
Identification of Novel Human Breast Carcinoma (MDA-MB-231) Cell Growth Modulators from a Carbohydrate-Based Diversity Oriented Synthesis Library


 ,
,  and
and 
Abstract
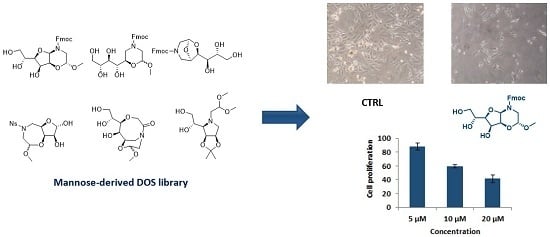
:
1. Introduction
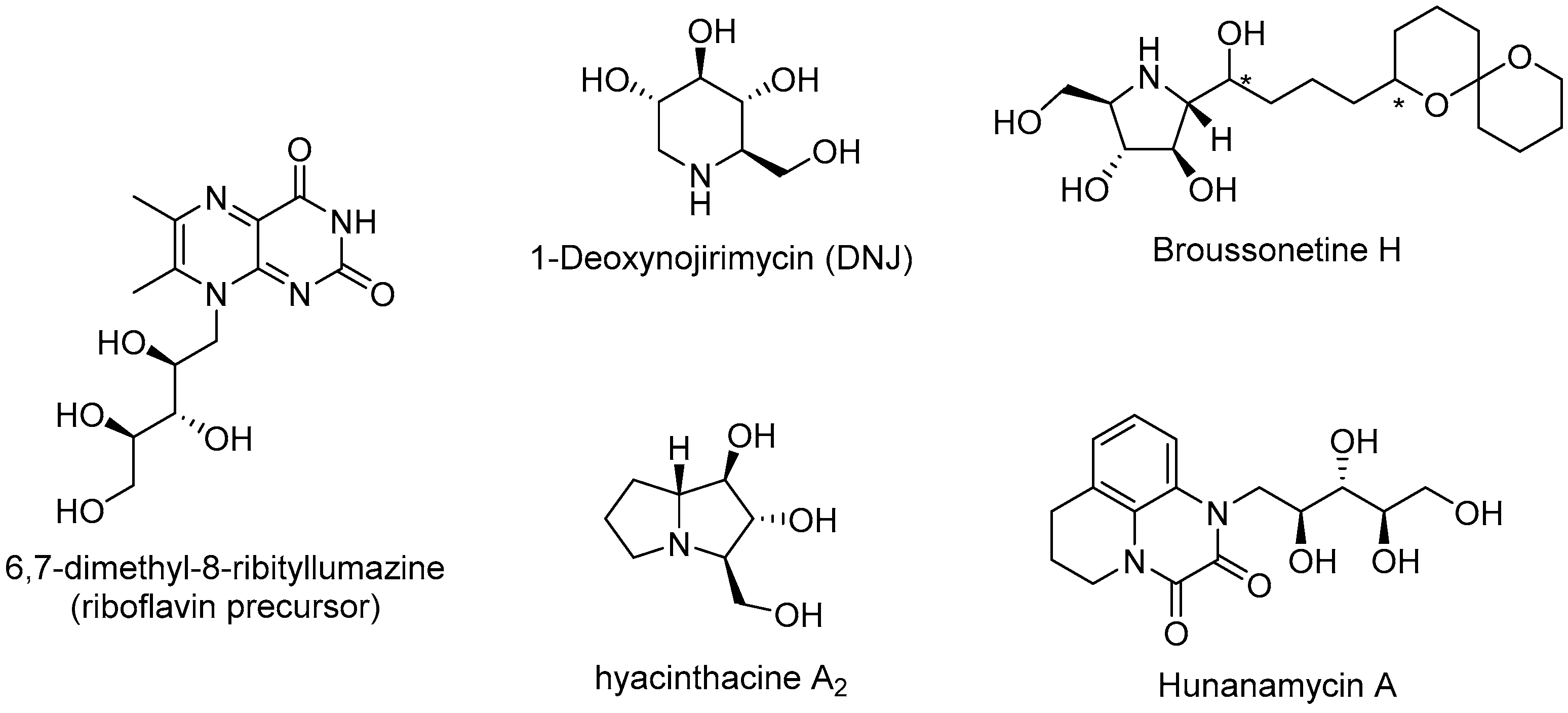
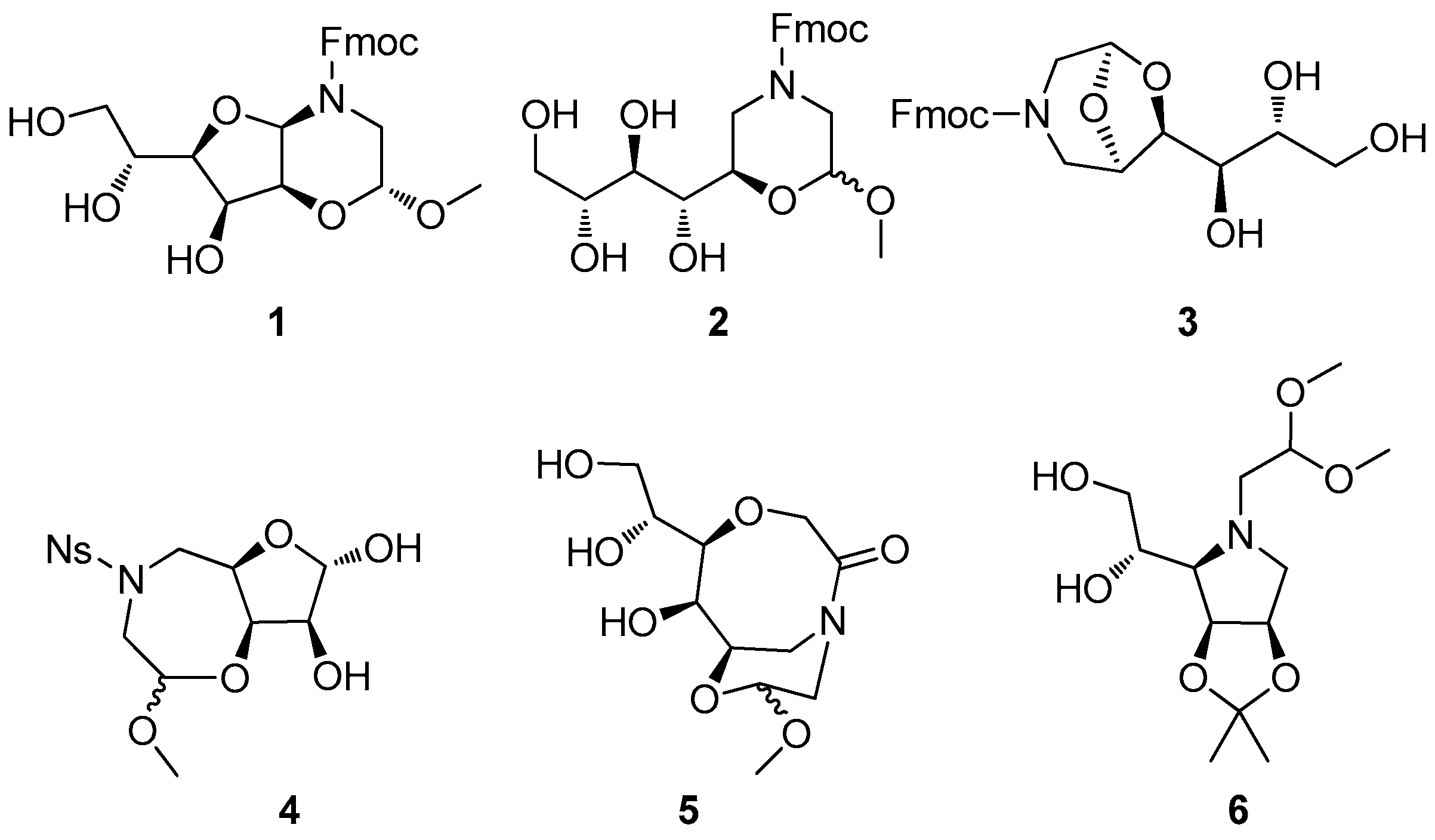
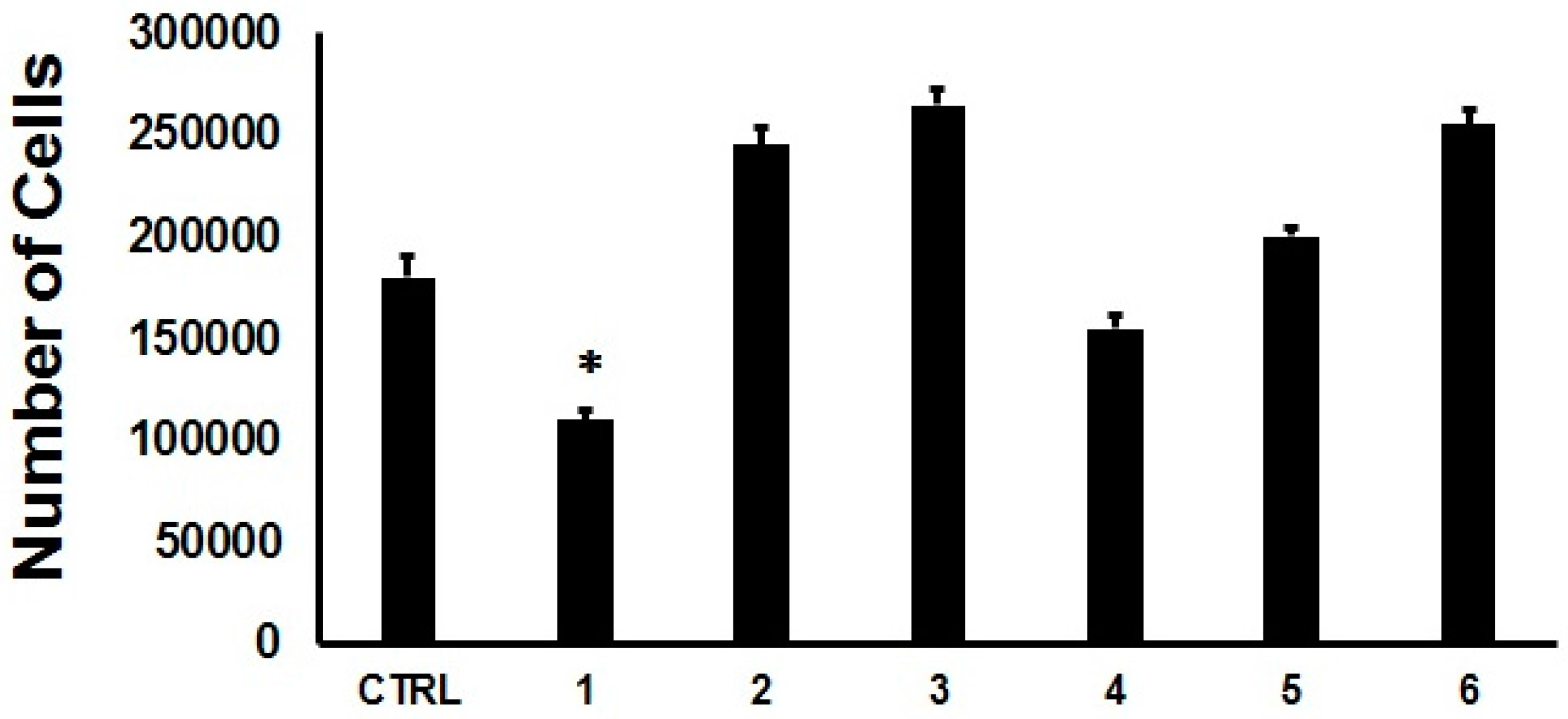
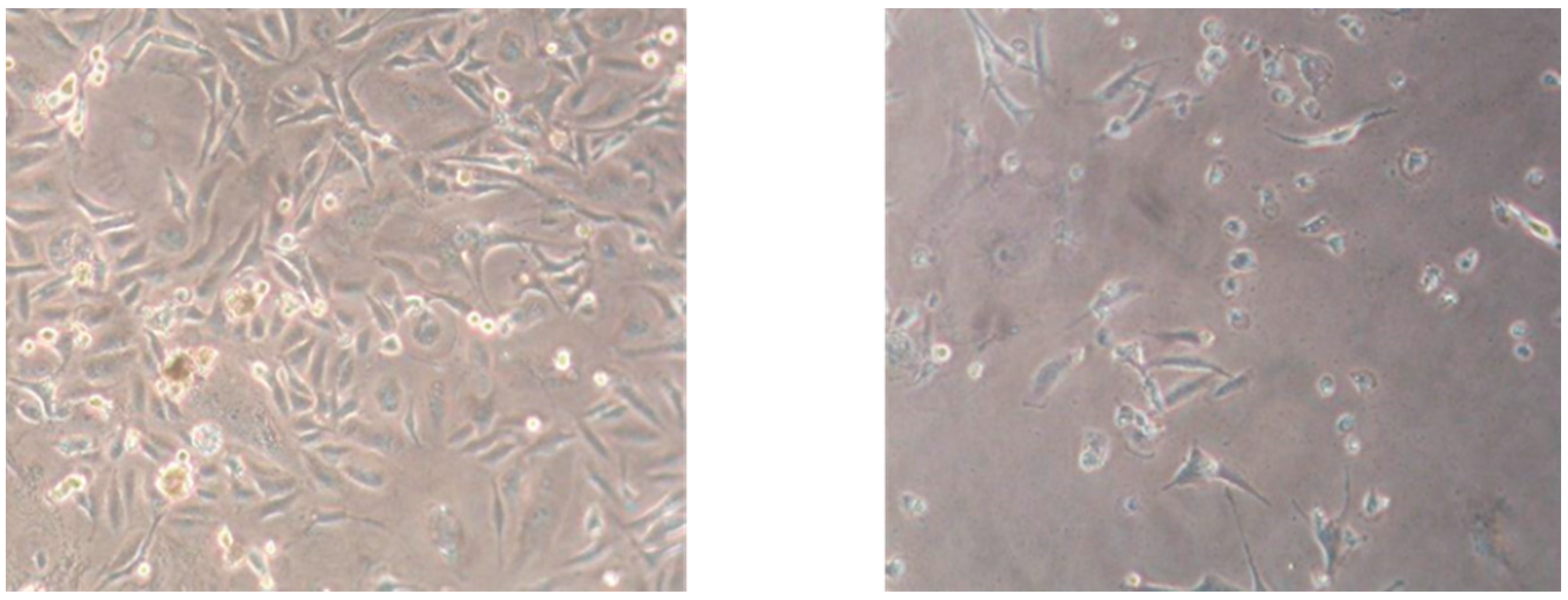
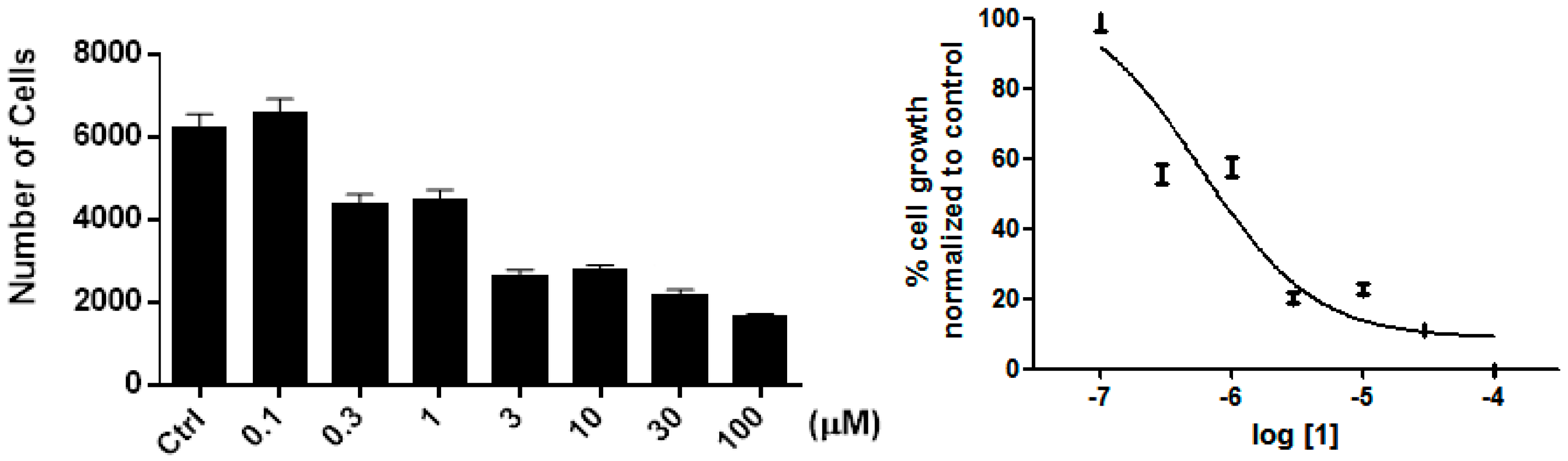
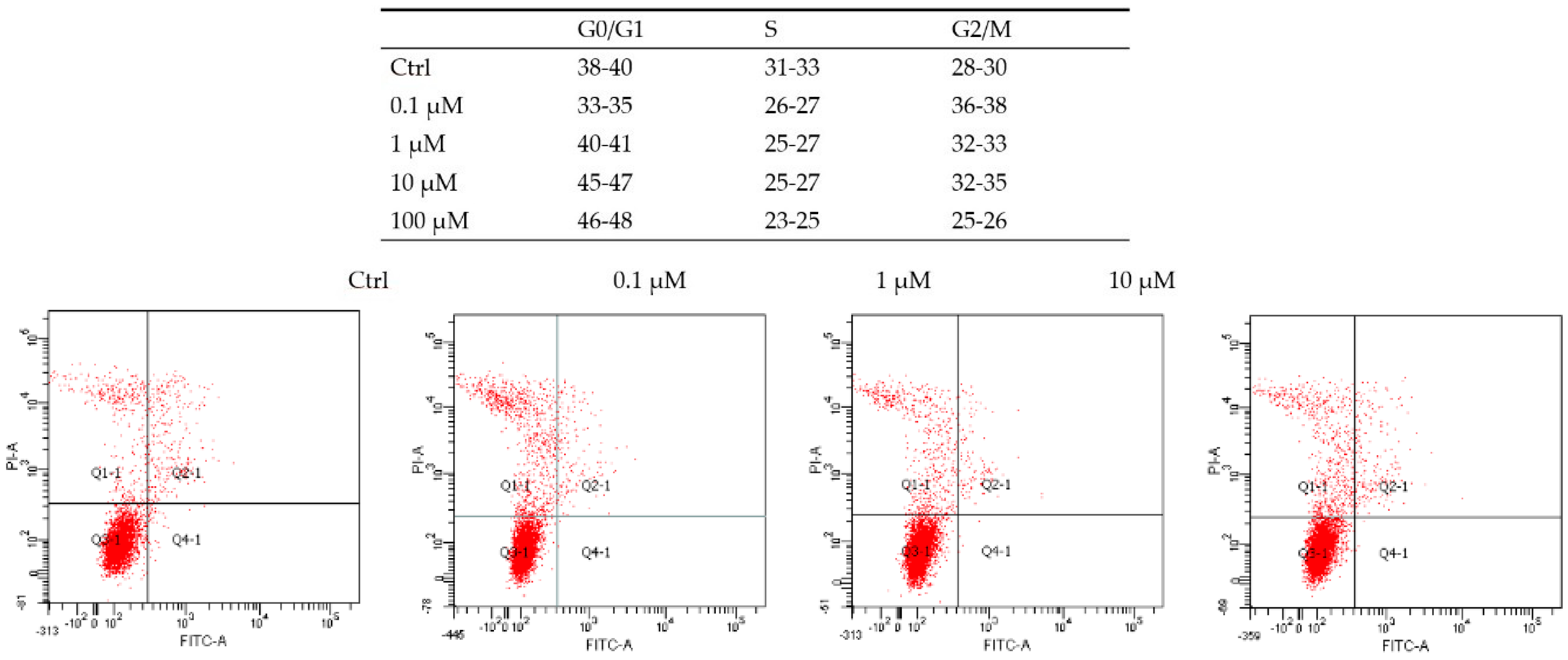
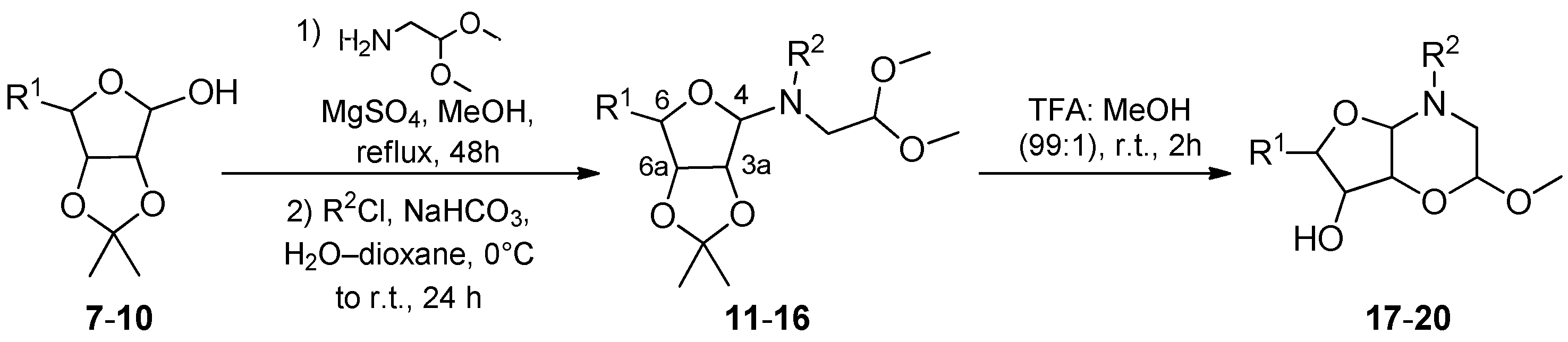
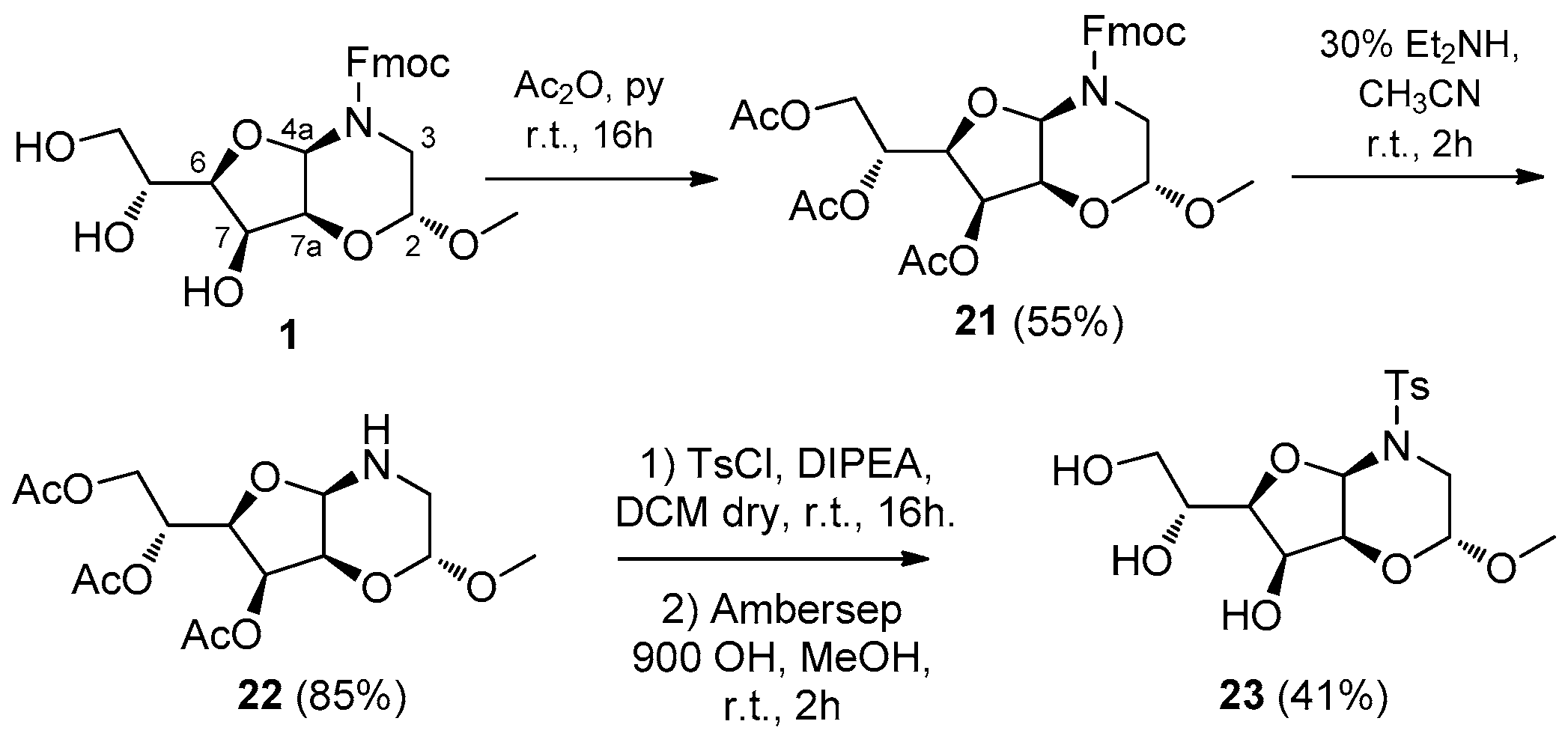
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. (9H-Fluoren-9-yl)methyl (2,2-dimethoxyethyl)((3aR,4S,6R,6aR)-6-(hydroxymethyl)-2,2-dimethyl-tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)carbamate (11α) and (9H-Fluoren-9-yl)-methyl-(2,2-dimethoxyethyl)-((3aR,4R,6R,6aR)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)carbamate (11β)
3.3. (9H-Fluoren-9-yl)methyl (2,2-dimethoxyethyl)((3aS,4S,6R,6aS)-6-(hydroxymethyl)-2,2-dimethyltetra-hydrofuro[3,4-d][1,3]dioxol-4-yl)carbamate (12α) and (9H-Fluoren-9-yl)-methyl-(2,2-dimethoxyethyl)-((3aS,4R,6R,6aS)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)carbamate (12β)
3.4. (9H-Fluoren-9-yl)methyl (2,2-dimethoxyethyl)((3aS,4S,6aS)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]-dioxol-4-yl)carbamate (13α)
3.5. N-(2,2-Dimethoxyethyl)-N-((3aS,4S,6R,6aS)-6-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetra-hydrofuro[3,4-d][1,3]dioxol-4-yl)benzamide (15α) and N-(2,2-Dimethoxyethyl)-N-((3aS,4R,6R,6aS)-6-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)benzamide (15β)
3.6. Benzyl (2,2-dimethoxyethyl)((3aS,4R,6R,6aS)-6-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyl-tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)carbamate (16α) and Benzyl (2,2-dimethoxyethyl)((3aS,4S,6R,6aS)-6-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)carbamate (16β)
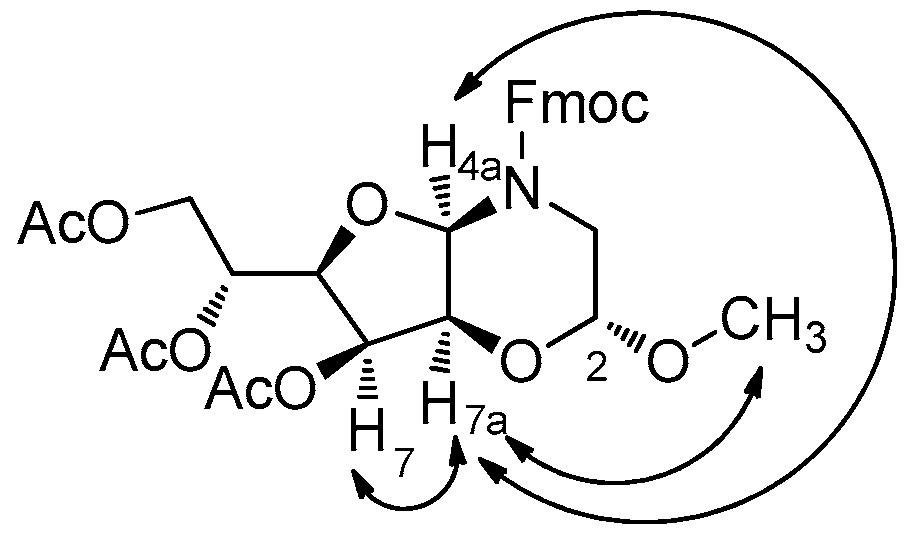
3.7. (2R,4aS,6R,7R,7aR)-(9H-Fluoren-9-yl)methyl 7-hydroxy-6-(hydroxymethyl)-2-methoxytetrahydro-2H-furo[3,2-b][1,4]oxazine-4(3H)-carboxylate (17)
3.8. (2R,4aR,6R,7S,7aS)-(9H-Fluoren-9-yl)methyl 7-hydroxy-6-(hydroxymethyl)-2-methoxytetrahydro-2H-furo[3,2-b][1,4]oxazine-4(3H)-carboxylate (18)
