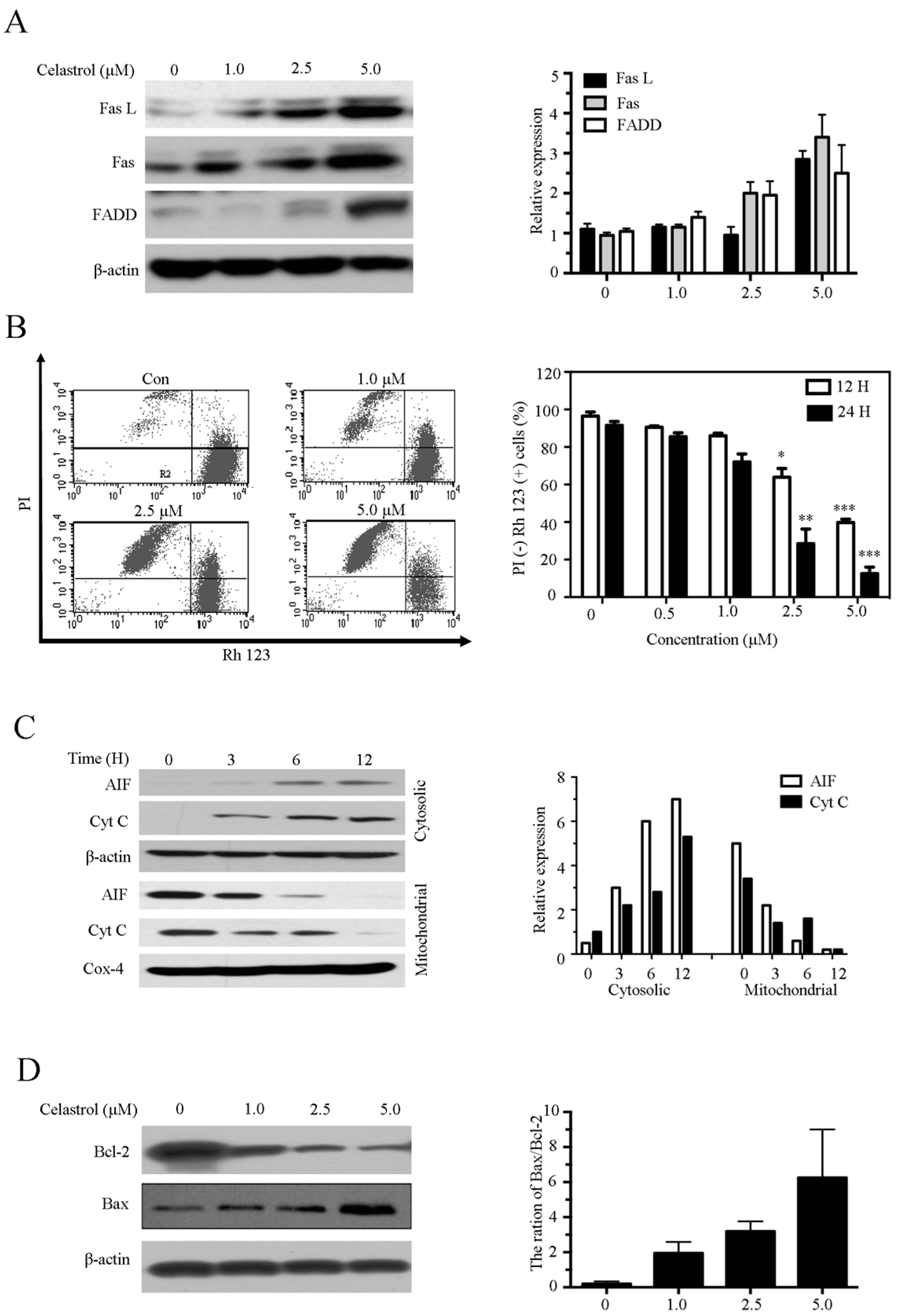
2.4. Celastrol Induces Apoptosis via the Extrinsic and Intrinsic Pathways
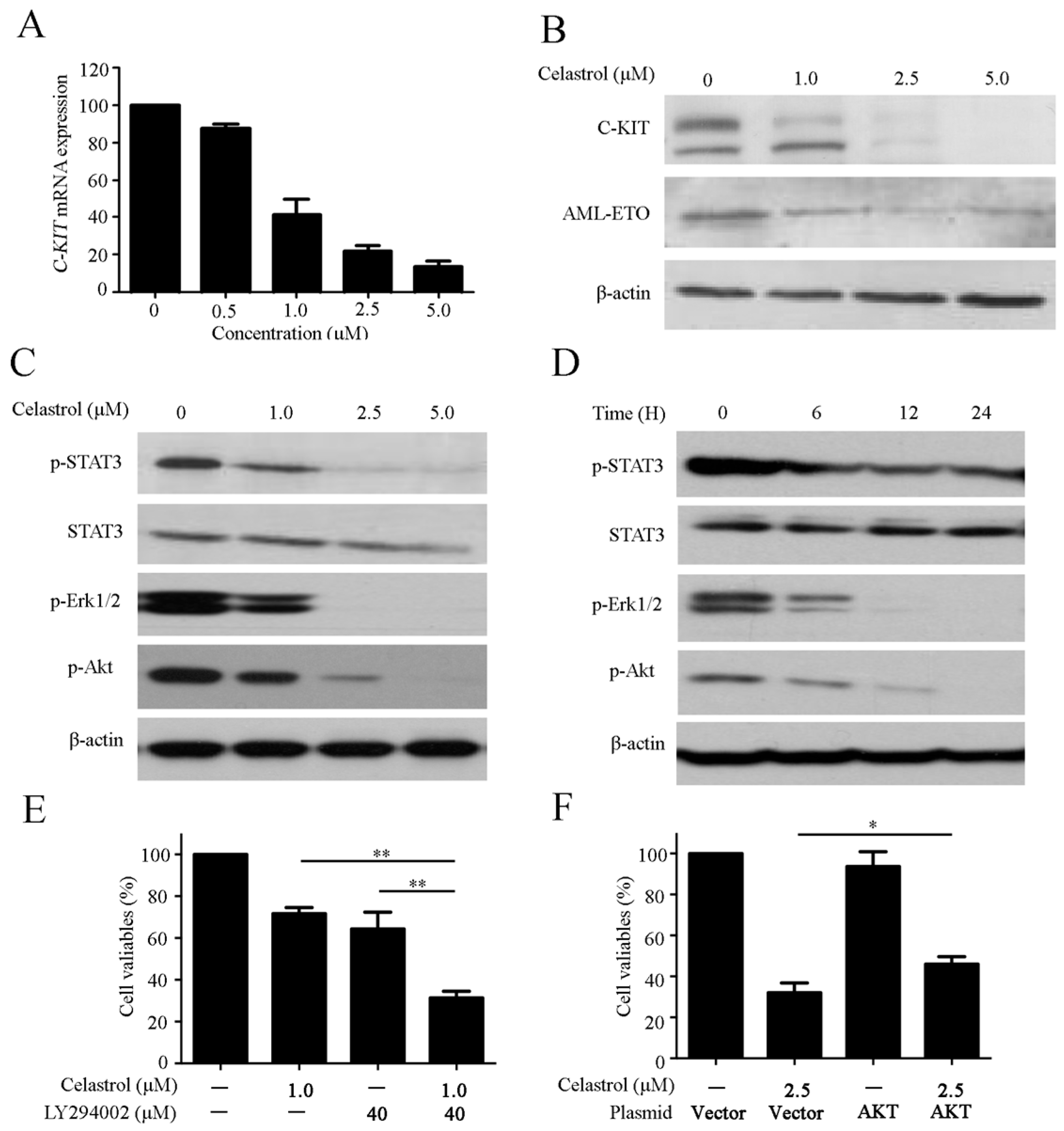
2.5. Celastrol Regulates the C-KIT/AML-ETO Oncoprotein and Downstream Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Assessment of Cell Viability, Apoptosis, Cell Morphology and DNA Fragmentation
4.4. Western Blot Assay
4.5. Measurement of Mitochondrial Membrane Integrity
4.6. Preparation of Cell Fractions and Western Blot Analysis
4.7. Quantitative Real-Time PCR Analysis
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Estey, E.; Dohner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Kuhnl, A.; Valk, P.J.; Sanders, M.A.; Ivey, A.; Hills, R.K.; Mills, K.I.; Gale, R.E.; Kaiser, M.F.; Dillon, R.; Joannides, M.; et al. Down-regulation of the Wnt inhibitor CXXC5 predicts a better prognosis in acute myeloid leukemia. Blood 2015, 125, 2985–2994. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Gilliland, D.G. Genetics of myeloid leukemias. Annu. Rev. Genom. Hum. Genet. 2002, 3, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Elagib, K.E.; Goldfarb, A.N. Oncogenic pathways of AML1-ETO in acute myeloid leukemia: Multifaceted manipulation of marrow maturation. Cancer Lett. 2007, 251, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.F.; Zhang, D.E. The 8;21 translocation in leukemogenesis. Oncogene 2004, 23, 4255–4262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Zhou, G.B.; Yin, T.; Chen, B.; Shi, J.Y.; Liang, W.X.; Jin, X.L.; You, J.H.; Yang, G.; Shen, Z.X.; et al. AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: Implication in stepwise leukemogenesis and response to Gleevec. Proc. Natl. Acad. Sci. USA 2005, 102, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, L.C.; Pang, J.; Santhanam, R.; Schwind, S.; Wu, Y.Z.; Hickey, C.J.; Yu, J.; Becker, H.; Maharry, K.; et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell 2010, 17, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Zhen, T.; Wu, C.F.; Liu, P.; Wu, H.Y.; Zhou, G.B.; Lu, Y.; Liu, J.X.; Liang, Y.; Li, K.K.; Wang, Y.Y.; et al. Targeting of AML1-ETO in t(8;21) leukemia by oridonin generates a tumor suppressor-like protein. Sci. Transl Med. 2012, 4, 127ra38. