
Novel Radioligands for Cyclic Nucleotide Phosphodiesterase Imaging with Positron Emission Tomography: An Update on Developments Since 2012


Abstract
:1. Introduction
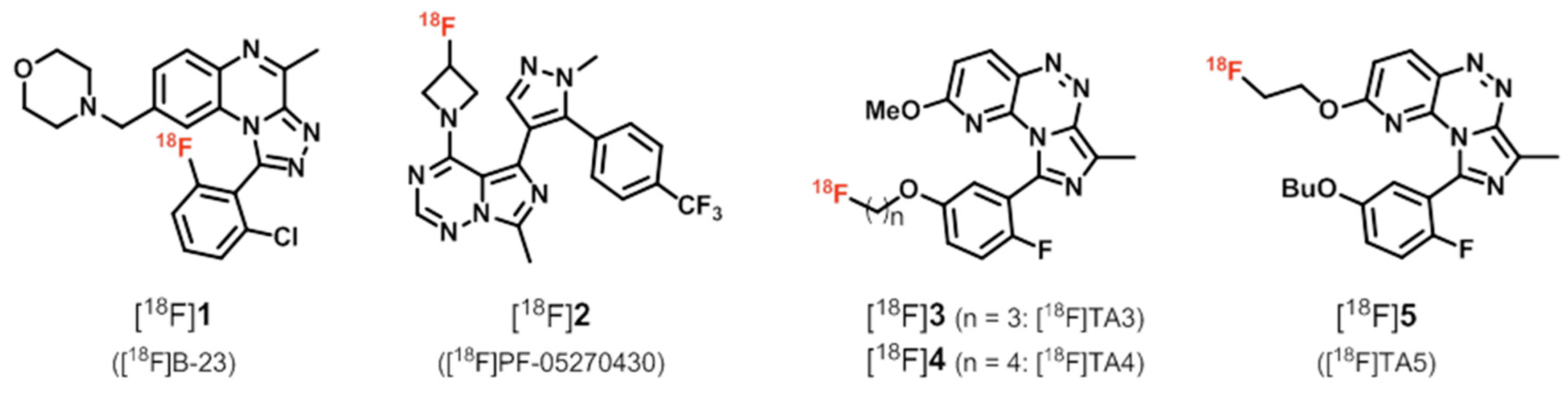
2. PDE2 Radioligands
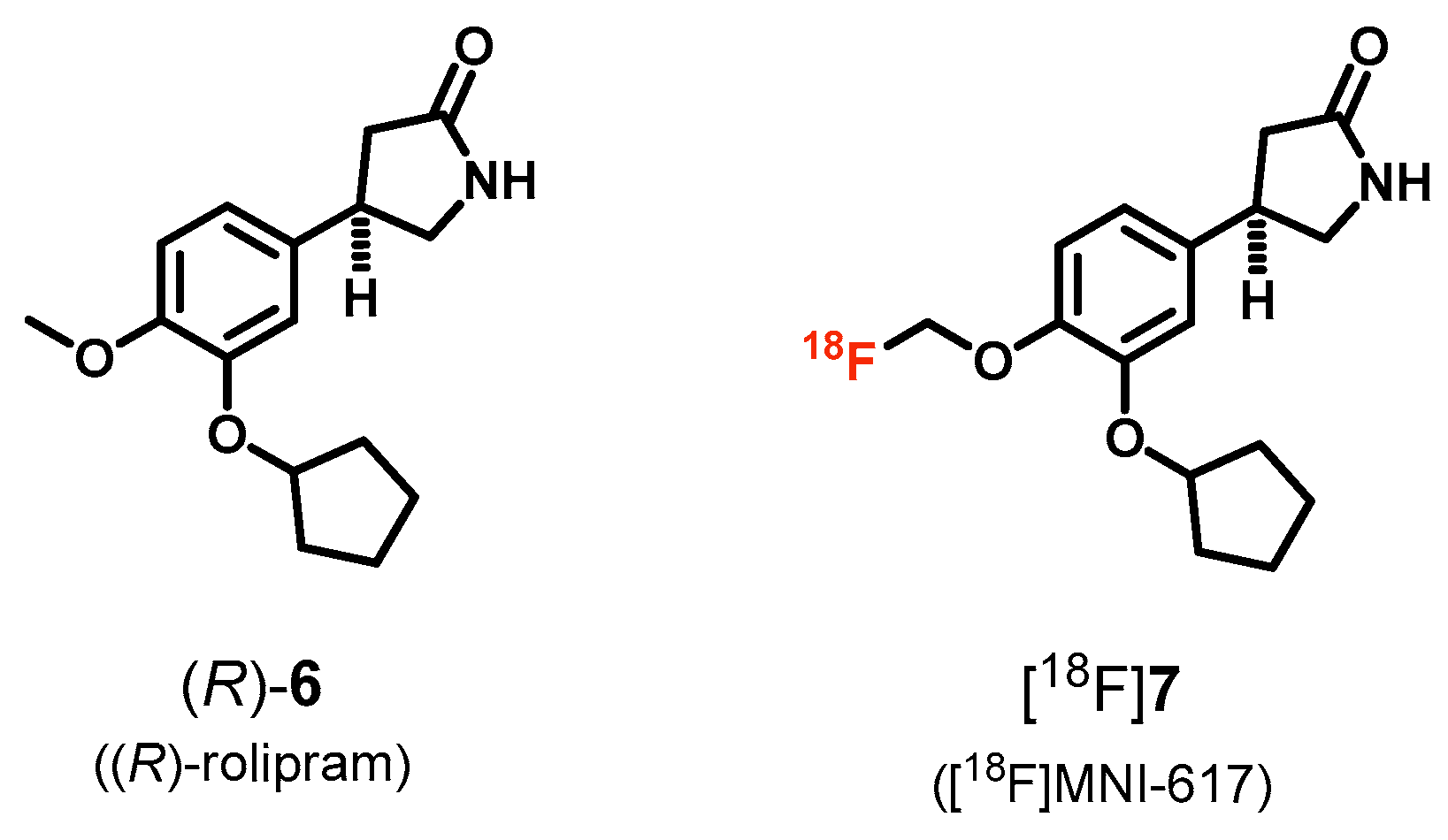
3. PDE4 Radioligands
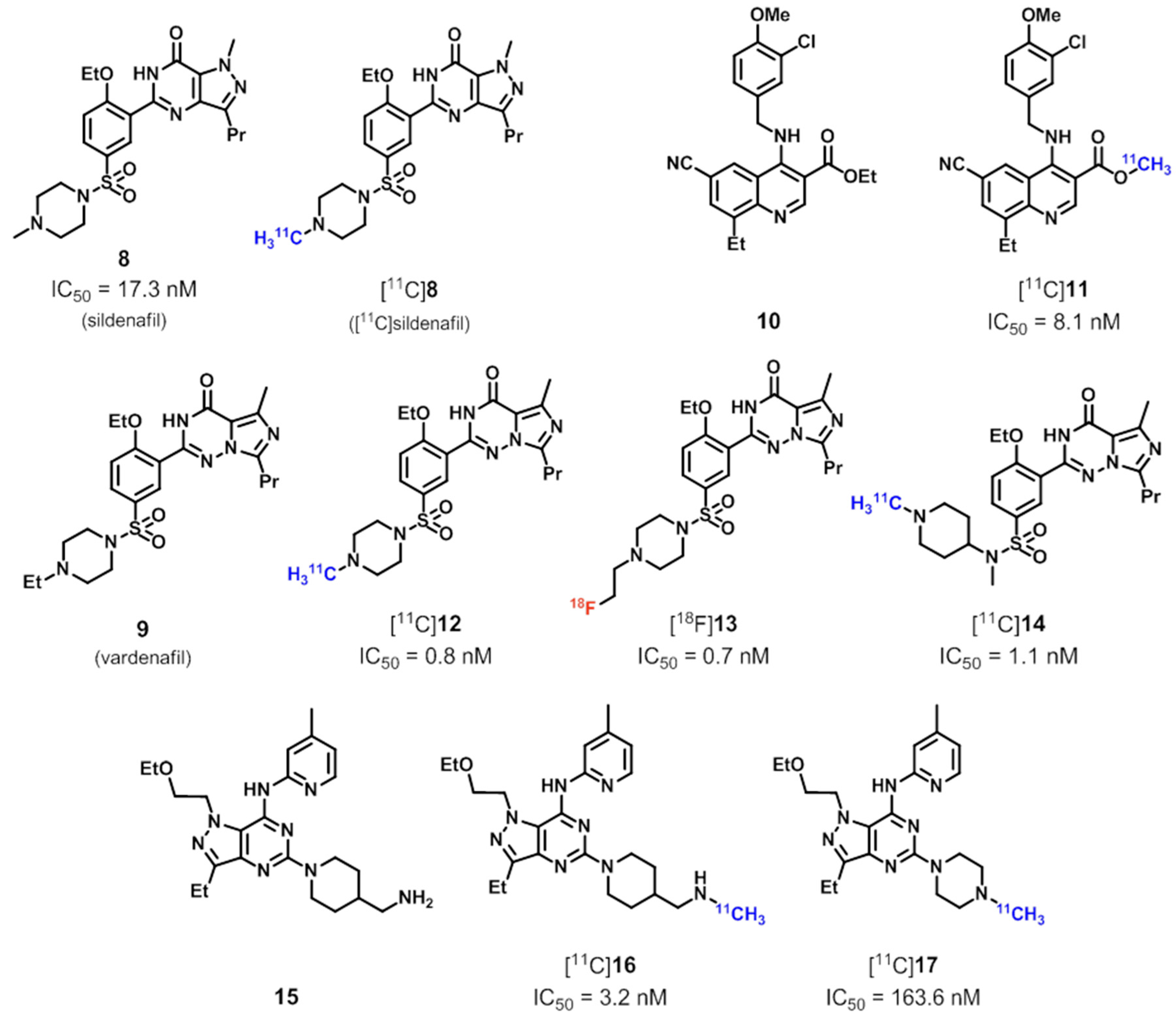
4. PDE5 Radioligands
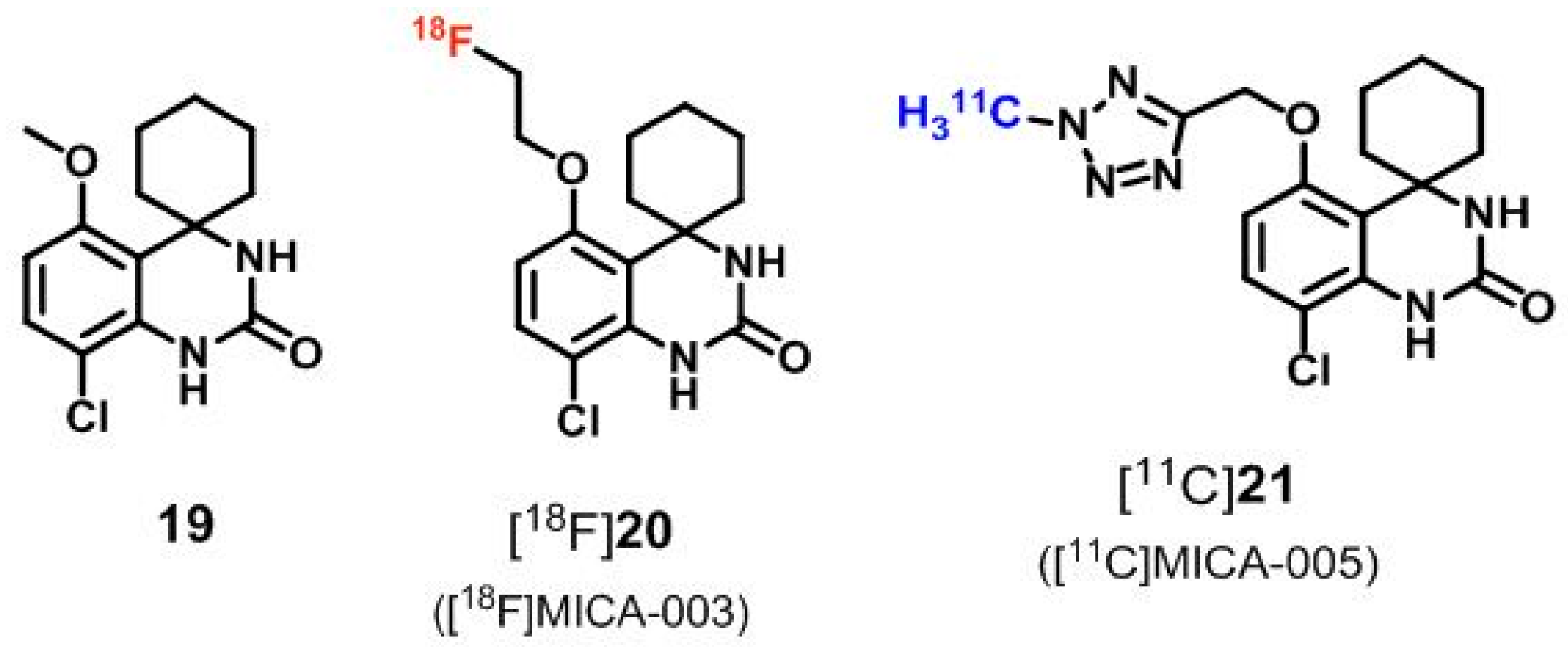
5. PDE7 Radioligands
6. PDE10 Radioligands
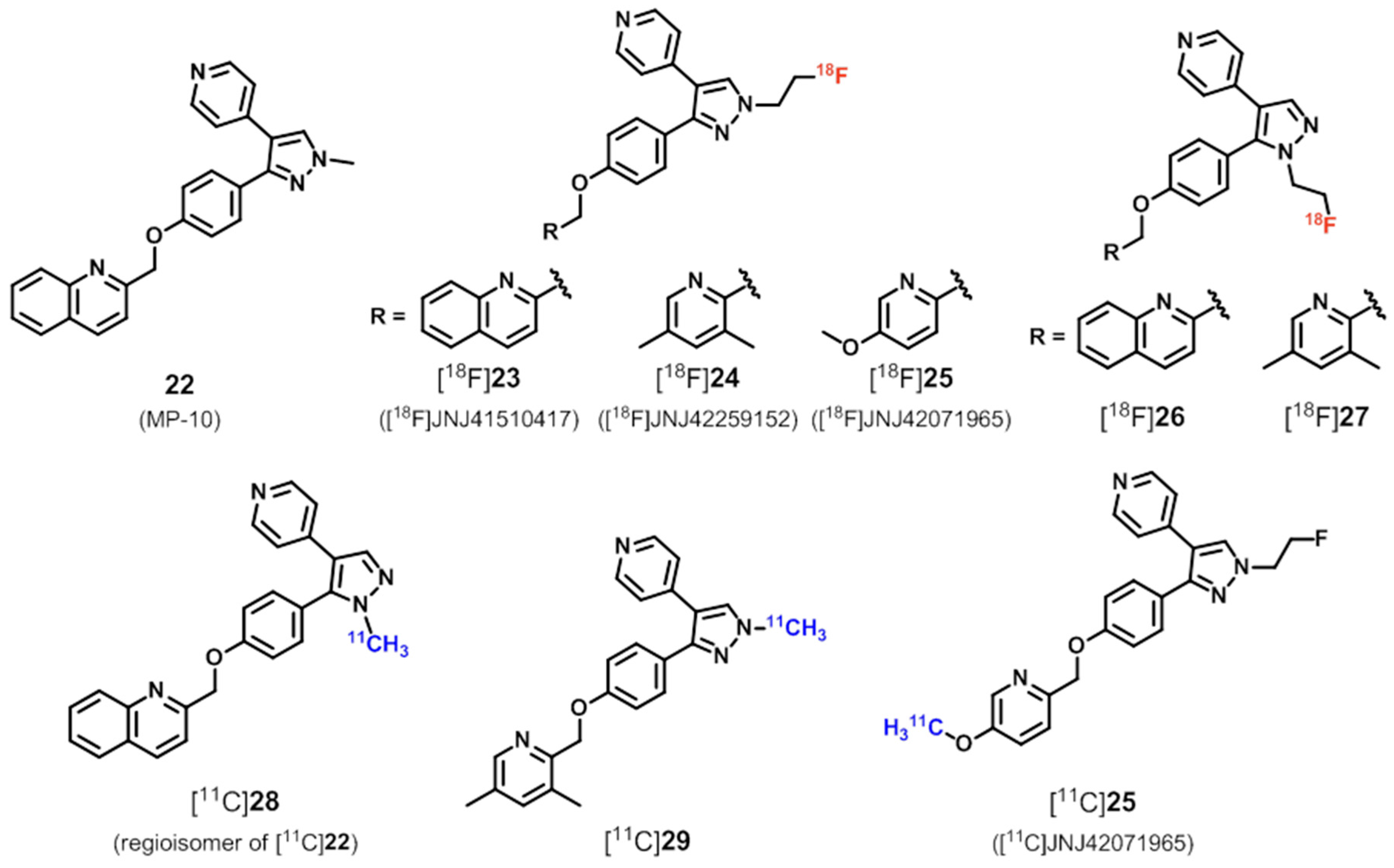
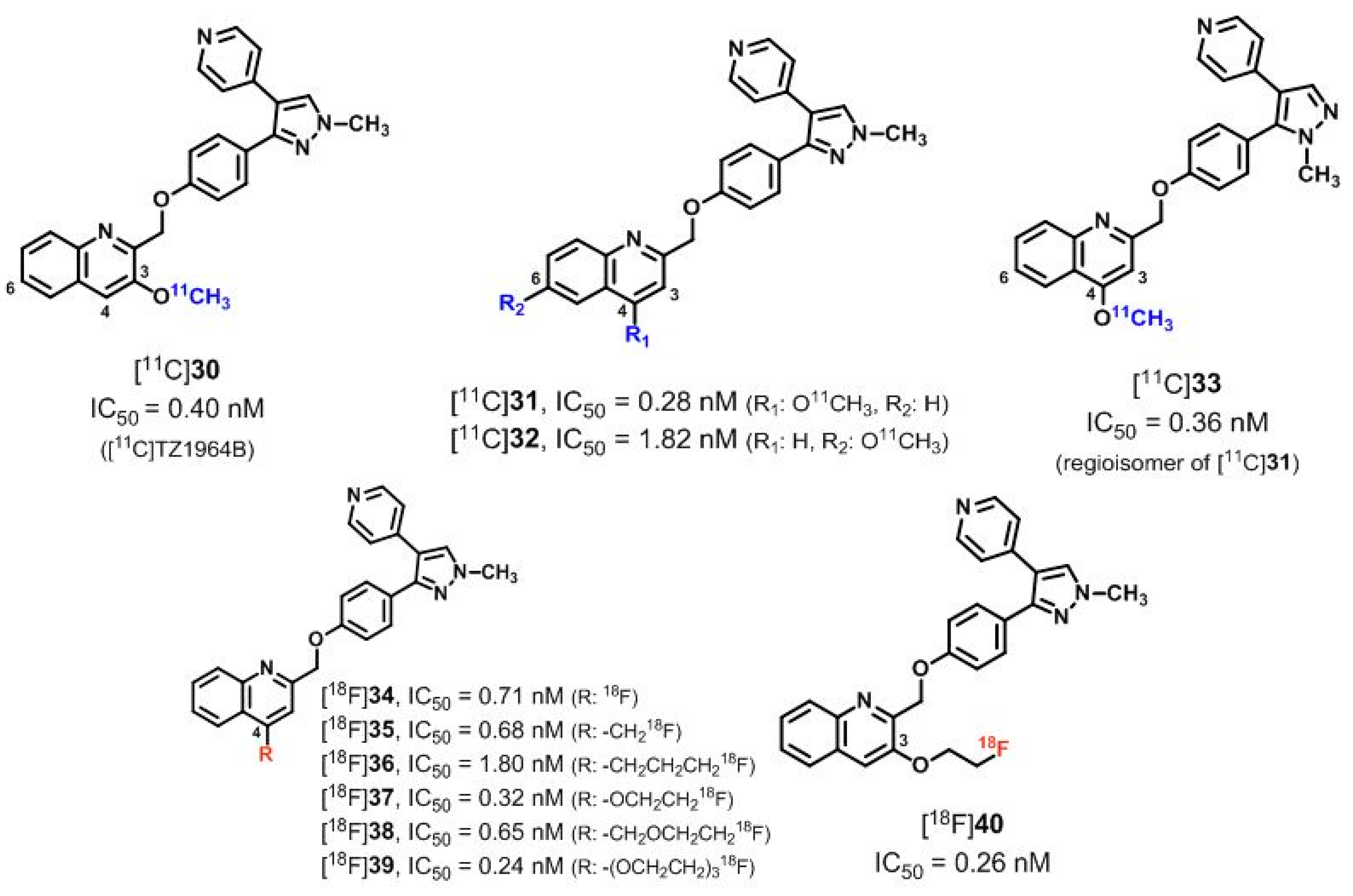
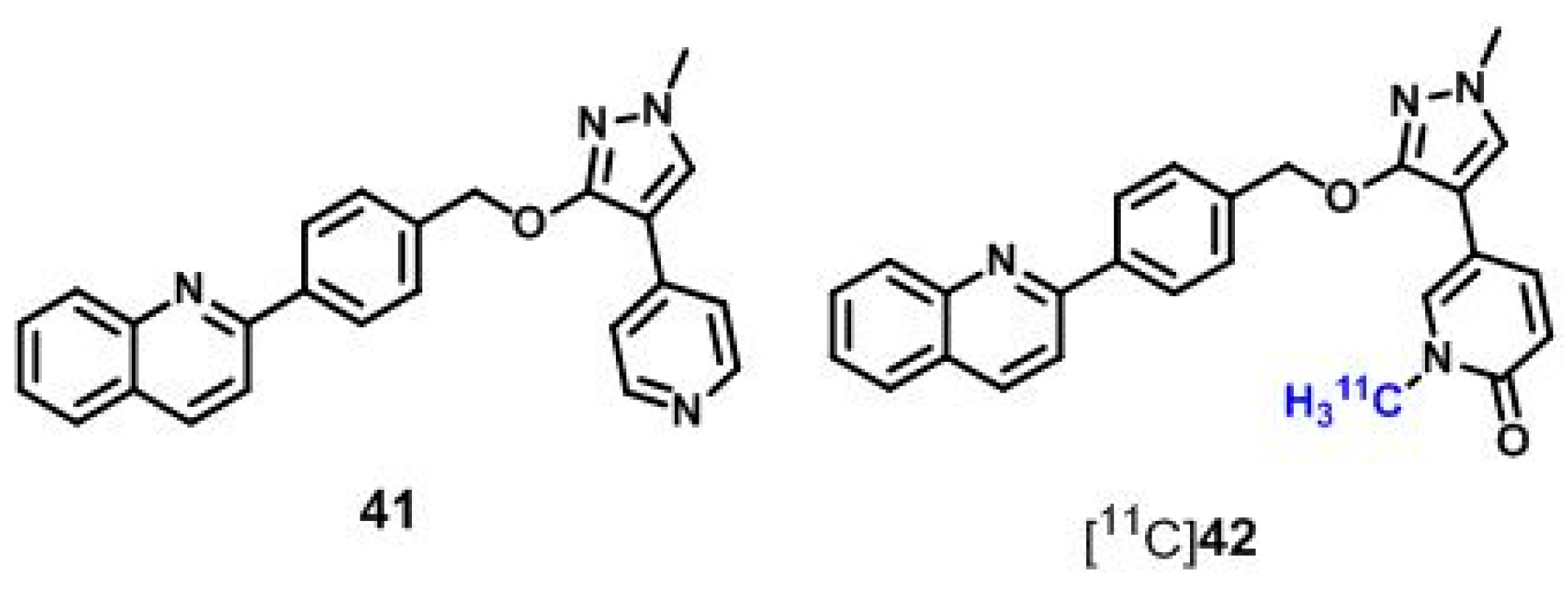
6.1. Radioligands Structurally Related to the PDE10A Inhibitor MP-10
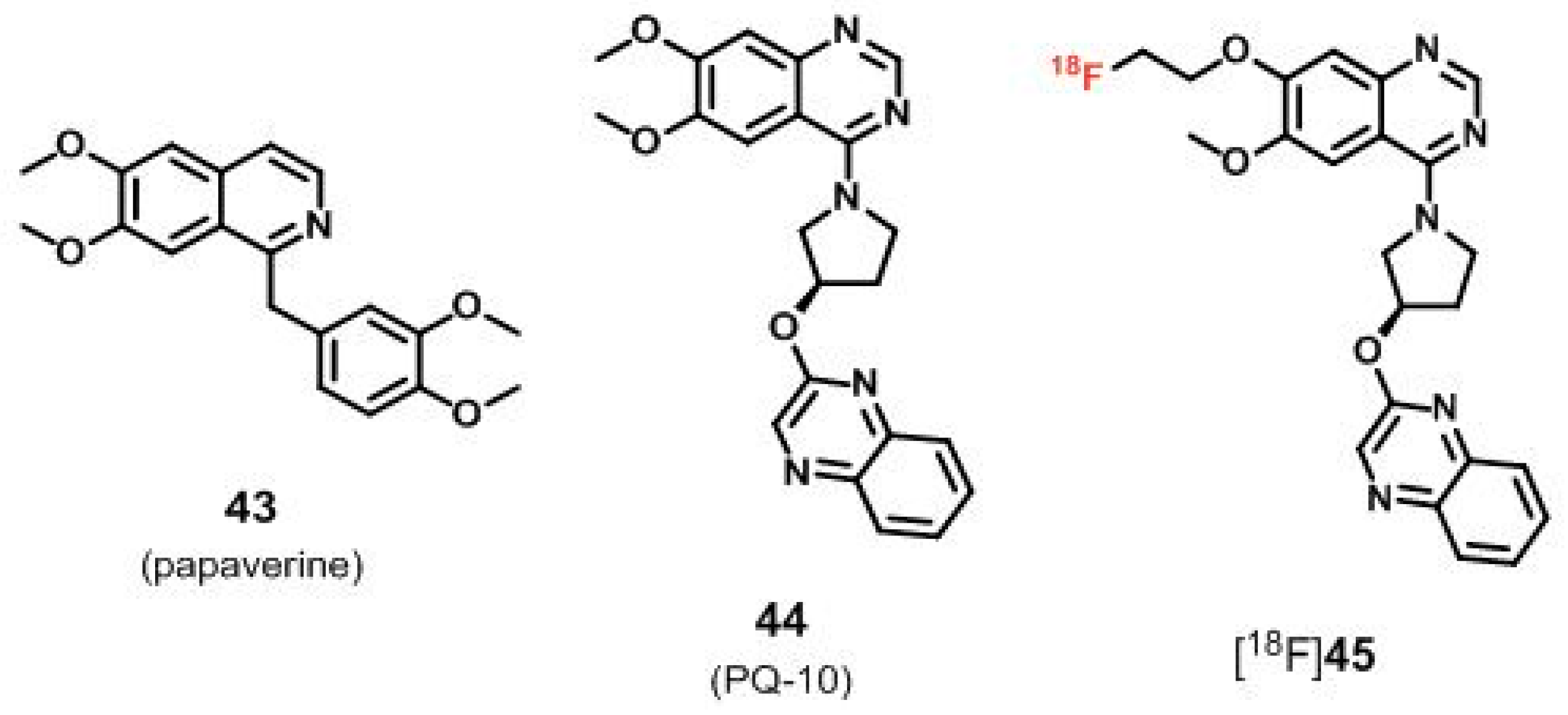
6.2. Radioligands Structurally not Derived from the PDE10A Inhibitor MP-10
7. Summary and Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Wahl, R.L.; Wagner, H.N. Principles and Practice of PET and PET/CT, 2nd ed.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009; p. 729. [Google Scholar]
- Brust, P.; van den Hoff, J.; Steinbach, J. Development of 18F-labeled radiotracers for neuroreceptor imaging with positron emission tomography. Neurosci. Bull. 2014, 30, 777–811. [Google Scholar] [CrossRef] [PubMed]
- Gallamini, A.; Zwarthoed, C.; Borra, A. Positron emission tomography (PET) in oncology. Cancers 2014, 6, 1821–1889. [Google Scholar] [CrossRef] [PubMed]
- Tee, S.S.; Keshari, K.R. Novel approaches to imaging tumor metabolism. Cancer J. 2015, 21, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Tarkin, J.M.; Joshi, F.R.; Rajani, N.K.; Rudd, J.H. PET imaging of atherosclerosis. Future Cardiol. 2015, 11, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Jivraj, N.; Phinikaridou, A.; Shah, A.M.; Botnar, R.M. Molecular imaging of myocardial infarction. Basic Res. Cardiol. 2014, 109, 397. [Google Scholar] [CrossRef] [PubMed]
- Mier, W.; Mier, D. Advantages in functional imaging of the brain. Front. Hum. Neurosci. 2015, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Jin, S.L. The molecular biology of cyclic nucleotide phosphodiesterases. Prog. Nucleic Acid Res. Mol. Biol. 1999, 63, 1–38. [Google Scholar] [PubMed]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Hardman, J.G.; Robison, G.A.; Sutherland, E.W. Cyclic nucleotides. Annu. Rev. Physiol. 1971, 33, 311–336. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Corbin, J.D. Cyclic nucleotide-dependent protein kinases: Intracellular receptors for cAMP and cGMP action. Crit. Rev. Clin. Lab. Sci. 1999, 36, 275–328. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Epac proteins: Multi-purpose cAMP targets. Trends Biochem. Sci. 2006, 31, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A.; Francis, S.H.; Houslay, M.D. Cyclic Nucleotide Phosphodiesterases in Health and Disease, 1st ed.; CRC Press: Boca Raton, FL, USA, 2006; p. 728. [Google Scholar]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.; Hait, W.N. Selective cyclic nucleotide phosphodiesterase inhibitors as potential therapeutic agents. Annu. Rev. Pharmacol. Toxicol. 1977, 17, 441–477. [Google Scholar] [CrossRef] [PubMed]
- Weishaar, R.E.; Cain, M.H.; Bristol, J.A. A new generation of phosphodiesterase inhibitors: Multiple molecular forms of phosphodiesterase and the potential for drug selectivity. J. Med. Chem. 1985, 28, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Schudt, C.; Winder, S.; Eltze, M.; Kilian, U.; Beume, R. Zardaverine: A cyclic AMP specific PDE III/IV inhibitor. Agents Actions Suppl. 1991, 34, 379–402. [Google Scholar] [PubMed]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, A.; Piazza, G.; Tinsley, H. The role of cyclic nucleotide signaling pathways in cancer: Targets for prevention and treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Z.; Zhang, Y.; Zhang, H.-T.; Li, Y.-F. Phosphodiesterase: An interface connecting cognitive deficits to neuropsychiatric and neurodegenerative diseases. Curr. Pharm. Des. 2015, 21, 303–316. [Google Scholar] [CrossRef]
- DaSilva, J.N.; Valente, C.M.; Wilson, A.A.; Warsh, J.J.; Houle, S. Carbon-11 labeling of the selective inhibitors of phosphodiesterase IV RO20–1724 and rolipram. J. Label. Comp. Radiopharm. 1997, 40, 678–680. [Google Scholar]
- DaSilva, J.N.; Lourenco, C.M.; Meyer, J.H.; Hussey, D.; Potter, W.Z.; Houle, S. Imaging cAMP-specific phosphodiesterase-4 in human brain with R-[11C]rolipram and positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Andrés, J.I.; De Angelis, M.; Alcazar, J.; Celen, S.; Bormans, G. Recent advances in positron emission tomography (PET) radiotracers for imaging phosphodiesterases. Curr. Top. Med. Chem. 2012, 12, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.J.; Mumby, M.C.; Beavo, J.A. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J. Biol. Chem. 1982, 257, 1973–1979. [Google Scholar] [PubMed]
- Pyne, N.J.; Cooper, M.E.; Houslay, M.D. Identification and characterization of both the cytosolic and particulate forms of cyclic GMP-stimulated cyclic AMP phosphodiesterase from rat liver. Biochem. J. 1986, 234, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, C.; Zoidl, G.; Koesling, D.; Russwurm, M. Dual acylation of PDE2A splice variant 3: Targeting to synaptic membranes. J. Biol. Chem. 2009, 284, 25782–25790. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.E.; Wu, A.Y.; Glavas, N.A.; Tang, X.B.; Turley, S.; Hol, W.G.; Beavo, J.A. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. USA 2002, 99, 13260–13265. [Google Scholar] [CrossRef] [PubMed]
- Iffland, A.; Kohls, D.; Low, S.; Luan, J.; Zhang, Y.; Kothe, M.; Cao, Q.; Kamath, A.V.; Ding, Y.-H.; Ellenberger, T. Structural determinants for inhibitor specificity and selectivity in PDE2A using the wheat germ in vitro translation system. Biochemistry 2005, 44, 8312–8325. [Google Scholar] [CrossRef] [PubMed]
- Pandit, J.; Forman, M.D.; Fennell, K.F.; Dillman, K.S.; Menniti, F.S. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc. Natl. Acad. Sci. USA 2009, 106, 18225–18230. [Google Scholar] [CrossRef] [PubMed]