3.9. (2R,4aR,7S,7aS)-(9H-Fluoren-9-yl)methyl 7-hydroxy-2-methoxytetrahydro-2H-furo[3,2-b][1,4]oxazine-4(3H)-carboxylate (19)
3.10. (2R,4aR,6R,7S,7aS)-Benzyl 6-((R)-1,2-dihydroxyethyl)-7-hydroxy-2-methoxytetrahydro-2H-furo[3,2-b][1,4]oxazine-4(3H)-carboxylate (20)
3.11. (R)-1-((2R,4aR,6R,7S,7aS)-7-Hydroxy-2-methoxy-4-tosylhexahydro-2H-furo[3,2-b][1,4]oxazin-6-yl)ethane-1,2-diol (23)
3.12. Cell Culture
3.13. Cell Growth Assay
3.14. Cell Viability
3.15. Cell Cycle Analysis
3.16. Data Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schirle, M.; Jenkins, J.L. Identifying compound efficacy targets in phenotypic drug discovery. Drug Discov. Today 2016, 21, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Trabocchi, A. Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis, Drug Discovery, and Chemical Biology; John Wiley and Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Spring, D.R. Diversity-oriented synthesis; A challenge for synthetic chemists. Org. Biomol. Chem. 2003, 1, 3867–3870. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.D.; Schreiber, S.L. A planning strategy for diversity-oriented synthesis. Angew. Chem. Int. Ed. 2004, 43, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [Google Scholar] [CrossRef] [PubMed]
- Cordier, C.; Morton, D.; Murrison, S.; Nelson, A.; O’Leary-Steele, C. Natural products as an inspiration in the diversity-oriented synthesis of bioactive compound libraries. Nat. Prod. Rep. 2008, 25, 719–737. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.B.; Robin, S.; Kumar, K.; Waldmann, H. Biology-oriented synthesis. Angew. Chem. Int. Ed. 2011, 50, 10800–10826. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Srivastava, S.; Gupta, G.; Chaturvedi, V.; Sinha, S.; Srivastava, R. Natural Product Inspired Diversity Oriented Synthesis of Tetrahydroquinoline Scaffolds as Antitubercular Agent. ACS Comb. Sci. 2011, 13, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Hirschmann, R.; Nicolaou, K.C.; Pietranico, S.; Leahy, E.M.; Salvino, J.; Arison, B.H.; Cichy, M.A.; Spoors, P.G.; Shakespeare, W.C.; Sprengeler, P.A.; et al. De novo design and synthesis of somatostatin non-peptide peptidomimetics utilizing.beta.-d-glucose as a novel scaffolding. J. Am. Chem. Soc. 1993, 115, 12550–12568. [Google Scholar] [CrossRef]