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.T.; Zhang, B.; Pan, X.F.; Gao, L.; Zhen, T.; Zhao, H.X.; Ma, L.; Xie, J.; Liu, Z.; Yu, X.J.; et al. Bortezomib interferes with C-KIT processing and transforms the t(8;21)-generated fusion proteins into tumor-suppressing fragments in leukemia cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2521–2526. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Chen, S.F.; Lv, Y.P.; Lu, L.N.; Hu, W.X.; Zhou, Y.L. ZGDHu-1 induces G(2)/M phase arrest and apoptosis in Kasumi-1 cells. Mol. Med. Rep. 2015, 11, 3398–3404. [Google Scholar] [CrossRef] [PubMed]
- Sawney, S.; Arora, R.; Aggarwal, K.K.; Saluja, D. Esculetin Downregulates the Expression of AML1-ETO and C-Kit in Kasumi-1 Cell Line by Decreasing Half-Life of mRNA. J. Oncol. 2015, 2015, 781473. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, L.; Zhou, G.B. The main anticancer bullets of the Chinese medicinal herb, thunder god vine. Molecules 2011, 16, 5283–5297. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Z.; Liu, Y.Q.; Cheng, X.; Zhou, G.B. Celastrol induces proteasomal degradation of FANCD2 to sensitize lung cancer cells to DNA crosslinking agents. Cancer Sci. 2015, 106, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, L.; Wen, Z.S.; Hu, Z.; Wu, F.Q.; Li, W.; Liu, J.; Zhou, G.B. Cancerous inhibitor of PP2A is targeted by natural compound celastrol for degradation in non-small-cell lung cancer. Carcinogenesis 2014, 35, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Jeengar, M.K.; Reddy, V.S.; Reddy, G.B.; Naidu, V.G. Anticancer effect of celastrol on human triple negative breast cancer: possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3K/Akt pathways. Exp. Mol. Pathol. 2015, 98, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huang, X.; Wang, H.; Yang, H. Celastrol Induces Autophagy by Targeting AR/miR-101 in Prostate Cancer Cells. PLoS ONE 2015, 10, e0140745. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Won, Y.S.; Park, K.H.; Lee, M.K.; Tachibana, H.; Yamada, K.; Seo, K.I. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis 2012, 17, 1275–1286. [Google Scholar] [CrossRef]
- Zhou, Y.X.; Huang, Y.L. Antiangiogenic effect of celastrol on the growth of human glioma: An in vitro and in vivo study. Chin. Med. J. 2009, 122, 1666–1673. [Google Scholar] [PubMed]