- DeNinno, M.P. Future directions in phosphodiesterase drug discovery. Bioorg. Med. Chem. Lett. 2012, 22, 6794–6800. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.P.; Adamowicz, W.; Bove, S.; Hartman, A.J.; Mariga, A.; Pathak, G.; Reinhart, V.; Romegialli, A.; Kleiman, R.J. Select 3′,5′-cyclic nucleotide phosphodiesterases exhibit altered expression in the aged rodent brain. Cell. Signal. 2014, 26, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Menniti, F.S.; Faraci, W.S.; Schmidt, C.J. Phosphodiesterases in the CNS: Targets for drug development. Nat. Rev. Drug Discov. 2006, 5, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, K.; Hensley, K.; Florio, V.A.; Wolda, S.L. Differential expression of the cyclic GMP-stimulated phosphodiesterase PDE2A in human venous and capillary endothelial cells. J. Histochem. Cytochem. 1999, 47, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Murata, T.; Shimizu, K.; Okumura, K.; Inui, M.; Tagawa, T. Characterization of phosphodiesterase 2A in human malignant melanoma PMP cells. Oncol. Rep. 2013, 29, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Drees, M.; Zimmermann, R.; Eisenbrand, G. 3′,5′-Cyclic nucleotide phosphodiesterase in tumor cells as potential target for tumor growth inhibition. Cancer Res. 1993, 53, 3058–3061. [Google Scholar] [PubMed]
- Durand, J.; Lampron, A.; Mazzuco, T.L.; Chapman, A.; Bourdeau, I. Characterization of differential gene expression in adrenocortical tumors harboring β-catenin (CTNNB1) mutations. J. Clin. Endocr. Metab. 2011, 96, E1206–E1211. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Claffey, K.P.; Brocke, S.; Epstein, P.M. Inhibition of breast cancer cell migration by activation of cAMP signaling. Breast Cancer Res. Treat. 2015, 152, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Coskran, T.M.; Wilhelms, M.B.; Adamowicz, W.O.; O’Donnell, M.M.; Muravnick, K.B.; Menniti, F.S.; Kleiman, R.J.; Morton, D. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J. Histochem. Cytochem. 2009, 57, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Coskran, T.M.; Kelly, M.P.; Kleiman, R.J.; Morton, D.; O’Neill, S.M.; Schmidt, C.J.; Weinberg, R.J.; Menniti, F.S. The distribution of phosphodiesterase 2A in the rat brain. Neuroscience 2012, 226, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Stange, H.; Langen, B.; Egerland, U.; Hoefgen, N.; Priebs, M.; Malamas, M.S.; Erdel, J.J.; Ni, Y. Triazine Derivatives as Inhibitors of Phosphodiesterases. WO 2010/054253 A1, PCT/US2009/063633, 14 May 2010. [Google Scholar]
- Van Staveren, W.C.G.; Markerink-van Ittersum, M.; Steinbusch, H.W.M.; de Vente, J. The effects of phosphodiesterase inhibition on cyclic GMP and cyclic AMP accumulation in the hippocampus of the rat. Brain Res. 2001, 888, 275–286. [Google Scholar] [CrossRef]
- Suvarna, N.U.; O′Donnell, J.M. Hydrolysis of N-methyl-d-aspartate receptor-stimulated cAMP and cGMP by PDE4 and PDE2 phosphodiesterases in primary neuronal cultures of rat cerebral cortex and hippocampus. J. Pharmacol. Exp. Ther. 2002, 302, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Blokland, A.; Schreiber, R.; Prickaerts, J. Improving memory: A role for phosphodiesterases. Curr. Pharm. Des. 2006, 12, 2511–2523. [Google Scholar] [CrossRef] [PubMed]
- Van Staveren, W.C.G.; Steinbusch, H.W.M.; Markerink-Van Ittersum, M.; Repaske, D.R.; Goy, M.F.; Kotera, J.; Omori, K.; Beavo, J.A.; de Vente, J. MRNA expression patterns of the cGMP-hydrolyzing phosphodiesterases types 2, 5, and 9 during development of the rat brain. J. Comp. Neurol. 2003, 467, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Breitenbucher, J.G. PDE2 inhibition: Potential for the treatment of cognitive disorders. Bioorg. Med. Chem. Lett. 2013, 23, 6522–6527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, Y.; Ruan, L.; Wang, C.; Pan, J.; Klabnik, J.; Lueptow, L.; Zhang, H.-T.; O’Donnell, J.M.; Xu, Y. The roles of phosphodiesterase 2 in the central nervous and peripheral systems. Curr. Pharm. Des. 2015, 21, 274–290. [Google Scholar] [CrossRef]
- Masood, A.; Huang, Y.; Hajjhussein, H.; Xiao, L.; Li, H.; Wang, W.; Hamza, A.; Zhan, C.-G.; O’Donnell, J.M. Anxiolytic effects of phosphodiesterase-2 inhibitors associated with increased cGMP signaling. J. Pharmacol. Exp. Ther. 2009, 331, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, D.; Rosenbrock, H.; Kroker, K.S. Inhibition of PDE2A, but not PDE9A, modulates presynaptic short-term plasticity measured by paired-pulse facilitation in the CA1 region of the hippocampus. Synapse 2015, 69, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Boess, F.G.; Hendrix, M.; van der Staay, F.J.; Erb, C.; Schreiber, R.; van Staveren, W.; de Vente, J.; Prickaerts, J.; Blokland, A.; Koenig, G. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 2004, 47, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Reneerkens, O.A.H.; Rutten, K.; Bollen, E.; Hage, T.; Blokland, A.; Steinbusch, H.W.M.; Prickaerts, J. Inhibition of phoshodiesterase type 2 or type 10 reverses object memory deficits induced by scopolamine or MK-801. Behav. Brain Res. 2013, 236, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Bales, K.; Plath, N.; Svenstrup, N.; Menniti, F. Phosphodiesterase inhibition to target the synaptic dysfunction in Alzheimer’s disease. In Neurodegenerative Diseases; Dominguez, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 6, pp. 57–90. [Google Scholar]
- Andrés, J.I.; Rombouts, F.J.R.; Trabanco, A.A.; Vanhoof, G.C.P.; De Angelis, M.; Buijnsters, P.J.J.A.; Guillemont, J.E.G.; Bormans, G.M.R.; Celen, S.J.L. 1-Aryl-4-methyl-[1,2,4]triazolo[4,3-a]quinoxaline derivatives. WO 2013/000924 A1, PCT/EP2012/062381, 3 January 2013. [Google Scholar]
- Zhang, L.; Villalobos, A.; Beck, E.M.; Bocan, T.; Chappie, T.A.; Chen, L.; Grimwood, S.; Heck, S.D.; Helal, C.J.; Hou, X.; et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef] [PubMed]
- Morley, T.; Massif, C.; Papin, C.; Carroll, V.; Alagille, D.; Baldwin, R.M.; Tamagnan, G. Improved production of [18F]PF-05270430 for clinical imaging of PDE2A in brain. In Proceedings of the 21st International Symposium on Radiopharmaceutical Sciences, Columbia, MO, USA, 26–31 May 2015; 199. p. S199.