- Abbenante, G.; Becker, B.; Blanc, S.; Clark, C.; Condie, G.; Fraser, G.; Grathwohl, M.; Halliday, J.; Henderson, S.; Lam, A.; et al. Biological diversity from a structurally diverse library: Systematically scanning conformational space using a pyranose scaffold. J. Med. Chem. 2010, 53, 5576–5586. [Google Scholar] [CrossRef] [PubMed]
- Hünger, U.; Ohnsmann, J.; Kunz, H. Carbohydrate scaffolds for combinatorial syntheses that allow selective deprotection of all four positions independent of the sequence. Angew. Chem. Int. Ed. 2004, 43, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Lenci, E.; Menchi, G.; Trabocchi, A. Carbohydrates in diversity-oriented synthesis: Challenges and opportunities. Org. Biomol. Chem. 2016, 14, 808–825. [Google Scholar] [CrossRef] [PubMed]
- Yadav, L.D.S.; Srivastava, V.P.; Rai, V.K.; Patel, R. Diversity oriented synthesis of fused-ring 1,3-oxazines from carbohydrates as biorenewable feedstocks. Tetrahedron 2008, 64, 4246–4253. [Google Scholar] [CrossRef]
- Aravind, A.; Kumar, P.S.; Sankar, M.G.; Baskaran, S. Diversity-Oriented Synthesis of Useful Chiral Building Blocks from d-Mannitol. Eur. J. Org. Chem. 2011, 6980–6988. [Google Scholar] [CrossRef]
- Lowe, J.T.; Lee, M.D.; Akella, L.B.; Davoine, E.; Donckele, E.J.; Durak, L.; Duvall, J.R.; Gerard, B.; Holson, E.B.; Joliton, A.; et al. Synthesis and Profiling of a Diverse Collection of Azetidine-Based Scaffolds for the Development of CNS-Focused Lead-like Libraries. J. Org. Chem. 2012, 77, 7187–7211. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.M.; Lobo, F.; Perez de las Vacas, D.; Valverde, S.; Lopez, J.C. Formation and reactivity of new Nicholas–Ferrier pyranosidic cations: Novel access to oxepanes via a 1,6-hydride shift/cyclization sequence. Chem. Commun. 2010, 6159–6161. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, S.; Prasad, S.S.; Kumar, P.S.; Baskaran, S. A diversity oriented one-pot synthesis of novel iminosugar C-glycosides. Chem. Commun. 2014, 1549–1551. [Google Scholar] [CrossRef] [PubMed]
- Manna, C.; Pathak, T. Diversity-Oriented Synthesis of Enantiopure Furofurans from Carbohydrates: An Expedient Approach with Built-in Michael Acceptor, Masked Aldehyde and Leaving Group in a Single Sugar Derivative. Eur. J. Org. Chem. 2013, 6084–6086. [Google Scholar] [CrossRef]