- Nicholson, D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999, 6, 1028–1042. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.J.; Vom, A.; Czabotar, P.E.; Lessene, G. Cell death and the mitochondria: therapeutic targeting of the BCL-2 family-driven pathway. Br. J. Pharmacol. 2014, 171, 1973–1987. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ. 2002, 9, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Amann, J.M.; Nip, J.; Strom, D.K.; Lutterbach, B.; Harada, H.; Lenny, N.; Downing, J.R.; Meyers, S.; Hiebert, S.W. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol. Cell Biol. 2001, 21, 6470–6483. [Google Scholar] [CrossRef] [PubMed]
- Hatlen, M.A.; Wang, L.; Nimer, S.D. AML1-ETO driven acute leukemia: Insights into pathogenesis and potential therapeutic approaches. Front. Med. 2012, 6, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Spirin, P.V.; Lebedev, T.D.; Orlova, N.N.; Gornostaeva, A.S.; Prokofjeva, M.M.; Nikitenko, N.A.; Dmitriev, S.E.; Buzdin, A.A.; Borisov, N.M.; Aliper, A.M.; et al. Silencing AML1-ETO gene expression leads to simultaneous activation of both pro-apoptotic and proliferation signaling. Leukemia 2014, 28, 2222–2228. [Google Scholar] [CrossRef] [PubMed]
- Jiao, B.; Wu, C.F.; Liang, Y.; Chen, H.M.; Xiong, S.M.; Chen, B.; Shi, J.Y.; Wang, Y.Y.; Wang, J.H.; Chen, Y.; et al. AML1-ETO9a is correlated with C-KIT overexpression/mutations and indicates poor disease outcome in t(8;21) acute myeloid leukemia-M2. Leukemia 2009, 23, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Kohl, T.M.; Haferlach, T.; Kern, W.; Hiddemann, W.; Spiekermann, K.; Schoch, C. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood 2006, 107, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Taki, T.; Tabuchi, K.; Tawa, A.; Horibe, K.; Tsuchida, M.; Hanada, R.; Tsukimoto, I.; Hayashi, Y. KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): A study of the Japanese Childhood AML Cooperative Study Group. Blood 2006, 107, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lin, J.; Gao, L.; Deng, A.; Lu, X.; Li, Y.; Wang, L.; Yu, L. High expression of c-kit mRNA predicts unfavorable outcome in adult patients with t(8;21) acute myeloid leukemia. PLoS ONE 2015, 10, e0124241. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.S.; Hsiau, C.W.; Serve, H.; Vosseller, K.; Besmer, P. Mechanism of down-regulation of c-kit receptor. Roles of receptor tyrosine kinase, phosphatidylinositol 3′-kinase, and protein kinase C. J. Biol. Chem. 1994, 269, 31991–31998. [Google Scholar] [PubMed]