- Naganawa, M.; Nabulsi, N.; Waterhouse, R.; Lin, S.-F.; Zhang, L.; Cass, T.; Ropchan, J.; McCarthy, T.; Huang, Y.; Carson, R. Human PET studies with [18F]PF-05270430, a PET radiotracer for imaging phosphodiesterase-2A. J. Nucl. Med. 2013, 54, 201. [Google Scholar]
- Naganawa, M.; Waterhouse, R.N.; Nabulsi, N.B.; Lin, S.-F.; Labaree, D.; Ropchan, J.; Tarabar, S.; DeMartinis, N.; Ogden, A.; Banerjee, A.; et al. First in human assessment of the novel PDE2A PET radiotracer 18F-PF-05270430. J. Nucl. Med. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Wenzel, B.; Kranz, M.; Egerland, U.; Teodoro, R.; Deuther-Conrad, W.; Fischer, S.; Höfgen, N.; Steinbach, J.; Brust, P. Development, synthesis and F-18 labelling of a fluoroalkylated triazine derivative for PET imaging of phosphodiesterase 2A. In Proceedings of the Annual Congress of the European Association of Nuclear Medicine, Gothenburg, Sweden, 18–22 October 2014; OP162. p. S197.
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Egerland, U.; Kranz, M.; Scheunemann, M.; Höfgen, N.; Steinbach, J.; Brust, P. Synthesis, 18F-radiolabelling and biological characterization of novel fluoroalkylated triazine derivatives for in vivo imaging of phosphodiesterase 2A in brain via positron emission tomography. Molecules 2015, 20, 9591–9615. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Egerland, U.; Kranz, M.; Fischer, S.; Höfgen, N.; Steinbach, J.; Brust, P. Novel 18F-labelled triazine derivatives for PET imaging of phosphodiesterase 2A. In Proceedings of the 21st International Symposium on Radiopharmaceutical Sciences, Columbia, MO, USA, 26–31 May 2015; 221. p. S221.
- Zoghbi, S.S.; Shetty, H.U.; Ichise, M.; Fujita, M.; Imaizumi, M.; Liow, J.-S.; Shah, J.; Musachio, J.L.; Pike, V.W.; Innis, R.B. PET imaging of the dopamine transporter with [18F]FECNT: A polar radiometabolite confounds brain radioligand measurements. J. Nucl. Med. 2006, 47, 520–527. [Google Scholar] [PubMed]
- Evens, N.; Vandeputte, C.; Muccioli, G.G.; Lambert, D.M.; Baekelandt, V.; Verbruggen, A.M.; Debyser, Z.; van Laere, K.; Bormans, G.M. Synthesis, in vitro and in vivo evaluation of fluorine-18 labelled FE-GW405833 as a PET tracer for type 2 cannabinoid receptor imaging. Bioorg. Med. Chem. 2011, 19, 4499–4505. [Google Scholar] [CrossRef] [PubMed]
- Bolger, G.B.; Rodgers, L.; Riggs, M. Differential CNS expression of alternative mRNA isoforms of the mammalian genes encoding cAMP-specific phosphodiesterases. Gene 1994, 149, 237–244. [Google Scholar] [CrossRef]
- Horton, Y.M.; Sullivan, M.; Houslay, M.D. Molecular cloning of a novel splice variant of human type IVA (PDE-IVA) cyclic AMP phosphodiesterase and localization of the gene to the p13.2-q12 region of human chromosome 19. Biochem. J. 1995, 308, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Kenk, M.; Thomas, A.; Lortie, M.; deKemp, R.; Beanlands, R.S.; DaSilva, J.N. PET measurements of cAMP-mediated phosphodiesterase-4 with (R)-[11C]rolipram. Curr. Radiopharm. 2011, 4, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, S.; Miró, X.; Palacios, J.M.; Cortés, R.; Puigdoménech, P.; Mengod, G. Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and [3H]rolipram binding autoradiography: Comparison with monkey and rat brain. J. Chem. Neuroanat. 2000, 20, 349–374. [Google Scholar] [CrossRef]
- Bureau, Y.; Handa, M.; Zhu, Y.; Laliberte, F.; Moore, C.S.; Liu, S.; Huang, Z.; MacDonald, D.; Xu, D.G.; Robertson, G.S. Neuroanatomical and pharmacological assessment of Fos expression induced in the rat brain by the phosphodiesterase-4 inhibitor 6-(4-pyridylmethyl)-8-(3-nitrophenyl)quinoline. Neuropharmacology 2006, 51, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Souness, J.E.; Griffin, M.; Maslen, C.; Ebsworth, K.; Scott, L.C.; Pollock, K.; Palfreyman, M.N.; Karlsson, J.A. Evidence that cyclic AMP phosphodiesterase inhibitors suppress TNF alpha generation from human monocytes by interacting with a ‘low-affinity’ phosphodiesterase 4 conformer. Br. J. Pharmacol. 1996, 118, 649–658. [Google Scholar] [CrossRef]
- Reyes-Irisarri, E.; Sanchez, A.J.; Garcia-Merino, J.A.; Mengod, G. Selective induction of cAMP phosphodiesterase PDE4B2 expression in experimental autoimmune encephalomyelitis. J. Neuropath. Exp. Neur. 2007, 66, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Barnette, M.S.; Bartus, J.O.; Burman, M.; Christensen, S.B.; Cieslinski, L.B.; Esser, K.M.; Prabhakar, U.S.; Rush, J.A.; Torphy, T.J. Association of the anti-inflammatory activity of phosphodiesterase 4 (PDE4) inhibitors with either inhibition of PDE4 catalytic activity or competition for [3H]rolipram binding. Biochem. Pharmacol. 1996, 51, 949–956. [Google Scholar] [PubMed]
- Johnson-Mills, K.; Arauz, E.; Coffey, R.G.; Krzanowski, J.J., Jr.; Polson, J.B. Effect of CI-930 (3-(2H)-pyridazinone-4,5-dihydro-6-(4-(1H-imidazolyl)phenyl)-5-methyl-monohydrochloride) and rolipram on human coronary artery smooth muscle cell proliferation. Biochem. Pharmacol. 1998, 56, 1065–1073. [Google Scholar] [CrossRef]
- Gurney, M.E.; D’Amato, E.C.; Burgin, A.B. Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer’s disease. Neurotherapeutics 2015, 12, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Dyke, H.J.; Montana, J.G. Update on the therapeutic potential of PDE4 inhibitors. Expert Opin. Investig. Drugs 2002, 11, 1–13. [Google Scholar] [PubMed]
- Saldou, N.; Obernolte, R.; Huber, A.; Baecker, P.A.; Wilhelm, R.; Alvarez, R.; Li, B.; Xia, L.; Callan, O.; Su, C.; et al. Comparison of recombinant human PDE4 isoforms: Interaction with substrate and inhibitors. Cell. Signal. 1998, 10, 427–440. [Google Scholar] [CrossRef]
- Wang, P.; Myers, J.G.; Wu, P.; Cheewatrakoolpong, B.; Egan, R.W.; Billah, M.M. Expression, purification, and characterization of human cAMP-specific phosphodiesterase (PDE4) subtypes A, B, C, and D. Biochem. Bioph. Res. Commun. 1997, 234, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.D.; Hofer, P.; Spina, D.; Seeds, E.A.; Banner, K.H.; Harrison, S.; Douglas, G.; Matsumoto, T.; Page, C.P.; Wong, R.H.; et al. Pharmacology of a new cyclic nucleotide phosphodiesterase type 4 inhibitor, V11294. Pulm. Pharmacol. Ther. 2003, 16, 97–104. [Google Scholar] [CrossRef]
- DaSilva, J.N.; Lourenco, C.M.; Wilson, A.A.; Houle, S. Syntheses of the phosphodiesterase-4 inhibitors [11C]Ro 20–1724, R-, R/S- and S-[11C]rolipram. J. Label. Comp. Radiopharm. 2001, 44, 373–384. [Google Scholar] [CrossRef]
- Fujita, M.; Hines, C.S.; Zoghbi, S.S.; Mallinger, A.G.; Dickstein, L.P.; Liow, J.S.; Zhang, Y.; Pike, V.W.; Drevets, W.C.; Innis, R.B.; et al. Downregulation of brain phosphodiesterase type IV measured with 11C-(R)-rolipram positron emission tomography in major depressive disorder. Biol. Psychiat. 2012, 72, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Lortie, M.; DaSilva, J.N.; Kenk, M.; Thorn, S.; Davis, D.; Birnie, D.; Beanlands, R.S.; deKemp, R.A. Analysis of (R)- and (S)-[11C]rolipram kinetics in canine myocardium for the evaluation of phosphodiesterase-4 with PET. Mol. Imaging Biol. 2012, 14, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Thomae, D.; Papin, C.; Morley, T.; Alagille, D.; Barret, O.; Lee, H.; Baldwin, R.M.; Tamagnan, G. Identification and in vivo evaluation of [18F]MNI-617 as a radioligand for PDE4 imaging in non human primate. In Proceedings of the 21st International Symposium on Radiopharmaceutical Sciences, Columbia, MO, USA, 26–31 May 2015; 235. p. S235.
- Thomae, D.; Morley, T.J.; Lee, H.S.; Barret, O.; Constantinescu, C.; Papin, C.; Baldwin, R.M.; Tamagnan, G.D.; Alagille, D. Identification and in vivo evaluation of a fluorine-18 rolipram analogue, [18F]MNI-617, as a radioligand for PDE4 imaging in mammalian brain. J. Label. Comp. Radiopharm. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kuchar, M.; Mamat, C. Methods to increase the metabolic stability of 18F-radiotracers. Molecules 2015, 20, 16186. [Google Scholar] [CrossRef] [PubMed]
- Sopory, S.; Kaur, T.; Visweswariah, S.S. The cGMP-binding, cGMP-specific phosphodiesterase (PDE5): Intestinal cell expression, regulation and role in fluid secretion. Cell. Signal. 2004, 16, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Turko, I.V.; Haik, T.L.; McAllister-Lucas, L.M.; Burns, F.; Francis, S.H.; Corbin, J.D. Identification of key amino acids in a conserved cGMP-binding site of cGMP-binding phosphodiesterases. A putative NKXnD motif for cGMP binding. J. Biol. Chem. 1996, 271, 22240–22244. [Google Scholar] [PubMed]
- Lin, C.S.; Lin, G.; Xin, Z.C.; Lue, T.F. Expression, distribution and regulation of phosphodiesterase 5. Curr. Pharm. Des. 2006, 12, 3439–3457. [Google Scholar] [CrossRef]
- Giovannoni, M.P.; Vergelli, C.; Graziano, A.; Dal Piaz, V. PDE5 inhibitors and their applications. Curr. Med. Chem. 2010, 17, 2564–2587. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.S.; Staib, R.L.; Wicks, L.M.; Feldman, J.P. Phosphodiesterase-5 inhibitors in management of pulmonary hypertension: Safety, tolerability, and efficacy. Drug Healthc. Patient Saf. 2010, 2, 151–161. [Google Scholar] [PubMed]
- Evans, J.D.; Hill, S.R. A comparison of the available phosphodiesterase-5 inhibitors in the treatment of erectile dysfunction: A focus on avanafil. Patient Prefer. Adher. 2015, 9, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Pokreisz, P.; Vandenwijngaert, S.; Bito, V.; Van den Bergh, A.; Lenaerts, I.; Busch, C.; Marsboom, G.; Gheysens, O.; Vermeersch, P.; Biesmans, L.; et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation 2009, 119, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Champion, H.C.; Li, M.; Belardi, D.; Ren, S.; Rodriguez, E.R.; Bedja, D.; Gabrielson, K.L.; Wang, Y.; Kass, D.A. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 2005, 11, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Salloum, F.N.; Ockaili, R.A.; Wittkamp, M.; Marwaha, V.R.; Kukreja, R.C. Vardenafil: A novel type 5 phosphodiesterase inhibitor reduces myocardial infarct size following ischemia/reperfusion injury via opening of mitochondrial KATP channels in rabbits. J. Mol. Cell. Cardiol. 2006, 40, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Salloum, F.N.; Xi, L.; Kukreja, R.C. PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol. Ther. 2015, 147, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Chekol, R.; Gheysens, O.; Cleyhens, J.; Janssens, S.; Verbruggen, A.; Bormans, G. Radiosynthesis and biological evaluation of a carbon-11 labelled vardenafil derivative as a PDE5 specific tracer. In Proceedings of the 19st International Symposium on Radiopharmaceutical Sciences, Amsterdam, The Netherlands, 28 August–2 September 2011; P-063. p. S152.
- Chekol, R.; Gheysens, O.; Cleyhens, J.; Janssens, S.; Verbruggen, A.; Bormans, G. Labelling and preliminary biological evaluation of [11C]RBQ08 as a specific PDE5 radiotracer. In Proceedings of the 19st International Symposium on Radiopharmaceutical Sciences, Amsterdam, The Netherlands, 28 August–2 September 2011; P-064. p. S153.
- Chekol, R.; Gheysens, O.; Cleynhens, J.; Pokreisz, P.; Vanhoof, G.; Ahamed, M.; Janssens, S.; Verbruggen, A.; Bormans, G. Evaluation of PET radioligands for in vivo visualization of phosphodiesterase 5 (PDE5). Nucl. Med. Biol. 2014, 41, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Stoy, P.; Adam, L.; He, B.; Krupinski, J.; Normandin, D.; Pongrac, R.; Seliger, L.; Watson, A.; Macor, J.E. Quinolines as extremely potent and selective PDE5 inhibitors as potential agents for treatment of erectile dysfunction. Bioorg. Med. Chem. Lett. 2004, 14, 1577–1580. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, M.B.; Acker, B.A.; Jacobsen, E.J.; Hughes, R.O.; Walker, J.K.; Fox, D.N.A.; Palmer, M.J.; Freeman, S.K.; Yu, Y.; Bond, B.R. 1-(2-Ethoxyethyl)-1H-pyrazolo[4,3-d]pyrimidines as potent phosphodiesterase 5 (PDE5) inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3120–3124. [Google Scholar] [CrossRef] [PubMed]
- Kotera, J.; Fujishige, K.; Omori, K. Immunohistochemical localization of cGMP-binding cGMP-specific phosphodiesterase (PDE5) in rat tissues. J. Histochem. Cytochem. 2000, 48, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Beavo, J.A. Specific localized expression of cGMP PDEs in Purkinje neurons and macrophages. Neurochem. Int. 2004, 45, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, C.A.; Nunes, A.K.S.; Garcia-Osta, A. Phosphodiesterase-5 inhibitors: Action on the signaling pathways of neuroinflammation, neurodegeneration, and cognition. Mediat. Inflamm. 2015, 2015, 940207. [Google Scholar] [CrossRef] [PubMed]
- Puerta, E.; Hervias, I.; Goni-Allo, B.; Lasheras, B.; Jordan, J.; Aguirre, N. Phosphodiesterase 5 inhibitors prevent 3,4-methylenedioxymethamphetamine-induced 5-HT deficits in the rat. J. Neurochem. 2009, 108, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Puerta, E.; Hervias, I.; Barros-Minones, L.; Jordan, J.; Ricobaraza, A.; Cuadrado-Tejedor, M.; Garcia-Osta, A.; Aguirre, N. Sildenafil protects against 3-nitropropionic acid neurotoxicity through the modulation of calpain, CREB, and BDNF. Neurobiol. Dis. 2010, 38, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Puerta, E.; Barros-Minones, L.; Hervias, I.; Gomez-Rodriguez, V.; Orejana, L.; Pizarro, N.; de la Torre, R.; Jordan, J.; Aguirre, N. Long-lasting neuroprotective effect of sildenafil against 3,4-methylenedioxymethamphetamine- induced 5-hydroxytryptamine deficits in the rat brain. J. Neurosci. Res. 2012, 90, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Barros-Minones, L.; Orejana, L.; Goni-Allo, B.; Suquia, V.; Hervias, I.; Aguirre, N.; Puerta, E. Modulation of the ASK1-MKK3/6-p38/MAPK signalling pathway mediates sildenafil protection against chemical hypoxia caused by malonate. Br. J. Pharmacol. 2013, 168, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Staniszewski, A.; Deng, S.X.; Privitera, L.; Leznik, E.; Liu, S.; Zhang, H.; Feng, Y.; Palmeri, A.; Landry, D.W.; et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. J. Neurosci. 2009, 29, 8075–8086. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Hervias, I.; Ricobaraza, A.; Puerta, E.; Perez-Roldan, J.M.; Garcia-Barroso, C.; Franco, R.; Aguirre, N.; Garcia-Osta, A. Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 2029–2041. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Vallejo, V.; Ugarte, A.; García-Barroso, C.; Cuadrado-Tejedor, M.; Szczupak, B.; Dopeso-Reyes, I.G.; Lanciego, J.L.; García-Osta, A.; Llop, J.; Oyarzabal, J.; et al. Pharmacokinetic investigation of sildenafil using positron emission tomography and determination of its effect on cerebrospinal fluid cGMP levels. J. Neurochem. 2016, 136, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wenzel, B.; Dukic-Stefanovic, S.; Teodoro, R.; Ludwig, F.-A.; Deuther-Conrad, W.; Schröder, S.; Chezal, J.-M.; Moreau, E.; Brust, P.; et al. Development of a new radiofluorinated quinoline analog for PET imaging of phosphodiesterase 5 (PDE5) in brain. Pharmaceuticals 2016, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Loughney, K.; Hill, T.R.; Florio, V.A.; Uher, L.; Rosman, G.J.; Wolda, S.L.; A. Jones, B.; Howard, M.L.; McAllister-Lucas, L.M.; Sonnenburg, W.K.; et al. Isolation and characterization of cDNAs encoding PDE5A, a human cGMP-binding, cGMP-specific 3′,5′-cyclic nucleotide phosphodiesterase. Gene 1998, 216, 139–147. [Google Scholar] [CrossRef]
- Cumming, P. A business of some heat: Molecular imaging of phosphodiesterase 5. J. Neurochem. 2016, 136, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Miro, X.; Perez-Torres, S.; Palacios, J.M.; Puigdomenech, P.; Mengod, G. Differential distribution of cAMP-specific phosphodiesterase 7A mRNA in rat brain and peripheral organs. Synapse 2001, 40, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.M.; Reyes-Irisarri, E.; Mengod, G. Comparison of cAMP-specific phosphodiesterase mRNAs distribution in mouse and rat brain. Neurosci. Lett. 2012, 525, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Irisarri, E.; Perez-Torres, S.; Mengod, G. Neuronal expression of cAMP-specific phosphodiesterase 7B mRNA in the rat brain. Neuroscience 2005, 132, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Nakata, A.; Ogawa, K.; Sasaki, T.; Koyama, N.; Wada, K.; Kotera, J.; Kikkawa, H.; Omori, K.; Kaminuma, O. Potential role of phosphodiesterase 7 in human T cell function: Comparative effects of two phosphodiesterase inhibitors. Clin. Exp. Immunol. 2002, 128, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Morales-Garcia, J.A.; Redondo, M.; Alonso-Gil, S.; Gil, C.; Perez, C.; Martinez, A.; Santos, A.; Perez-Castillo, A. Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PLoS ONE 2011, 6, e17240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demopulos, G.A.; Gaitanaris, G.A. Treatment of Addiction and Impulse-Control Disorders Using PDE7 Inhibitors. WO2012/064667 A3, 14 November 2013. [Google Scholar]
- Gil, C.; Campillo, N.E.; Perez, D.I.; Martinez, A. PDE7 inhibitors as new drugs for neurological and inflammatory disorders. Expert Opin. Ther. Pat. 2008, 18, 1127–1139. [Google Scholar] [CrossRef]
- De Gortari, P.; Mengod, G. Dopamine D1, D2 and mu-opioid receptors are co-expressed with adenylyl cyclase 5 and phosphodiesterase 7B mRNAs in striatal rat cells. Brain Res. 2010, 1310, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomae, D.; Servaes, S.; Vazquez, N.; Wyffels, L.; Dedeurwaerdere, S.; van der Veken, P.; Joossens, J.; Augustyns, K.; Stroobants, S.; Staelens, S. Synthesis and preclinical evaluation of two novel radioligands for PDE7 imaging in the brain. In Proceedings of the 21st International Symposium on Radiopharmaceutical Sciences, Columbia, MO, USA, 26–31 May 2015; 295. p. S295.