- Lenci, E.; Menchi, G.; Guarna, A.; Trabocchi, A. Skeletal Diversity from Carbohydrates: Use of Mannose for the Diversity-Oriented Synthesis of Polyhydroxylated Compounds. J. Org. Chem. 2015, 80, 2182–2191. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.A.; Fleet, G.W.J.; Asano, N.; Molyneux, R.J.; Nash, R.J. Polyhydroxylated alkaloids—Natural occurrence and therapeutic applications. Phytochemistry 2001, 56, 265–295. [Google Scholar] [CrossRef]
- Yamashita, T.; Yasuda, K.; Kizu, H.; Kameda, Y.; Watson, A.A.; Nash, R.J.; Fleet, G.W.J.; Asano, N.J. New Polyhydroxylated Pyrrolidine, Piperidine, and Pyrrolizidine Alkaloids from Scilla sibirica. J. Nat. Prod. 2002, 65, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Yamauchi, T.; Kagamifuchi, K.; Shimizu, N.; Takahashi, S.; Takatsuka, H.; Ikeda, K.; Kizu, H.; Chuakul, W.; Kettawan, A.; et al. Iminosugar-Producing Thai Medicinal Plants. J. Nat. Prod. 2005, 68, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Pyrrole, pyrrolidine, pyridine, piperidine and tropane alkaloids. Nat. Prod. Rep. 2000, 17, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-Y.; Kinami, K.; Kato, A.; Jia, Y.-M.; Li, Y.-X.; Fleet, G.W.J.; Yu, C.-Y. First total synthesis of (+)-broussonetine W: Glycosidase inhibition of natural product & analogs. Org. Biomol. Chem. 2016, 14, 5157–5174. [Google Scholar] [PubMed]
- Winchester, B.; Fleet, G.W.J. Amino-sugar glycosidase inhibitors: Versatile tools for glycobiologists. Glycobiology 1992, 2, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Nash, R.J.; Molyneux, R.J.; Fleet, G.W.J. Sugar-mimic glycosidase inhibitors: Natural occurrence, biological activity and prospects for therapeutic application. Tetrahedron Asymmetry 2000, 11, 1645–1844. [Google Scholar] [CrossRef]
- Compain, P.; Martin, O.R. Design, synthesis and biological evaluation of iminosugar-based glycosyltransferase inhibitors. Curr. Top. Med. Chem. 2003, 3, 541–560. [Google Scholar] [CrossRef] [PubMed]
- Asano, N. Naturally Occurring Iminosugars and Related Compounds: Structure, Distribution, and Biological Activity. Curr. Top. Med. Chem. 2003, 3, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Greimel, P.; Spreitz, J.; Stütz, A.E.; Wrodnigg, T.M. Iminosugars and Relatives as Antiviral and Potential Anti-infective Agents. Curr. Top. Med. Chem. 2003, 3, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Martin, O.R. Iminosugars: From Synthesis to Therapeutic Applications; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Horne, G.; Wilson, F.X. Therapeutic applications of iminosugars: Current perspectives and future opportunities. Prog. Med. Chem. 2011, 50, 135–176. [Google Scholar] [PubMed]