- Ueda, S.; Mizuki, M.; Ikeda, H.; Tsujimura, T.; Matsumura, I.; Nakano, K.; Daino, H.; Honda Zi, Z.; Sonoyama, J.; Shibayama, H.; et al. Critical roles of c-Kit tyrosine residues 567 and 719 in stem cell factor-induced chemotaxis: contribution of src family kinase and PI3-kinase on calcium mobilization and cell migration. Blood 2002, 99, 3342–3349. [Google Scholar] [CrossRef] [PubMed]
- Harir, N.; Boudot, C.; Friedbichler, K.; Sonneck, K.; Kondo, R.; Martin-Lanneree, S.; Kenner, L.; Kerenyi, M.; Yahiaoui, S.; Gouilleux-Gruart, V.; et al. Oncogenic Kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood 2008, 112, 2463–2473. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.S.; Hu, Z.; Fang, H.T.; Zhang, F.X.; Pan, X.F.; Chen, X.Q.; Hu, A.M.; Xu, L.; Zhou, G.B. Biologic activity of triptolide in t(8;21) acute myeloid leukemia cells. Leuk Res. 2011, 35, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.J.; Han, Q.B.; Wen, Z.S.; Ma, L.; Gao, J.; Zhou, G.B. Gambogenic acid induces G1 arrest via GSK3beta-dependent cyclin D1 degradation and triggers autophagy in lung cancer cells. Cancer Lett. 2012, 322, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, W.L.; Yan, J.S.; Liu, P.; Sun, H.P.; Zhou, G.B.; Weng, Z.Y.; Wu, W.L.; Weng, X.Q.; Sun, X.J.; et al. Eriocalyxin B induces apoptosis of t(8;21) leukemia cells through NF-kappaB and MAPK signaling pathways and triggers degradation of AML1-ETO oncoprotein in a caspase-3-dependent manner. Cell Death Differ. 2007, 14, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of celastrol are available from the authors.





© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Ruan, X.; Zhang, J.; Zhao, Q. Celastrol Induces Cell Apoptosis and Inhibits the Expression of the AML1-ETO/C-KIT Oncoprotein in t(8;21) Leukemia. Molecules 2016, 21, 574. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050574
Yu X, Ruan X, Zhang J, Zhao Q. Celastrol Induces Cell Apoptosis and Inhibits the Expression of the AML1-ETO/C-KIT Oncoprotein in t(8;21) Leukemia. Molecules. 2016; 21(5):574. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050574
Chicago/Turabian StyleYu, Xianjun, Xuzhi Ruan, Jingxuan Zhang, and Qun Zhao. 2016. "Celastrol Induces Cell Apoptosis and Inhibits the Expression of the AML1-ETO/C-KIT Oncoprotein in t(8;21) Leukemia" Molecules 21, no. 5: 574. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050574