- Thomae, D.; Servaes, S.; Vazquez, N.; Wyffels, L.; Dedeurwaerdere, S.; Van der Veken, P.; Joossens, J.; Augustyns, K.; Stroobants, S.; Staelens, S. Synthesis and preclinical evaluation of an 18F labeled PDE7 inhibitor for PET neuroimaging. Nucl. Med. Biol. 2015, 42, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Bernardelli, P.; Lorthiois, E.; Vergne, F.; Oliveira, C.; Mafroud, A.K.; Proust, E.; Pham, N.; Ducrot, P.; Moreau, F.; Idrissi, M.; et al. Spiroquinazolinones as novel, potent, and selective PDE7 inhibitors. Part 2: Optimization of 5,8-disubstituted derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 4627–4631. [Google Scholar] [CrossRef] [PubMed]
- Fujishige, K.; Kotera, J.; Omori, K. Striatum- and testis-specific phosphodiesterase PDE10A. Eur. J. Biochem. 1999, 266, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Fujishige, K.; Kotera, J.; Michibata, H.; Yuasa, K.; Takebayashi, S.-I.; Okumura, K.; Omori, K. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J. Biol. Chem. 1999, 274, 18438–18445. [Google Scholar] [CrossRef] [PubMed]
- Kotera, J.; Fujishige, K.; Yuasa, K.; Omori, K. Characterization and phosphorylation of PDE10A2, a novel alternative splice variant of human phosphodiesterase that hydrolyzes cAMP and cGMP. Biochem. Biophys. Res. Commun. 1999, 261, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Kotera, J.; Sasaki, T.; Kobayashi, T.; Fujishige, K.; Yamashita, Y.; Omori, K. Subcellular localization of cyclic nucleotide phosphodiesterase type 10A variants, and alteration of the localization by cAMP-dependent protein kinase-dependent phosphorylation. J. Biol. Chem. 2004, 279, 4366–4375. [Google Scholar] [CrossRef] [PubMed]
- Jäger, R.; Russwurm, C.; Schwede, F.; Genieser, H.-G.; Koesling, D.; Russwurm, M. Activation of PDE10 and PDE11 phosphodiesterases. J. Biol. Chem. 2012, 287, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Gross-Langenhoff, M.; Hofbauer, K.; Weber, J.; Schultz, A.; Schultz, J.E. CAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J. Biol. Chem. 2006, 281, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.F.; Bartlett, B.; Coskran, T.M.; Culp, J.S.; James, L.C.; Krull, D.L.; Lanfear, J.; Ryan, A.M.; Schmidt, C.J.; Strick, C.A.; et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003, 985, 113–126. [Google Scholar] [CrossRef]
- Coskran, T.M.; Morton, D.; Menniti, F.S.; Adamowicz, W.O.; Kleiman, R.J.; Ryan, A.M.; Strick, C.A.; Schmidt, C.J.; Stephenson, D.T. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J. Histochem. Cytochem. 2006, 54, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Bollen, E.; Prickaerts, J. Phosphodiesterases in neurodegenerative disorders. IUBMB Life 2012, 64, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Kehler, J.; Nielsen, J. PDE10A inhibitors: Novel therapeutic drugs for schizophrenia. Curr. Pharm. Des. 2011, 17, 137–150. [Google Scholar] [CrossRef]
- Chappie, T.A.; Helal, C.J.; Hou, X. Current landscape of phosphodiesterase 10A (PDE10A) inhibition. J. Med. Chem. 2012, 55, 7299–7331. [Google Scholar] [CrossRef] [PubMed]
- Dedeurwaerdere, S.; Wintmolders, C.; Vanhoof, G.; Langlois, X. Patterns of brain glucose metabolism induced by phosphodiesterase 10A inhibitors in the mouse: A potential translational biomarker. J. Pharmacol. Exp. Ther. 2011, 339, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Hoefgen, N.; Grunwald, C.; Langen, B. Targeting PDE10A in schizophrenia. Drug Future 2012, 37, 577–589. [Google Scholar] [CrossRef]
- Kehler, J.; Kilburn, J.P. Patented PDE10A inhibitors: Novel compounds since 2007. Expert Opin. Ther. Pat. 2009, 19, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Kehler, J. Phosphodiesterase 10A inhibitors: A 2009–2012 patent update. Expert Opin. Ther. Pat. 2013, 23, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Grauer, S.M.; Pulito, V.L.; Navarra, R.L.; Kelly, M.P.; Kelley, C.; Graf, R.; Langen, B.; Logue, S.; Brennan, J.; Jiang, L.; et al. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J. Pharmacol. Exp. Ther. 2009, 331, 574–590. [Google Scholar] [CrossRef] [PubMed]
- Verhoest, P.R.; Chapin, D.S.; Corman, M.; Fonseca, K.; Harms, J.F.; Hou, X.; Marr, E.S.; Menniti, F.S.; Nelson, F.; O’Connor, R.; et al. Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2-(4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl)- quinoline (PF-2545920) for the treatment of schizophrenia. J. Med. Chem. 2009, 52, 5188–5196. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Fan, J.; Li, S.; Jones, L.A.; Cui, J.; Padakanti, P.K.; Xu, J.; Zeng, D.; Shoghi, K.I.; Perlmutter, J.S.; et al. Radiosynthesis and in vivo evaluation of [11C]MP-10 as a PET probe for imaging PDE10A in rodent and non-human primate brain. Bioorg. Med. Chem. 2011, 19, 1666–1673. [Google Scholar] [CrossRef] [PubMed]
- Plisson, C.; Weinzimmer, D.; Jakobsen, S.; Natesan, S.; Salinas, C.; Lin, S.F.; Labaree, D.; Zheng, M.Q.; Nabulsi, N.; Marques, T.R.; et al. Phosphodiesterase 10A PET radioligand development program: From pig to human. J. Nucl. Med. 2014, 55, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Ooms, M.; Celen, S.; Koole, M.; Langlois, X.; Schmidt, M.; de Angelis, M.; Andrés, J.I.; Verbruggen, A.; van Laere, K.; Bormans, G. Synthesis and biological evaluation of carbon-11 and fluorine-18 labeled tracers for in vivo visualization of PDE10A. Nucl. Med. Biol. 2014, 41, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Ooms, M.; Celen, S.; Verbruggen, A.; van Laere, K.; Bormans, G. Synthesis and preliminary biological evaluation of [11C]-MP10 and its regio-isomer as potential radioligands for positron emission tomography imaging of phosphodiesterase-10A in the brain. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, S206. [Google Scholar]
- Plisson, C.; Salinas, C.; Weinzimmer, D.; Labaree, D.; Lin, S.-F.; Ding, Y.-S.; Jakobsen, S.; Smith, P.W.; Eiji, K.; Carson, R.E.; et al. Radiosynthesis and in vivo evaluation of [11C]MP-10 as a positron emission tomography radioligand for phosphodiesterase 10A. Nucl. Med. Biol. 2011, 38, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Natesan, S.; Ashworth, S.; Nielsen, J.; Tang, S.P.; Salinas, C.; Kealey, S.; Lauridsen, J.B.; Stensbol, T.B.; Gunn, R.N.; Rabiner, E.A.; et al. Effect of chronic antipsychotic treatment on striatal phosphodiesterase 10A levels: A [11C]MP-10 PET rodent imaging study with ex vivo confirmation. Transl. Psychiatry 2014, 4, e376. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-F.; Labaree, D.; Chen, M.-K.; Holden, D.; Gallezot, J.-D.; Kapinos, M.; Teng, J.-K.; Najafzadeh, S.; Plisson, C.; Rabiner, E.A.; et al. Further evaluation of [11C]MP-10 as a radiotracer for phosphodiesterase 10A: PET imaging study in rhesus monkeys and brain tissue metabolite analysis. Synapse 2015, 69, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Gil, J.I.; Bormans, G.; De Angelis, M.; Celen, S. Radiolabelled PDE10 Ligands. WO 2010/097367 A1, 2 September 2010. [Google Scholar]
- Celen, S.; Koole, M.; De Angelis, M.; Sannen, I.; Chitneni, S.K.; Alcazar, J.; Dedeurwaerdere, S.; Moechars, D.; Schmidt, M.; Verbruggen, A.; et al. Preclinical evaluation of 18F-JNJ41510417 as a radioligand for PET imaging of phosphodiesterase-10A in the brain. J. Nucl. Med. 2010, 51, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Andrés, J.I.; de Angelis, M.; Alcázar, J.; Iturrino, L.; Langlois, X.; Dedeurwaerdere, S.; Lenaerts, I.; Vanhoof, G.; Celen, S.; Bormans, G. Synthesis, in vivo occupancy, and radiolabeling of potent phosphodiesterase subtype-10 inhibitors as candidates for positron emission tomography imaging. J. Med. Chem. 2011, 54, 5820–5835. [Google Scholar] [CrossRef] [PubMed]
- Celen, S.; Koole, M.; Ooms, M.; De Angelis, M.; Sannen, I.; Cornelis, J.; Alcazar, J.; Schmidt, M.; Verbruggen, A.; Langlois, X.; et al. Preclinical evaluation of [18F]JNJ42259152 as a PET tracer for PDE10A. Neuroimage 2013, 82, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, K.; Ahmad, R.U.; Hudyana, H.; Celen, S.; Dubois, K.; Schmidt, M.E.; Bormans, G.; Koole, M. Human biodistribution and dosimetry of [18F]JNJ42259152, a radioligand for phosphodiesterase 10A imaging. Eur. J. Nucl. Med. Mol. Imaging 2012, 40, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, K.; Ahmad, R.U.; Hudyana, H.; Dubois, K.; Schmidt, M.E.; Celen, S.; Bormans, G.; Koole, M. Quantification of [18F]JNJ42259152, a novel phosphodiesterase 10A PET tracer: Kinetic modeling and test–retest study in human brain. J. Nucl. Med. 2013, 54, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Bourgeois, S.; Postnov, A.; Schmidt, M.E.; Bormans, G.; Van Laere, K.; Vandenberghe, W. PET imaging shows loss of striatal PDE10A in patients with Huntington disease. Neurology 2014, 82, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Ooms, M.; Rietjens, R.; Rangarajan, J.R.; Vunckx, K.; Valdeolivas, S.; Maes, F.; Himmelreich, U.; Fernandez-Ruiz, J.; Bormans, G.; Van Laere, K.; et al. Early decrease of type 1 cannabinoid receptor binding and phosphodiesterase 10A activity in vivo in R6/2 Huntington mice. Neurobiol. Aging 2014, 35, 2858–2869. [Google Scholar] [CrossRef] [PubMed]
- Ooms, M.; Celen, S.; De Hoogt, R.; Lenaerts, I.; Liebregts, J.; Vanhoof, G.; Langlois, X.; Postnov, A.; Koole, M.; Verbruggen, A.; et al. Striatal phosphodiesterase 10A availability is altered secondary to chronic changes in dopamine neurotransmission. EJNMMI Radiopharm. Chem. 2016, 1, 1–17. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, X.; Li, J.; Jin, H.; Padakanti, P.K.; Jones, L.A.; Flores, H.P.; Su, Y.; Perlmutter, J.S.; Tu, Z. Radiosyntheses and in vivo evaluation of carbon-11 PET tracers for PDE10A in the brain of rodent and nonhuman primate. Bioorg. Med. Chem. 2014, 22, 2648–2654. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, X.; Jin, H.; Fan, J.; Flores, H.; Perlmutter, J.S.; Tu, Z. Synthesis of fluorine-containing phosphodiesterase 10A (PDE10A) inhibitors and the in vivo evaluation of F-18 labeled PDE10A PET tracers in rodent and nonhuman primate. J. Med. Chem. 2015, 58, 8584–8600. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jin, H.; Zhou, H.; Rothfuss, J.; Tu, Z. Synthesis and in vitro biological evaluation of pyrazole group-containing analogues for PDE10A. Med. Chem. Comm. 2013, 4, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jin, H.; Yue, X.; Zhang, X.; Yang, H.; Li, J.; Flores, H.; Su, Y.; Perlmutter, J.S.; Tu, Z. Preclinical evaluation of a promising C-11 labeled PET tracer for imaging phosphodiesterase 10A in the brain of living subject. Neuroimage 2015, 121, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, W.; Masuda, N.; Miyamoto, S.; Shiina, Y.; Kikuchi, S.; Mihara, T.; Moriguchi, H.; Fushiki, H.; Murakami, Y.; Amano, Y.; et al. Synthesis, SAR study, and biological evaluation of novel quinoline derivatives as phosphodiesterase 10A inhibitors with reduced CYP3A4 inhibition. Bioorg. Med. Chem. 2015, 23, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, W.; Masuda, N.; Samizu, K.; Mihara, T.; Takama, K.; Watanabe, T. Synthesis and in vivo evaluation of novel quinoline derivatives as phosphodiesterase 10A inhibitors. Chem. Pharm. Bull. 2014, 62, 1200–1213. [Google Scholar] [CrossRef] [PubMed]
- Funke, U.; Schwan, G.; Scheunemann, M.; Maisonial, A.; Hiller, A.; Fischer, S.; Deuther-Conrad, W.; Egerland, U.; Briel, D.; Nieber, K.; et al. Radiosynthesis, in vitro and in vivo evaluation of a 7-(2-[18F]fluoroethoxy)-6-methoxyquinazoline derivative for imaging PDE10A with PET. In Proceedings of the 19st International Symposium on Radiopharmaceutical Sciences, Amsterdam, The Netherlands, 28 August–2 September 2011; P-179. p. S268.
- Funke, U.; Deuther-Conrad, W.; Schwan, G.; Maisonial, A.; Scheunemann, M.; Fischer, S.; Hiller, A.; Briel, D.; Brust, P. Radiosynthesis and radiotracer properties of a 7-(2-[18F]fluoroethoxy)-6-methoxy-pyrrolidinylquinazoline for imaging of phosphodiesterase 10A with PET. Pharmaceuticals 2012, 5, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Chappie, T.A.; Humphrey, J.M.; Allen, M.P.; Estep, K.G.; Fox, C.B.; Lebel, L.A.; Liras, S.; Marr, E.S.; Menniti, F.S.; Pandit, J.; et al. Discovery of a series of 6,7-dimethoxy-4-pyrrolidylquinazoline PDE10A Inhibitors. J. Med. Chem. 2007, 50, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Schwan, G.; Barbar Asskar, G.; Höfgen, N.; Kubicova, L.; Funke, U.; Egerland, U.; Zahn, M.; Nieber, K.; Scheunemann, M.; Sträter, N.; et al. Fluorine-containing 6,7-dialkoxybiaryl-based inhibitors for phosphodiesterase 10 A: Synthesis and in vitro evaluation of inhibitory potency, selectivity, and metabolism. Chem. Med. Chem. 2014, 9, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, R.R.; Bondy, C.A. Differential cellular pattern of gene expression for two distinct cGMP-inhibited cyclic nucleotide phosphodiesterases in developing and mature rat brain. Neuroscience 1996, 72, 567–578. [Google Scholar] [CrossRef]
- Nieber, K.; Erdmann, S.; Briel, D.; Schwan, G.; Kubicova, L.; Barbar Asskar, G.; Sträter, N.; Zahn, M.; Brust, P.; Funke, U. Neue Halogenalkoxychinazoline, deren Herstellung und Verwendung. 00401P0051DE, 20 October 2010. [Google Scholar]
- Malamas, M.S.; Ni, Y.; Erdei, J.; Stange, H.; Schindler, R.; Lankau, H.-J.; Grunwald, C.; Fan, K.Y.; Parris, K.; Langen, B.; et al. Highly potent, selective, and orally active phosphodiesterase 10A inhibitors. J. Med. Chem. 2011, 54, 7621–7638. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Scheunemann, M.; Fischer, S.; Egerland, U.; Ludwig, F.-A.; Höfgen, N.; Steinbach, J.; Brust, P. 1-Arylimidazo[1,5a]quinoxalines as lead compounds for a PDE10A PET tracer. In Proceedings of Annual Congress of the European Association of Nuclear Medicine, Gothenburg, Sweden, 18–22 October 2014; OP163. p. S197.
- Wagner, S.; Scheunemann, M.; Dipper, K.; Egerland, U.; Hoefgen, N.; Steinbach, J.; Brust, P. Development of highly potent phosphodiesterase 10A (PDE10A) inhibitors: Synthesis and in vitro evaluation of 1,8-dipyridinyl- and 1-pyridinyl-substituted imidazo[1,5-a]quinoxalines. Eur. J. Med. Chem. 2016, 107, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Kranz, M.; Hankir, M.; Deuther-Conrad, W.; Scheunemann, M.; Teodoro, R.; Wenzel, B.; Fischer, S.; Egerland, U.; Fenske, W.K.; et al. Evaluation of the new radioligand [18F]AQ-28A by small animal PET/MR demonstrates increse of PDE10A expression in striatum and brown adipose tissue (BAT) of obese mice. In Proceedings of the 21st International Symposium on Radiopharmaceutical Sciences, Columbia, MO, USA, 26–31 May 2015; 52. p. S52.