- Nash, R.J.; Kato, A.; Yu, C.-Y.; Fleet, G.W.J. Iminosugars as therapeutic agents: Recent advances and promising trends. Fut. Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, S.; O’Donnell, R.A.; Koussis, K.; Dluzewski, A.R.; Ansel, K.H.; Osborne, S.A.; Hackett, F.; Withers-Martinez, C.; Mitchell, G.H.; Bannister, L.H. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 2007, 131, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Arastu-Kapur, S.; Ponder, E.L.; Fonović, U.P.; Yeoh, S.; Yuan, F.; Fonović, M.; Grainger, M.; Phillips, C.; Powers, J.C.; Bogyo, M. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat. Chem. Biol. 2008, 4, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.; Foley, M.A.; Shair, M.D.; Schreiber, S.L. Stereoselective Synthesis of over Two Million Compounds Having Structural Features Both Reminiscent of Natural Products and Compatible with Miniaturized Cell-Based Assays. J. Am. Chem. Soc. 1998, 120, 8565–8566. [Google Scholar] [CrossRef]
- Lenci, E.; Guarna, A.; Trabocchi, A. Diversity-Oriented Synthesis as a Tool for Chemical Genetics. Molecules 2014, 19, 16506–16528. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.G.; Rudolph, J.; Bailey, D. Phenotypic screening in cancer drug discovery—past, present and future. Nat. Rev. Drug Discov. 2014, 13, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.; Park, J.; Koo, J.Y.; Lim, D.; Cha, M.Y.; Jo, A.; Choi, J.H.; Park, S.B. Phenotypic screening to identify small-molecule enhancers for glucose uptake: Target identification and rational optimization of their efficacy. Angew. Chem. Int. Ed. 2014, 53, 5102–5106. [Google Scholar]
- Gregori-Puigjané, E.; Setola, V.; Hert, J.; Crews, B.A.; Irwin, J.J.; Lounkine, E.; Marnett, L.; Roth, B.L.; Shoichet, B.K. Identifying mechanism-of-action targets for drugs and probes. Proc. Natl. Acad. Sci. USA 2012, 109, 11178–11183. [Google Scholar]