- Wagner, S.; Teodoro, R.; Deuther-Conrad, W.; Kranz, M.; Scheunemann, M.; Fischer, S.; Wenzel, B.; Egerland, U.; Hoefgen, N.; Steinbach, J.; et al. Radiosynthesis and biological evaluation of the new PDE10A radioligand [18F]AQ28A. J. Label. Comp. Radiopharm. 2015. submitted. [Google Scholar]
- Nawrocki, A.R.; Rodriguez, C.G.; Toolan, D.M.; Price, O.; Henry, M.; Forrest, G.; Szeto, D.; Keohane, C.A.; Pan, Y.; Smith, K.M.; et al. Genetic deletion and pharmacological inhibition of phosphodiesterase 10A protects mice from diet-induced obesity and insulin resistance. Diabetes 2014, 63, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Barret, O.; Thomae, D.; Alagille, D.; Lee, H.; Papin, C.; Baldwin, R.; Jennings, D.; Marek, K.; Seibyl, J.; Tamagnan, G. First in vivo assessment of two PDE10 tracers [18F]MNI654 and [18F]MNI659. J. Nucl. Med. 2012, 53, 361. [Google Scholar]
- Barret, O.; Thomae, D.; Tavares, A.; Alagille, D.; Papin, C.; Waterhouse, R.; McCarthy, T.; Jennings, D.; Marek, K.; Russell, D.; et al. In vivo assessment and dosimetry of 2 novel PDE10A PET radiotracers in humans: 18F-MNI-659 and 18F-MNI-654. J. Nucl. Med. 2014, 55, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.S.; Barret, O.; Jennings, D.L.; Friedman, J.H.; Tamagnan, G.D.; Thomae, D.; Alagille, D.; Morley, T.J.; Papin, C.; Papapetropoulos, S.; et al. The phosphodiesterase 10 positron emission tomography tracer, [18F]MNI-659, as a novel biomarker for early huntington disease. J. Am. Med. Assoc. Neurol. 2014, 71, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.S.; Jennings, D.L.; Barret, O.; Tamagnan, G.D.; Carroll, V.M.; Caille, F.; Alagille, D.; Morley, T.J.; Papin, C.; Seibyl, J.P.; et al. Change in PDE10 across early Huntington disease assessed by [18F]MNI-659 and PET imaging. Neurology 2016, 86, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Niccolini, F.; Haider, S.; Marques, T.R.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Piccini, P.; Kapur, S.; Rabiner, E.A.; et al. Altered PDE10A expression detectable early before symptomatic onset in Huntington’s disease. Brain 2015, 138, 3016–3029. [Google Scholar] [CrossRef] [PubMed]
- Niccolini, F.; Foltynie, T.; Marques, T.R.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Kapur, S.; Rabiner, E.A.; Gunn, R.N.; et al. Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson’s disease. Brain 2015, 138, 3003–3015. [Google Scholar] [CrossRef] [PubMed]
- Marques, T.R.; Natesan, S.; Niccolini, F.; Politis, M.; Gunn, R.N.; Searle, G.E.; Howes, O.; Rabiner, E.A.; Kapur, S. Phosphodiesterase 10A in schizophrenia: A PET study using [11C]IMA107. Am. J. Psychiat. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kehler, J.; Kilburn, J.P.; Estrada, S.; Christensen, S.R.; Wall, A.; Thibblin, A.; Lubberink, M.; Bundgaard, C.; Brennum, L.T.; Steiniger-Brach, B.; et al. Discovery and development of 11C-Lu AE92686 as a radioligand for PET imaging of phosphodiesterase 10A in the brain. J. Nucl. Med. 2014, 55, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Bang-Andersen, B.; Kehler, J. Radiolabelled Phenylimidazole-Based Ligands. WO 2012/062319 A1, 18 May 2012. [Google Scholar]