- Elliott, W.J.; Ram, C.V. Calcium Channel Blockers. J. Clin. Hypertens. 2011, 13, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Triggle, D.J. Calcium channel antagonists: Clinical uses—Past, present and future. Biochem. Pharmacol. 2007, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Thorne, N.; McKew, J.C. Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 2013, 18, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Carey, L.A. Biology, Metastatic Patterns, and Treatment of Patients with Triple-Negative Breast Cancer. Clin. Breast Cancer 2009, 9, S73–S81. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.; Siegel, R.; Bandi, P.; Jemal, A. Breast cancer statistics, 2011. CA Cancer J. Clin. 2011, 61, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Brenton, J.D.; Carey, L.A.; Ahmed, A.A.; Caldas, C. Molecular Classification and Molecular Forecasting of Breast Cancer: Ready for Clinical Application? J. Clin. Oncol. 2005, 23, 7350–7360. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.B. Triple-negative breast cancers: An updated review on treatment options. Curr. Oncol. 2011, 18, e173–e179. [Google Scholar] [CrossRef] [PubMed]
- Chavez, K.J.; Garimella, S.V.; Lipkow, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, E.M.; Goncalves-Pereira, R.; Risquez-Cuadro, R.; Plata, G.B.; Padron, J.M.; García Fernández, J.M.; Mellet, C.M. Influence of the configurational pattern of sp2-iminosugar pseudo N-, S-, O- and C-glycosides on their glycoside inhibitory and antitumor properties. Carbohydr. Res. 2016, 429, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, E.M.; Risquez-Cuadro, R.; Chasseraud, M.; Ahidouch, A.; Ortiz Mellet, C.; Ouadid-Ahidouch, H.; García Fernández, J.M. Synthesis of N-, S-, and C-glycoside castanospermine analogues with selective neutral α-glucosidase inhibitory activity as antitumour agents. Chem. Commun. 2010, 46, 5328–5330. [Google Scholar]
- Hottin, A.; Dubar, F.; Steenackers, A.; Delannoy, P.; Biot, C.; Behr, J.B. Iminosugar–ferrocene conjugates as potential anticancer agents. Org. Biomol. Chem. 2012, 10, 5592–5597. [Google Scholar] [CrossRef] [PubMed]