- Ritzén, A.; Kehler, J.; Langgârd, M.; Nielsen, J.; Kilburn, J.P.; Farah, M.M. Novel Phenylimidazole Derivatives as PDE10A Enzyme Inhibitors. WO 2009/152825 A1, 23 December 2009. [Google Scholar]
- Hwang, D.-R.; Hu, E.; Rumfelt, S.; Easwaramoorthy, B.; Castrillon, J.; Davis, C.; Allen, J.R.; Chen, H.; Treanor, J.; Abi-Dargham, A.; et al. Initial characterization of a PDE10A selective positron emission tomography tracer [11C]AMG 7980 in non-human primates. Nucl. Med. Biol. 2014, 41, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.-R.; Hu, E.; Allen, J.R.; Davis, C.; Treanor, J.; Miller, S.; Chen, H.; Shi, B.; Narayanan, T.K.; Barret, O.; et al. Radiosynthesis and initial characterization of a PDE10A specific PET tracer [18F]AMG 580 in non-human primates. Nucl. Med. Biol. 2015, 42, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lester-Zeiner, D.; Shi, J.; Miller, S.; Glaus, C.; Hu, E.; Chen, N.; Able, J.; Biorn, C.; Wong, J.; et al. AMG 580: A novel small molecule phosphodiesterase 10A (PDE10A) positron emission tomography tracer. J. Pharmacol. Exp. Ther. 2015, 352, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Ma, J.; Biorn, C.; Lester-Zeiner, D.; Cho, R.; Rumfelt, S.; Kunz, R.K.; Nixey, T.; Michelsen, K.; Miller, S.; et al. Rapid identification of a novel small molecule phosphodiesterase 10A (PDE10A) tracer. J. Med. Chem. 2012, 55, 4776–4787. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Miura, S.; Hasui, T.; Halldin, C.; Stepanov, V.; Takano, A. Radiolabeled Compounds and Their Use as Radiotracers for Quantitative Imaging of Phosphodiesterase (PDE10A) in Mammals. WO 2013/027845 A1, 28 February 2013. [Google Scholar]
- Stepanov, V.; Miura, S.; Takano, A.; Amini, N.; Nakao, R.; Hasui, T.; Nakashima, K.; Taniguchi, T.; Kimura, H.; Kuroita, T.; et al. Development of a series of novel carbon-11 labeled PDE10A inhibitors. J. Label. Comp. Radiopharm. 2015, 58, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Suzuki, K.; Miura, S.; Hasui, T.; Kamiguchi, N.; Ishii, T.; Taniguchi, T.; Kuroita, T.; Takano, A.; Stepanov, V.; et al. Characterization of the binding properties of T-773 as a PET radioligand for phosphodiesterase 10A. Nucl. Med. Biol. 2015, 42, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Takano, A.; Stepanov, V.; Gulyás, B.; Nakao, R.; Amini, N.; Miura, S.; Kimura, H.; Taniguchi, T.; Halldin, C. Evaluation of a novel PDE10A PET radioligand, [11C]T-773, in nonhuman primates: Brain and whole