- Schley, P.D.; Jijon, H.B.; Robinson, L.E.; Field, C.J. Mechanisms of omega-3 fatty acid-induced growth inhibition in MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2005, 92, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Clarion, L.; Jacquard, C.; Sainte-Catherine, O.; Decoux, M.; Loiseau, S.; Rolland, M.; Lecouvey, M.; Hugnot, J.-P.; Volle, J.-N.; Virieux, D.; et al. C-Glycoside Mimetics Inhibit Glioma Stem Cell Proliferation, Migration, and Invasion. J. Med. Chem. 2014, 57, 8293–8306. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Hsung, R.P. An unusual stereoselectivity in the anomeric substitution with carbamates promoted by HNTf2. Org. Biomol. Chem. 2007, 5, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Senese, S.; Lo, Y.C.; Huang, D.; Zangle, T.A.; Gholkar, A.A.; Robert, L.; Homet, B.; Ribas, A.; Summers, M.K.; Teitell, M.A.; et al. Chemical dissection of the cell cycle: Probes for cell biology and anti-cancer drug development. Cell Death Dis. 2014, 5, e1462. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Mayer, T.U.; Miyamoto, D.T.; Fathi, R.; King, R.W.; Mitchison, T.J.; Schreiber, S.L. Dissecting cellular processes using small molecules: Identification of colchicine-like, taxol-like and other small molecules that perturb mitosis. Chem. Biol. 2000, 7, 275–286. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Fahlman, R.P. Phosphorylation impacts N-end rule degradation of the proteolytically activated form of BMX kinase. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.; Fahlman, R. The-N-end rule: The beginning determines the end. Protein Pept. Lett. 2016, 23, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The N-end rule and regulation of apoptosis. Nat. Cell Biol. 2003, 5, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.A.; Fahlman, R.P. The anti-apoptotic form of tyrosine kinase Lyn that is generated by proteolysis is degraded by the N-end rule pathway. Oncotarget 2014, 5, 2714–2722. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Liu, P.; Wang, J.; Baker, R.; Huggins, J.; Chu, C.K. Practical Synthesis of d- and l-2-Cyclopentenone and Their Utility for the Synthesis of Carbocyclic Antiviral Nucleosides against Orthopox Viruses (Smallpox, Monkeypox, and Cowpox Virus). J. Org. Chem. 2003, 68, 9012–9018. [Google Scholar] [CrossRef] [PubMed]
- Mahankali, B.; Srihari, P. A Carbohydrate Approach for the First Total Synthesis of Cochliomycin C: Stereoselective Total Synthesis of Paecilomycin E, Paecilomycin F and 6′-epi-Cochliomycin C. Eur. J. Org. Chem. 2015, 3983–3993. [Google Scholar] [CrossRef]