body PET and brain autoradiography. Synapse 2015, 69, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Tóth, M.; Haggkvist, J.; Stepanov, V.; Takano, A.; Nakao, R.; Amini, N.; Miura, S.; Kimura, H.; Taniguchi, T.; Gulyas, B.; et al. Molecular imaging of PDE10A knockout mice with a novel PET radiotracer: [11C]T-773. Mol. Imaging Biol. 2015, 17, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Takano, A.; Stepanov, V.; Nakao, R.; Amini, N.; Gulyás, B.; Kimura, H.; Halldin, C. Brain PET measurement of PDE10A occupancy by TAK-063, a new PDE10A inhibitor, using [11C]T-773 in nonhuman primates. Synapse 2016. [Google Scholar] [CrossRef] [PubMed]
- Kunitomo, J.; Yoshikawa, M.; Fushimi, M.; Kawada, A.; Quinn, J.F.; Oki, H.; Kokubo, H.; Kondo, M.; Nakashima, K.; Kamiguchi, N.; et al. Discovery of 1-[2-fluoro-4-(1H-pyrazol-1-yl)phenyl]-5-methoxy-3-(1-phenyl-1H-pyrazol-5-yl)pyridazin-4(1H)-one (TAK-063), a highly potent, selective, and orally active phosphodiesterase 10A (PDE10A) inhibitor. J. Med. Chem. 2014, 57, 9627–9643. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.D.; Hostetler, E.D.; Flores, B.A.; Evelhoch, J.L.; Fan, H.; Gantert, L.; Holahan, M.; Eng, W.; Joshi, A.; McGaughey, G.; et al. Discovery of [11C]MK-8193 as a PET tracer to measure target engagement of phosphodiesterase 10A (PDE10A) inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 4893–4898. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, E.; Cox, C.D.; Fan, H. Radiolabeled PDE10 Inhibitors. WO 2010/138577 A1, 2 December 2010. [Google Scholar]
- Raheem, I.T.; Breslin, M.J.; Fandozzi, C.; Fuerst, J.; Hill, N.; Huszar, S.; Kandebo, M.; Kim, S.H.; Ma, B.; McGaughey, G.; et al. Discovery of tetrahydropyridopyrimidine phosphodiesterase 10A inhibitors for the treatment of schizophrenia. Bioorg. Med. Chem. Lett. 2012, 22, 5903–5908. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, E.D.; Fan, H.; Joshi, A.D.; Zeng, Z.; Eng, W.; Gantert, L.; Holahan, M.; Meng, X.; Miller, P.; O’Malley, S.; et al. Preclinical characterization of the phosphodiesterase 10A PET tracer [11C]MK-8193. Mol. Imaging Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lu, A.Y.H. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–449. [Google Scholar] [PubMed]
- Jakobsen, S.; Kodahl, G.M.; Olsen, A.K.; Cumming, P. Synthesis, radiolabeling and in vivo evaluation of [11C]RAL-01, a potential phosphodiesterase 5 radioligand. Nucl. Med. Biol. 2006, 33, 593–597. [Google Scholar] [CrossRef] [PubMed]



































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
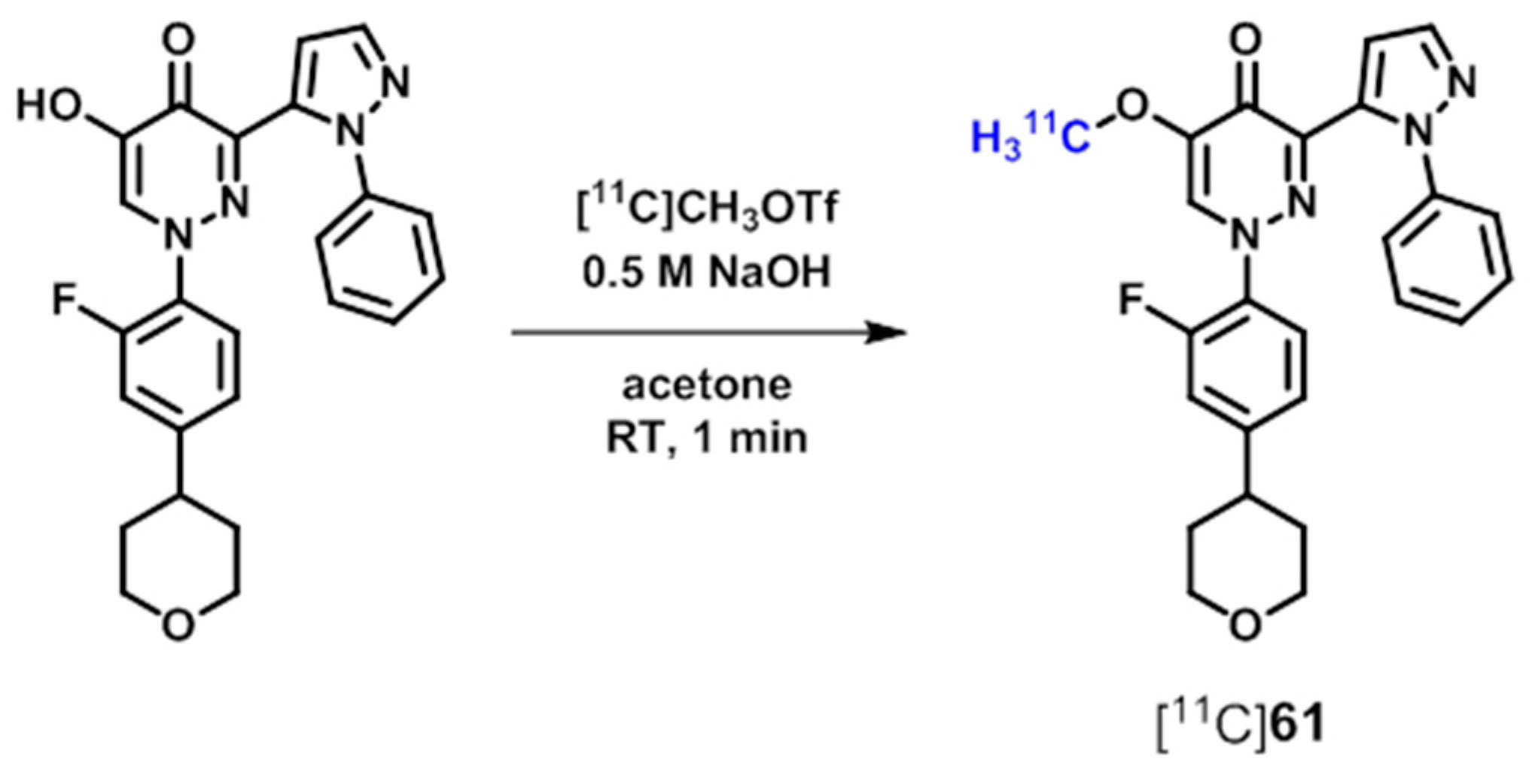{kind=link}
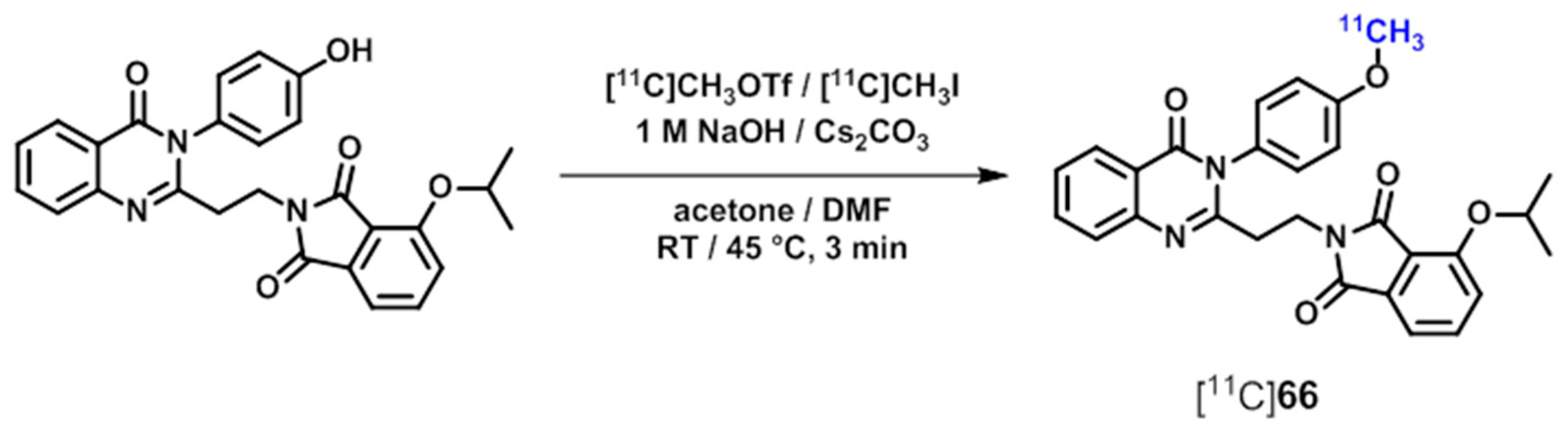{kind=link}
| Phosphodiesterase | Radioligands Review Compound No. (Code No. Given) | References |
|---|---|---|
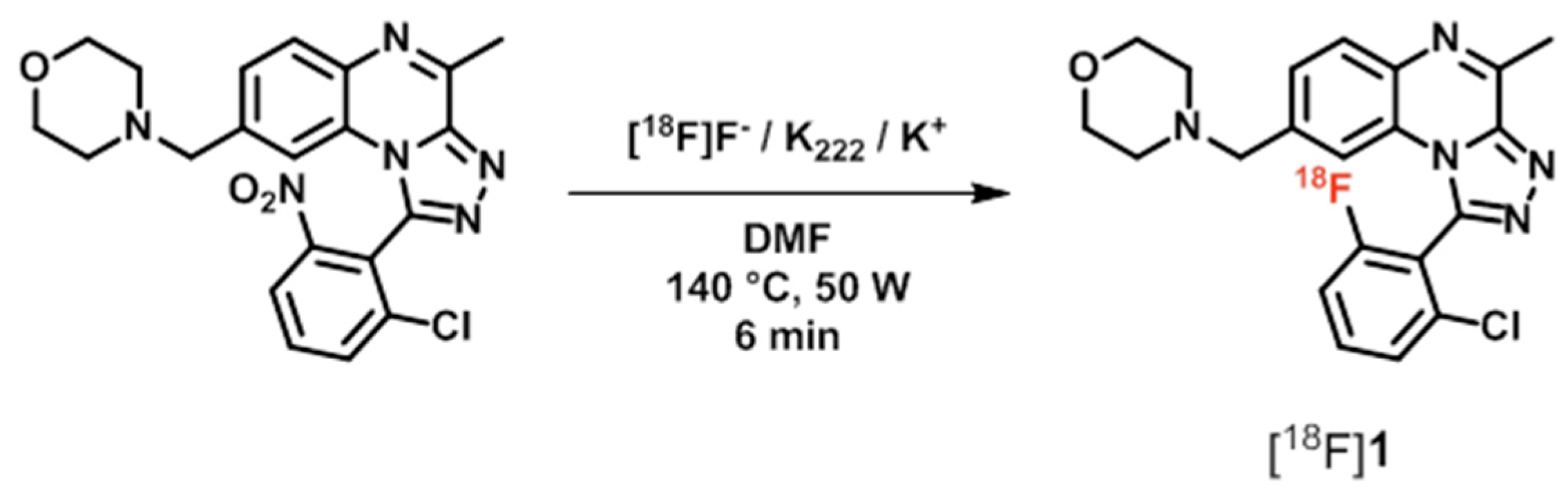
| PDE2 | [18F]1 ([18F]B-23) | [48,55] |
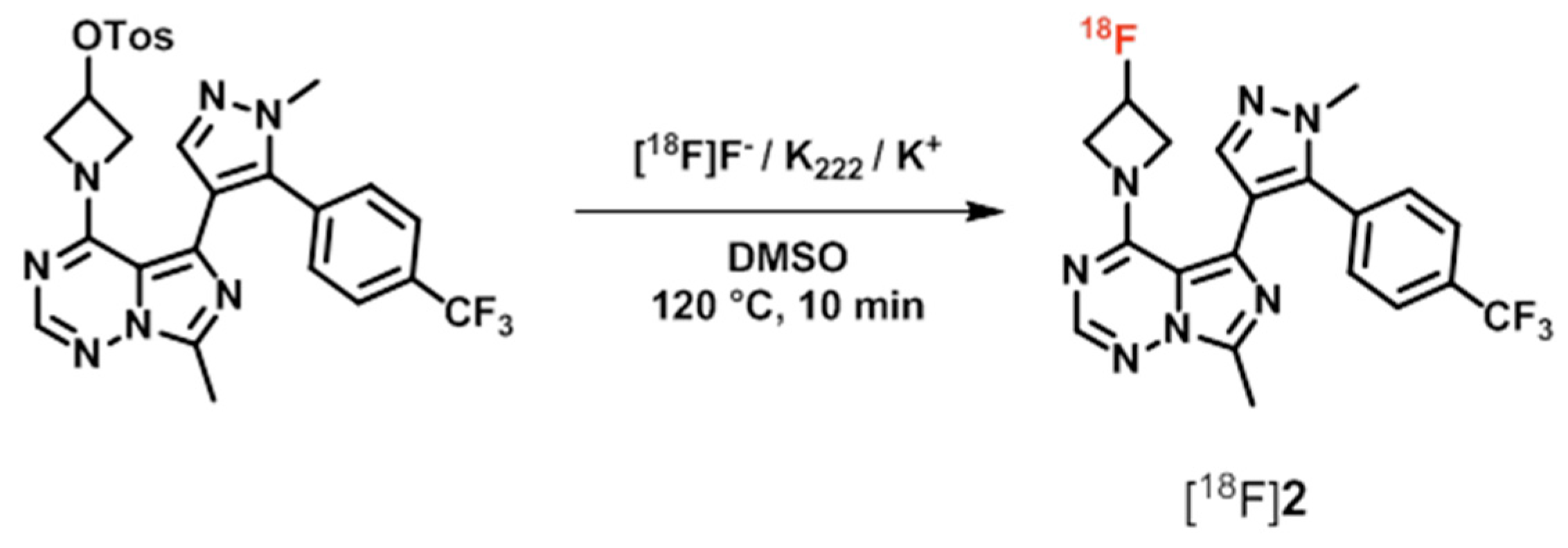
| [18F]2 ([18F]PF-05270430) | [48,56,57,58] | |
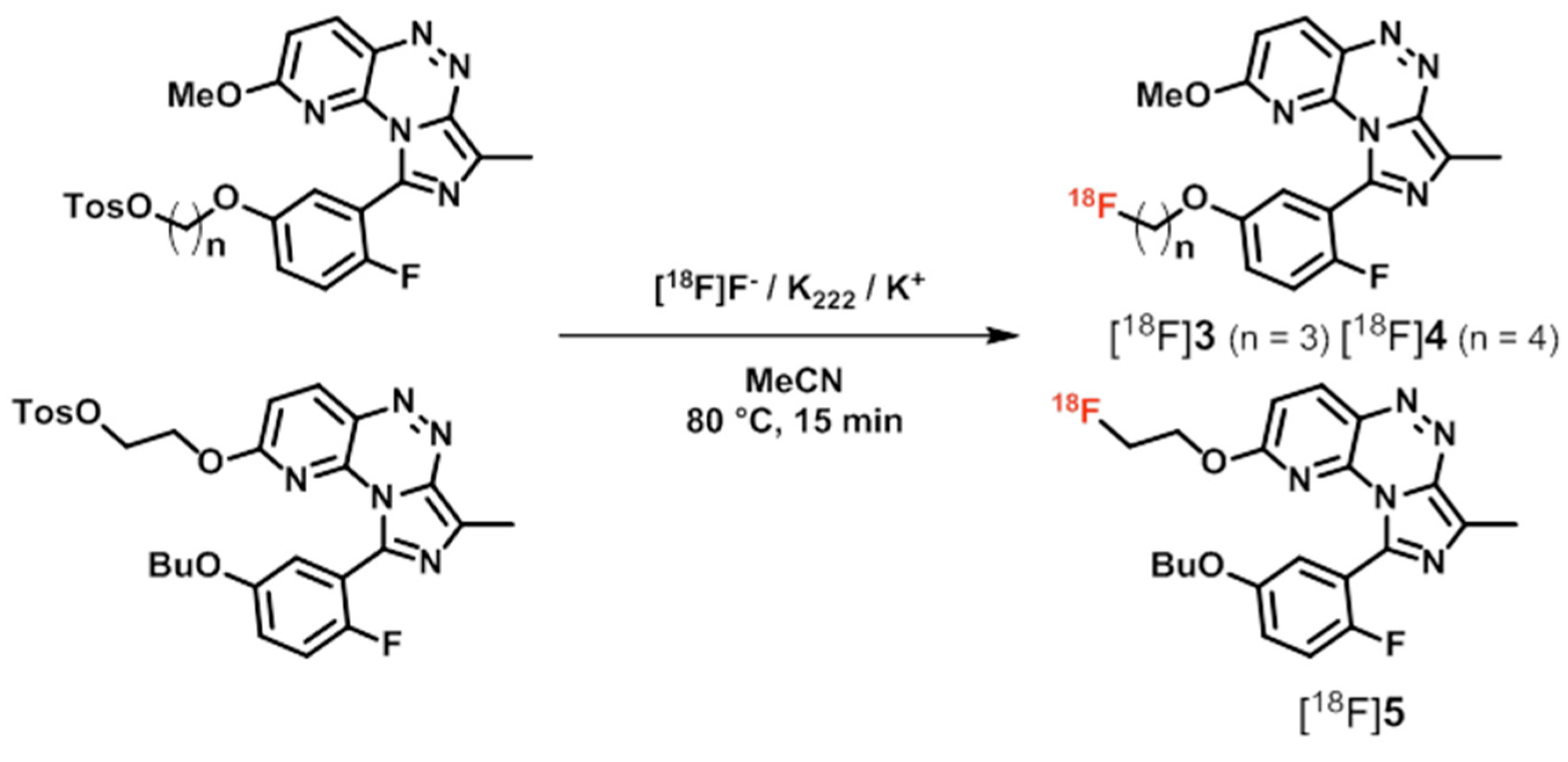
| [18F]3, [18F]4, [18F]5 ([18F]TA3–5) | [60,61,62] | |
| PDE4 | [18F]7 ([18F]MNI-617) | [82] |
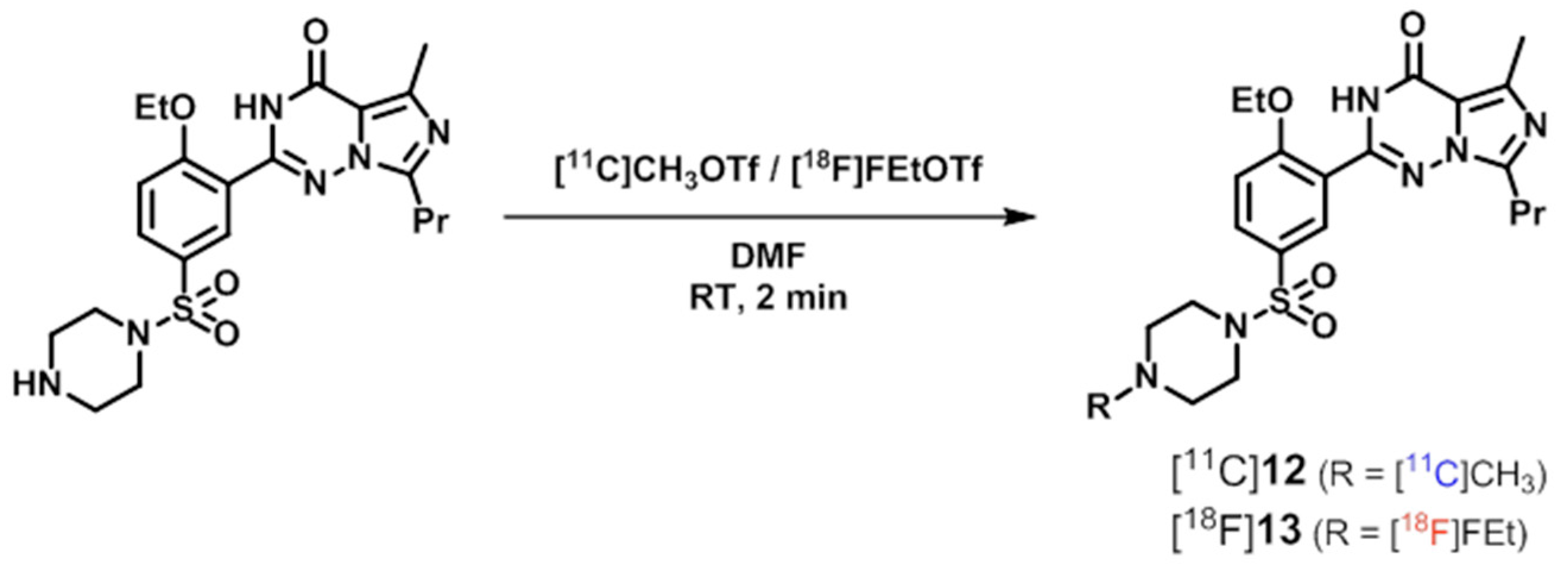
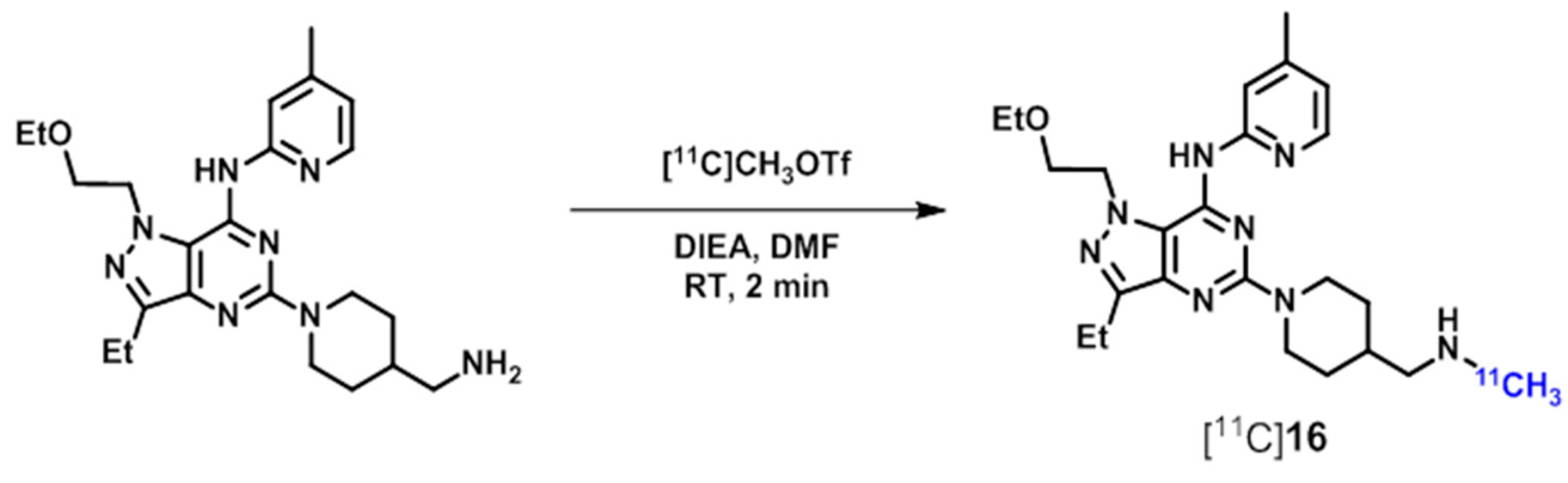
| PDE5 | [11C]8 ([11C]sildenafil), [18F]13, [11C]14, [11C]16, [11C]17 | [97] |
| [18F]18 ([18F]ICF24027) | [110] | |
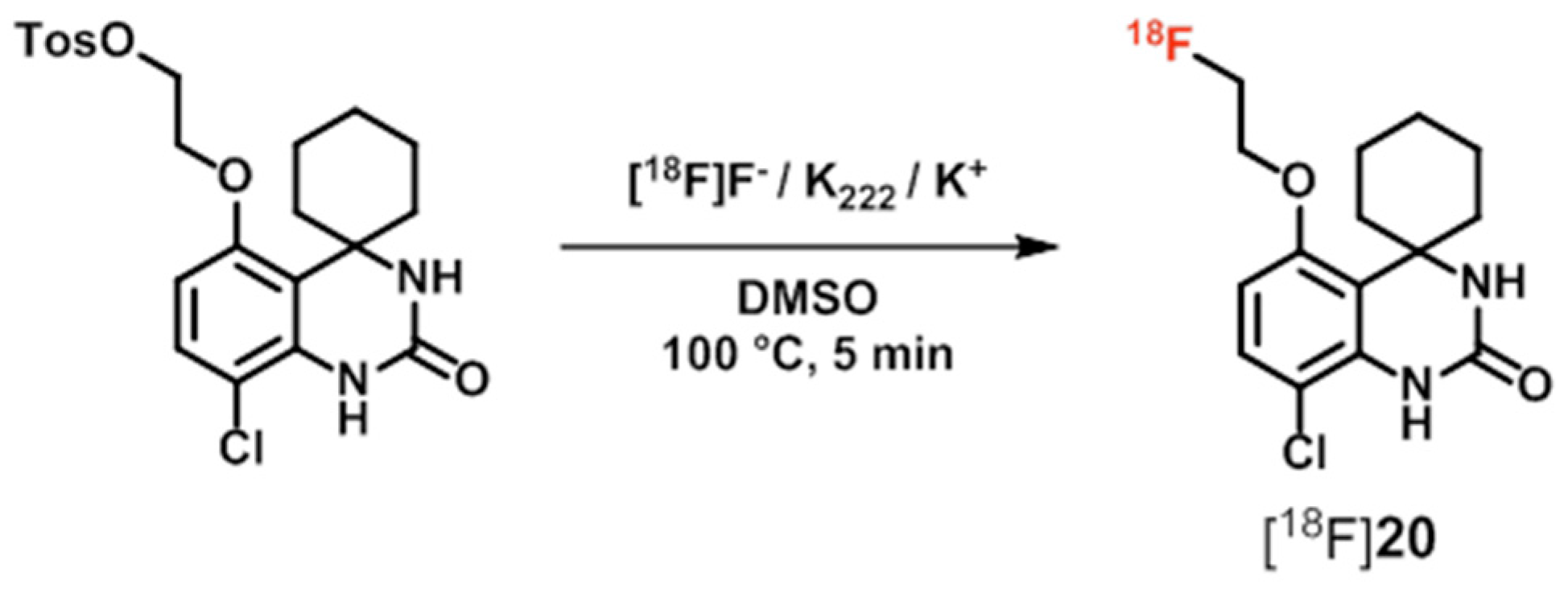
| PDE7 | [18F]20 ([18F]MICA-003), [11C]21 ([11C]MICA-005) | [121,122] |
| PDE10 | Structurally related to compound 22 (MP-10) | |
| [18F]26, [11C]29 | [143] | |
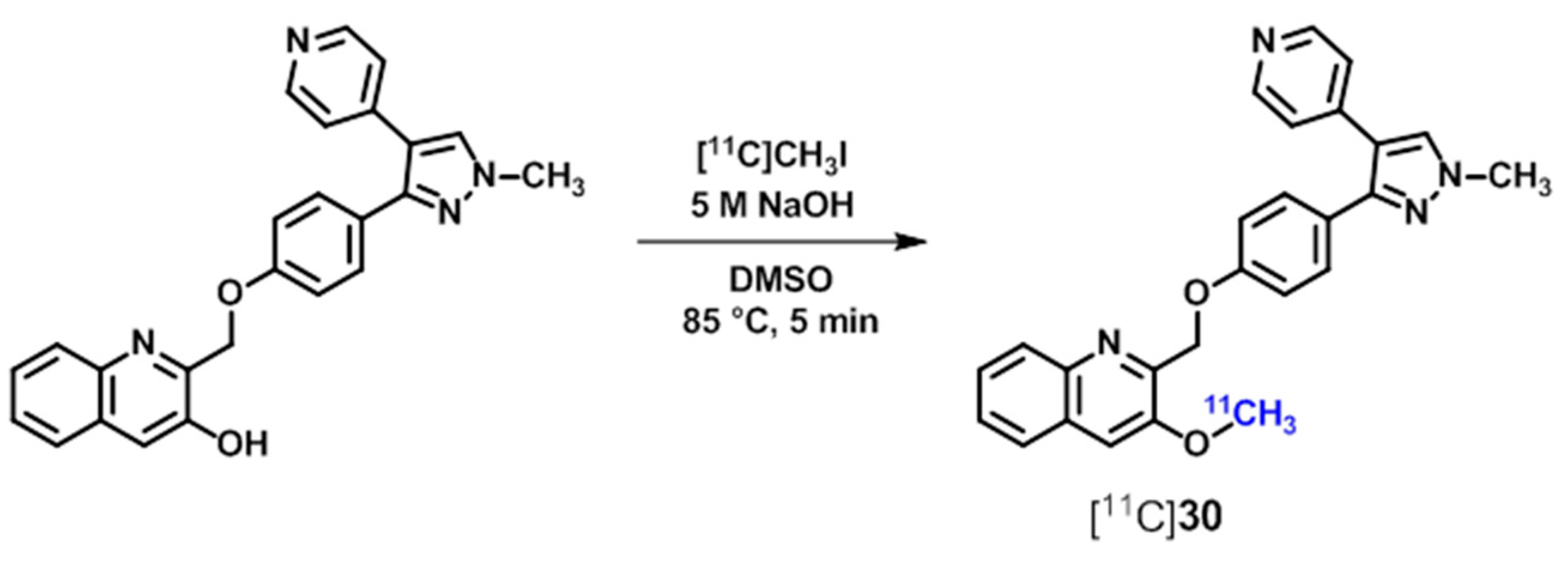
| [11C]30 ([11C]TZ1964B), [11C]31–33 | [157,160] | |
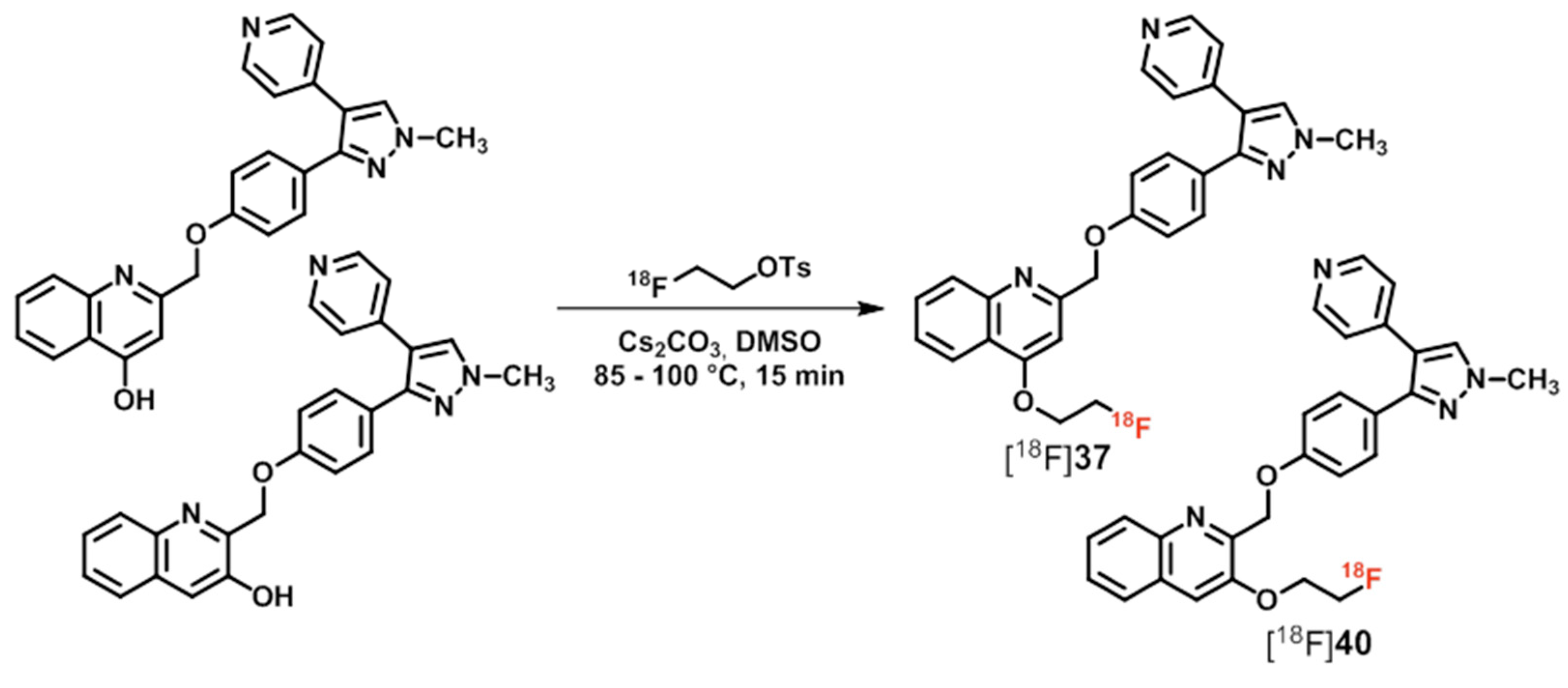
| [18F]34–40 | [158] | |
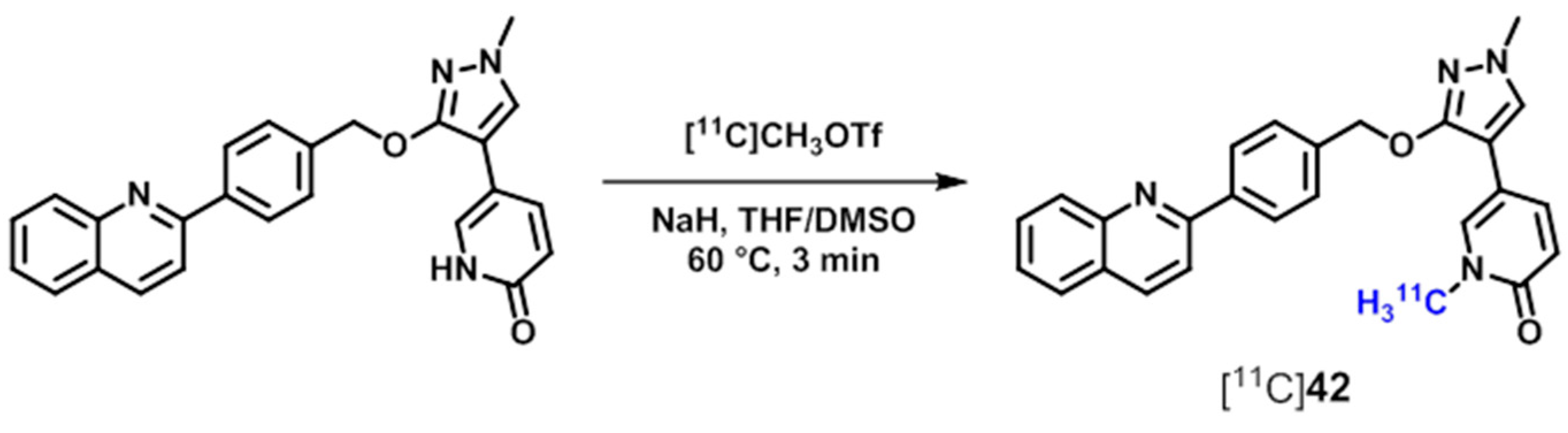
| [11C]42 | [161] | |
| Structurally not derived from compound 22 (MP-10) | ||
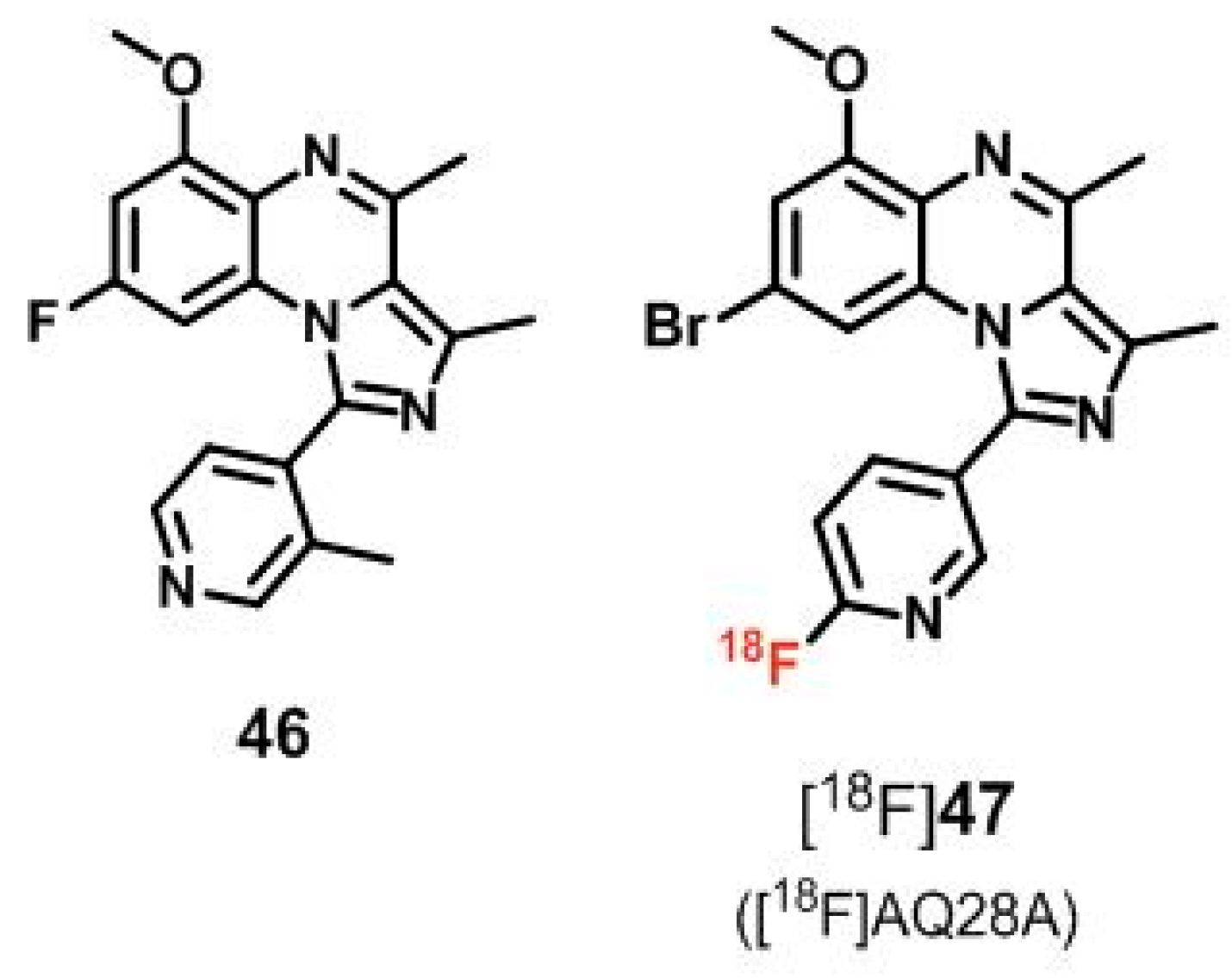
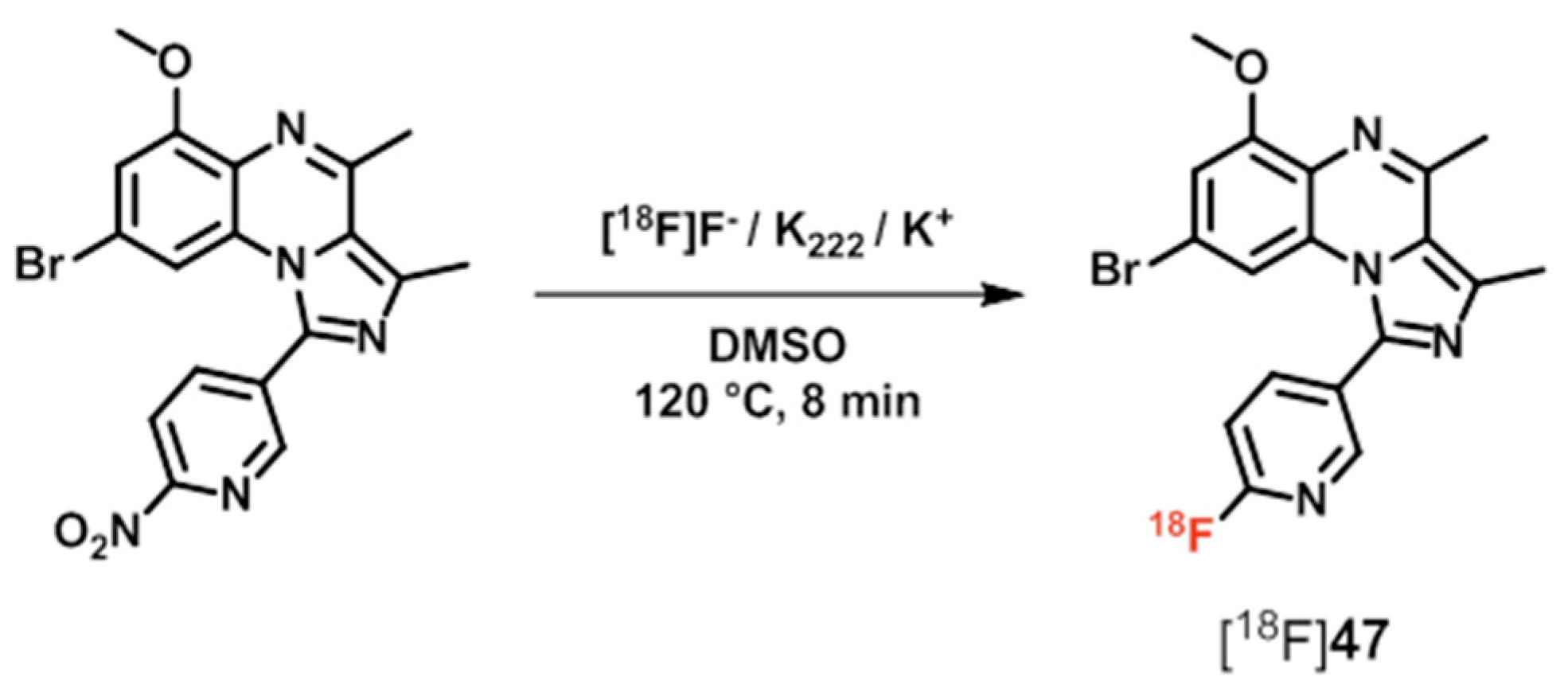
| [18F]47 ([18F]AQ28A) | [170,172] | |
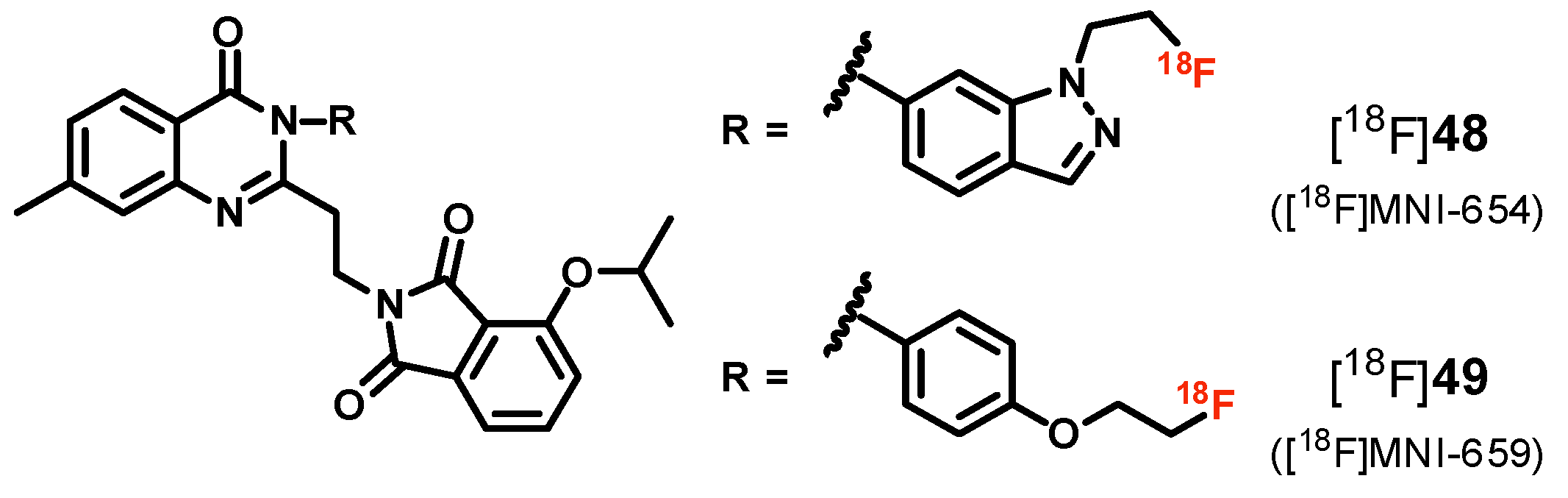
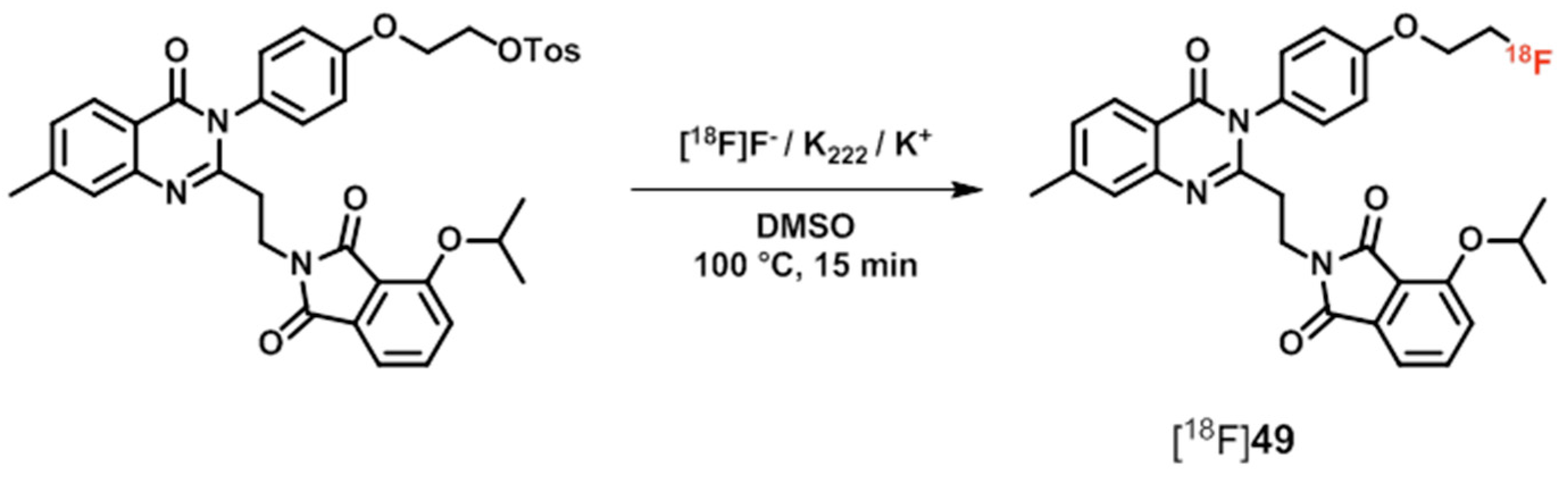
| [18F]48 ([18F]MNI-654), [18F]49 ([18F]MNI-659) | [175,176,177,178] | |
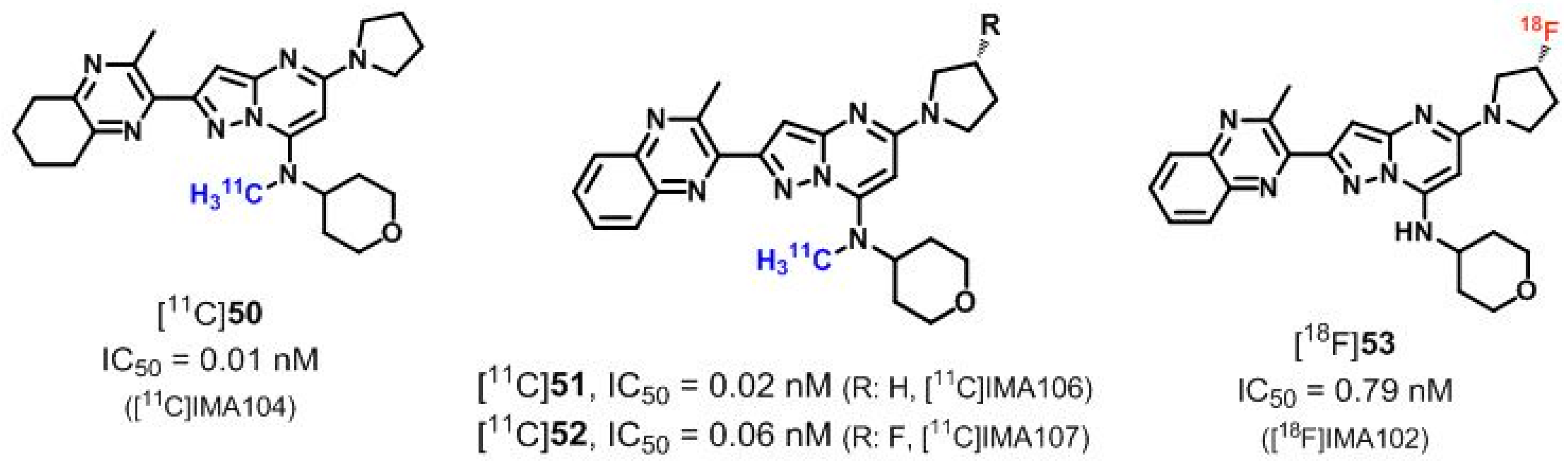
| [11C]50 ([11C]IMA104), [11C]51 ([11C]IMA106) | [142,181] | |
| [11C]52 ([11C]IMA107), [18F]53 ([18F]IMA102) | ||
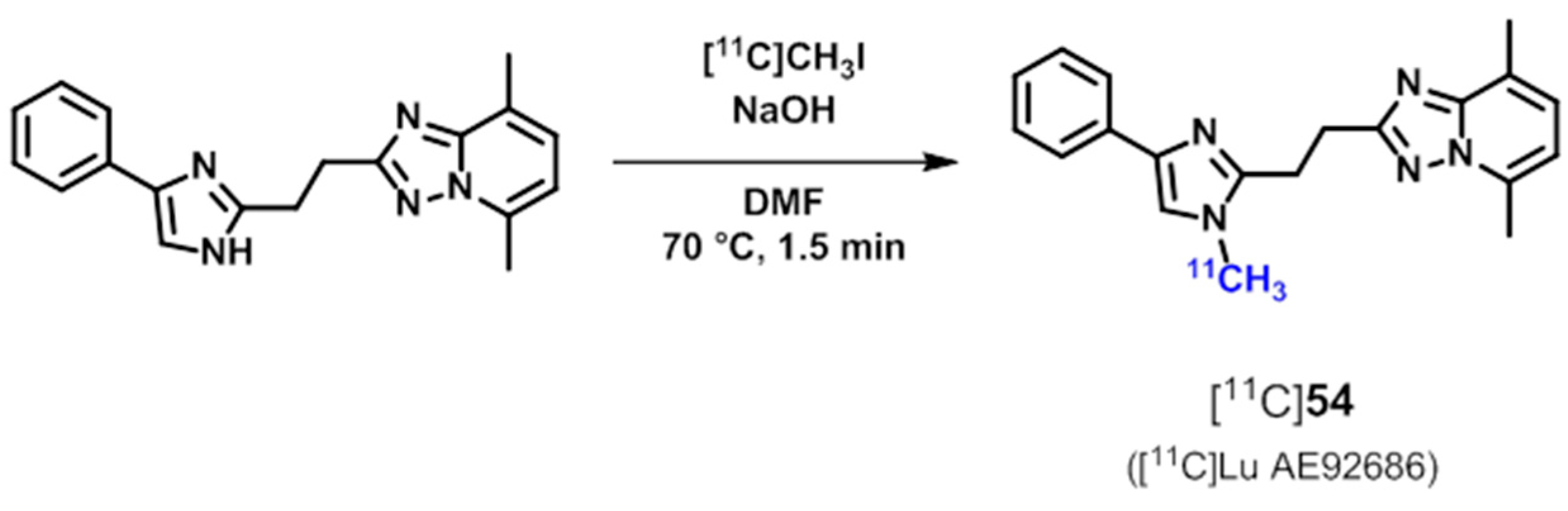
| [11C]54 ([11C]Lu AE92686) | [182,183] | |
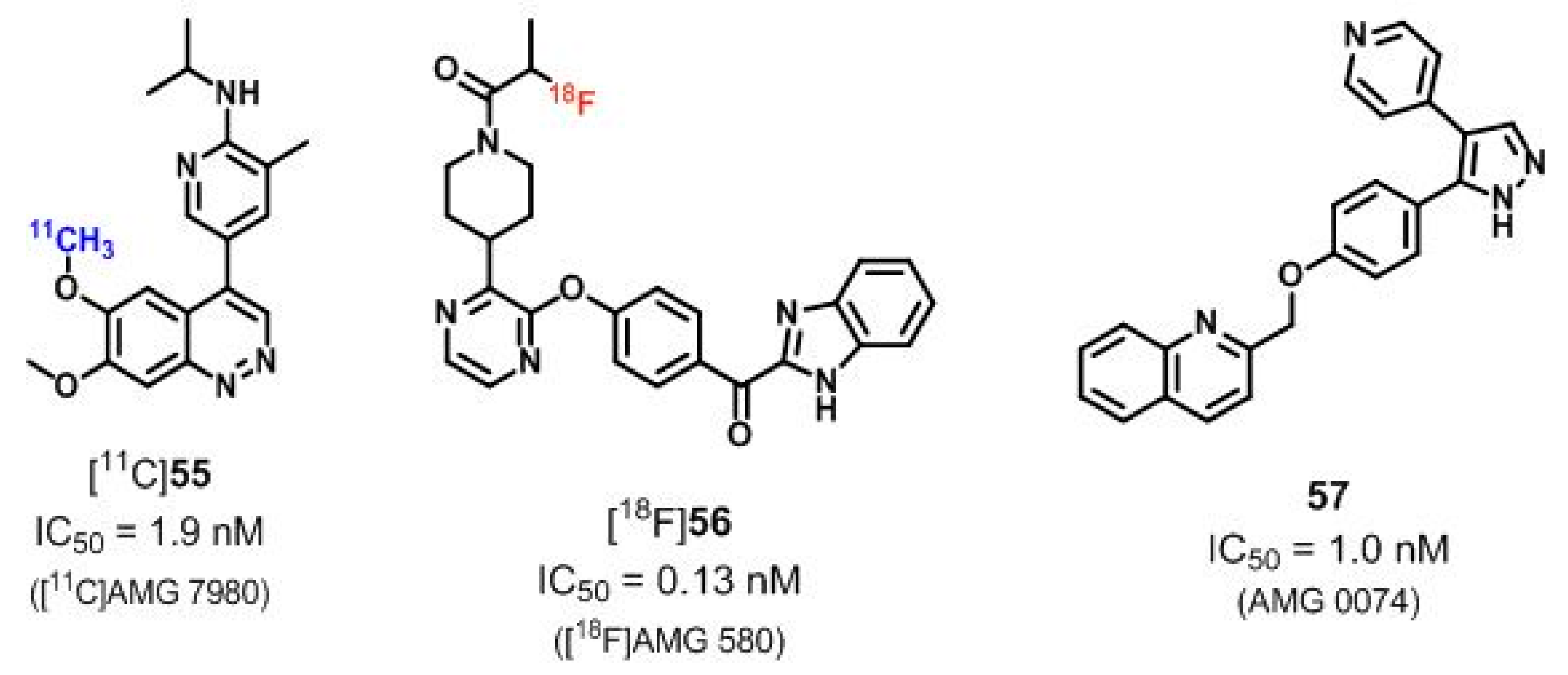
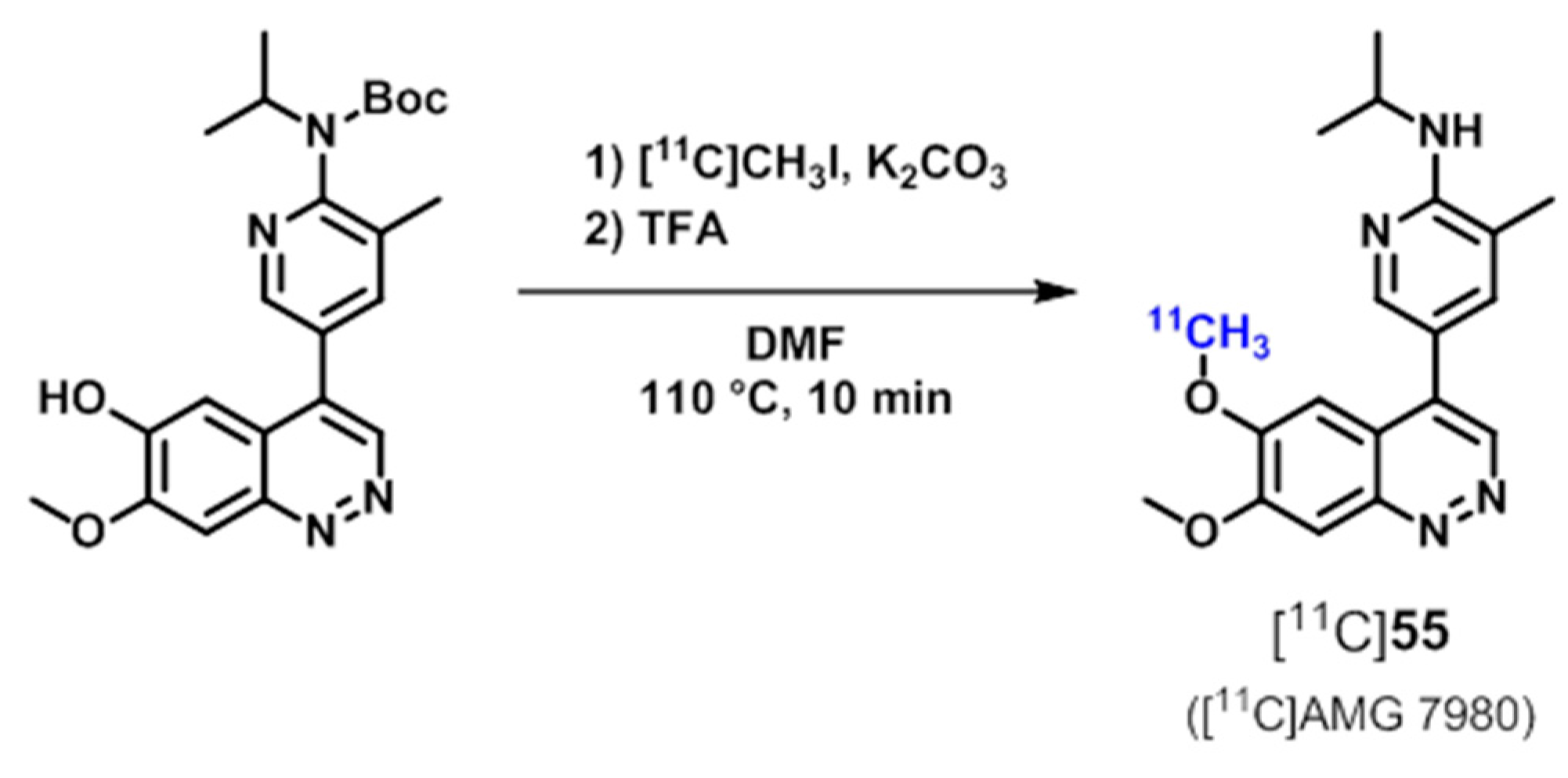
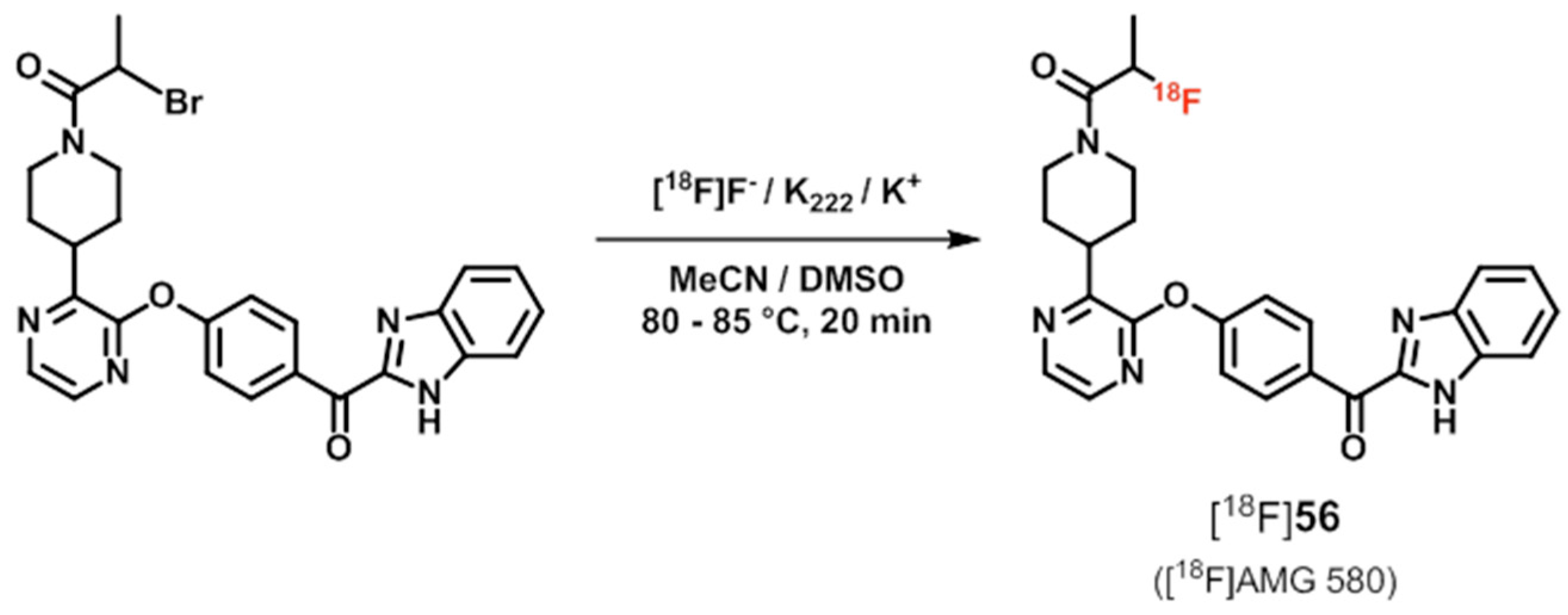
| [11C]55 ([11C]AMG 7980), [18F]56 ([18F]AMG 580) | [185,186,187] | |
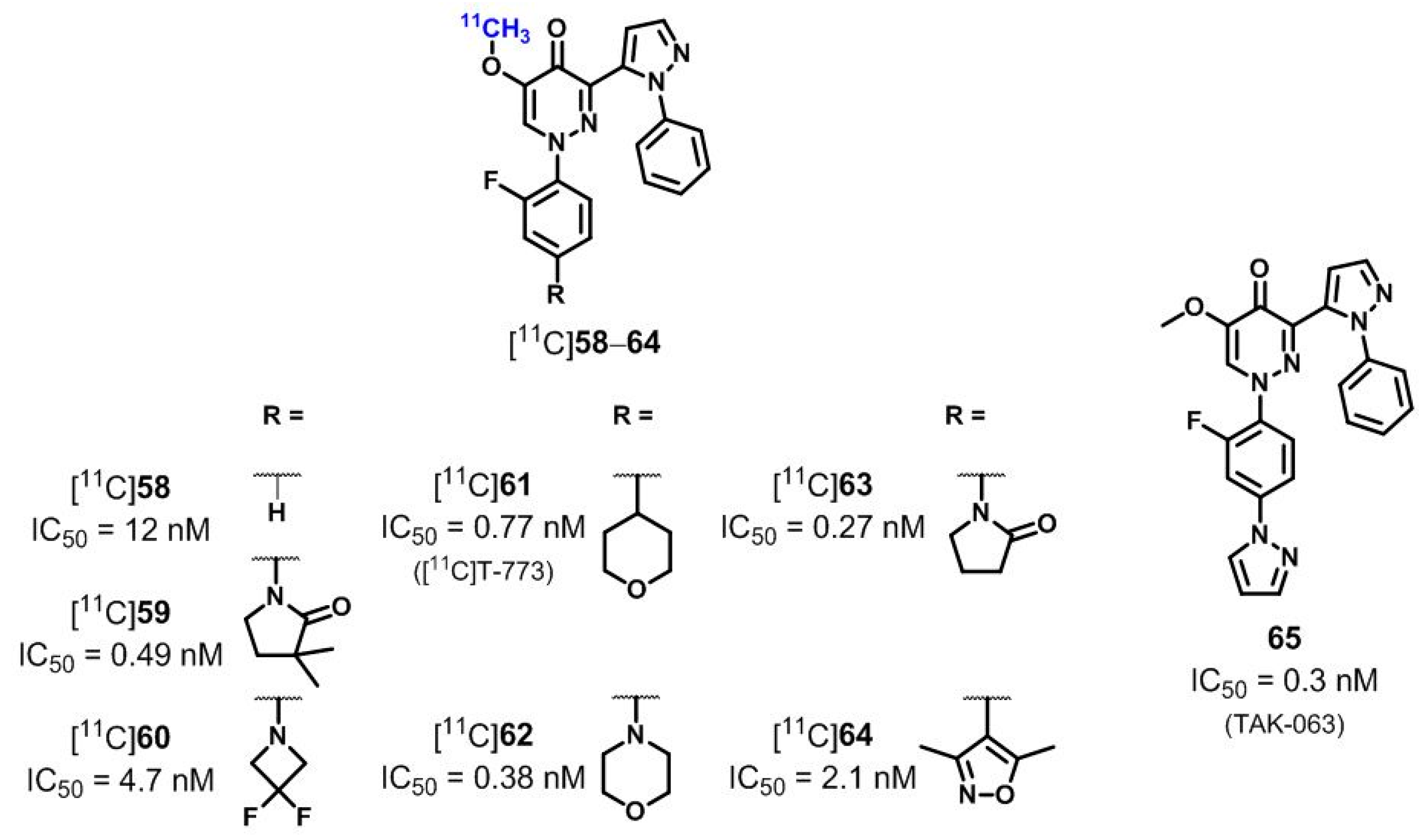
| [11C]58–60, [11C]61 ([11C]T-773), [11C]62–64 | [190,191,192,193,194] | |
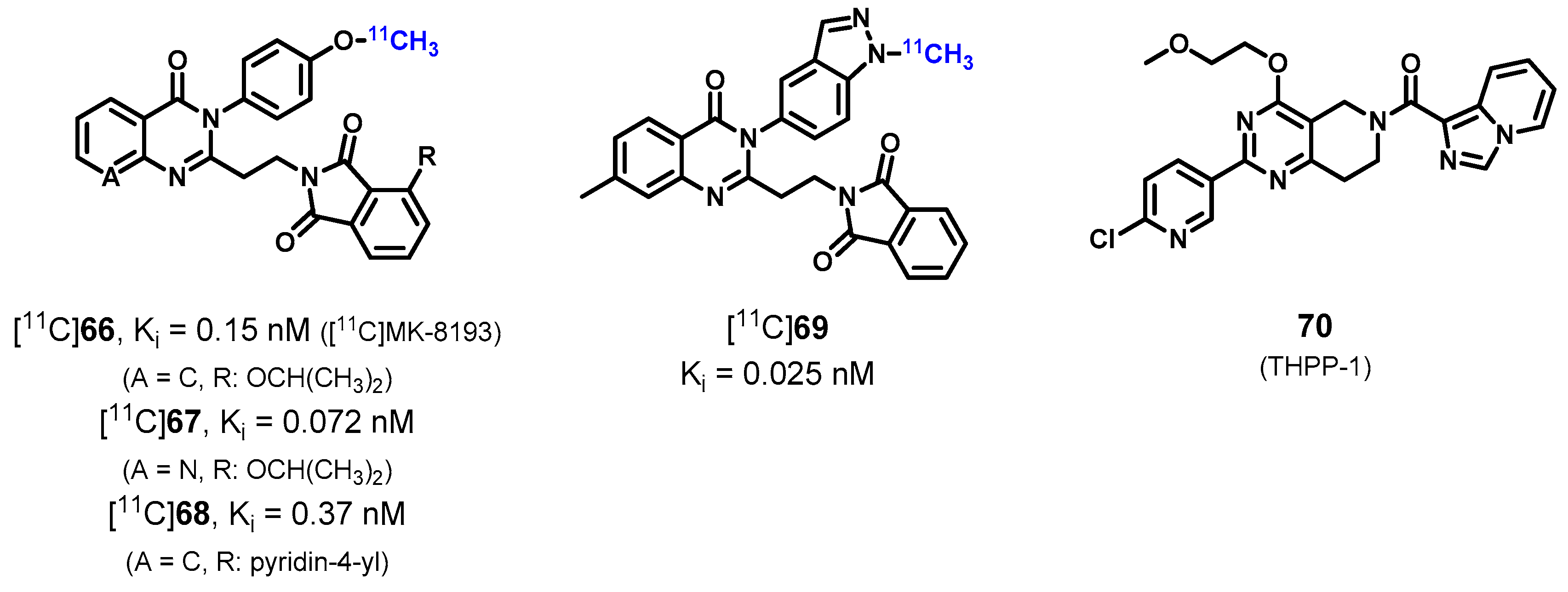
| [11C]66 ([11C]MK-8193), [11C]67–69 | [196,197,199] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Scheunemann, M.; Brust, P. Novel Radioligands for Cyclic Nucleotide Phosphodiesterase Imaging with Positron Emission Tomography: An Update on Developments Since 2012. Molecules 2016, 21, 650. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050650
Schröder S, Wenzel B, Deuther-Conrad W, Scheunemann M, Brust P. Novel Radioligands for Cyclic Nucleotide Phosphodiesterase Imaging with Positron Emission Tomography: An Update on Developments Since 2012. Molecules. 2016; 21(5):650. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050650
Chicago/Turabian StyleSchröder, Susann, Barbara Wenzel, Winnie Deuther-Conrad, Matthias Scheunemann, and Peter Brust. 2016. "Novel Radioligands for Cyclic Nucleotide Phosphodiesterase Imaging with Positron Emission Tomography: An Update on Developments Since 2012" Molecules 21, no. 5: 650. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050650