- Thompson, D.K.; Hubert, C.N.; Wightman, R.H. Hydroxylated pyrrolidines. Synthesis of 1,4-dideoxy-1,4-imino-l-lyxitol, 1,4,5-trideoxy-1,4-imino-d- and -l-lyxo-hexitol, 2,3,6-trideoxy-3,6-imino-d-glycero-l-altro- and -d-glycero-l-galacto-octitols, and of a chiral potential precursor of carbapenem. Tetrahedron 1993, 49, 3827–3840. [Google Scholar] [CrossRef]
- Stockwell, B.R. Chemical genetics: Ligand-based discovery of gene function. Nat. Rev. Genet. 2000, 1, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.P.; Chang, Y.-T. Chemical Genetics. Chem. Rev. 2006, 106, 2476–2530. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Sugar Derivative | Coupling Intermediate | Hexahydro-2H-furo[3,2-b][1,4]oxazine |
|---|---|---|---|
| 1 |  7 |  11α, 38%; 11β, 20% |  17, 55% |
| 2 |  8 |  12α, 32%; 12β, 31% |  18, 68% |
| 3 |  9 |  13α, 32%; 13β, traces |  19, 47% |
| 4 |  10 |  14α, 65%; 14β, 22% |  1, 76% |
| 5 |  10 |  15α, 49%; 15β, 33% | ___ |
| 6 |  10 |  16α, 50%; 16β, 22% |  20, 65% |
| Compoud | Structure | % Inhibition a |
|---|---|---|
| 1 |  | 41 ± 3 |
| 17 |  | 13 ± 1 |
| 23 |  | 13 ± 3 |
| 21 |  | 13 ± 2 |
| 18 |  | 15 ± 11 |
| 19 |  | 12 ± 6 |
| 20 |  | 10 ± 4 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenci, E.; Innocenti, R.; Biagioni, A.; Menchi, G.; Bianchini, F.; Trabocchi, A. Identification of Novel Human Breast Carcinoma (MDA-MB-231) Cell Growth Modulators from a Carbohydrate-Based Diversity Oriented Synthesis Library. Molecules 2016, 21, 1405. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21101405
Lenci E, Innocenti R, Biagioni A, Menchi G, Bianchini F, Trabocchi A. Identification of Novel Human Breast Carcinoma (MDA-MB-231) Cell Growth Modulators from a Carbohydrate-Based Diversity Oriented Synthesis Library. Molecules. 2016; 21(10):1405. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21101405
Chicago/Turabian StyleLenci, Elena, Riccardo Innocenti, Alessio Biagioni, Gloria Menchi, Francesca Bianchini, and Andrea Trabocchi. 2016. "Identification of Novel Human Breast Carcinoma (MDA-MB-231) Cell Growth Modulators from a Carbohydrate-Based Diversity Oriented Synthesis Library" Molecules 21, no. 10: 1405. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21101405