Access to New Cytotoxic Quinone-Amino Acid Conjugates Linked through A Vinylic Spacer from 2-Acylnaphthoquinones and Methyl 3-Aminocrotonate

 and
and
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Preparation of Enaminone-Amino Acid Derivatives. General Procedure
3.3. Preparation of Compounds 5a,b and 6–14. General Procedure
3.4. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thompson, R.H. Naturally Occurring Quinones IV: Recent Advances, 4th ed.; Blackie Academic & Professional: London, UK, 1997. [Google Scholar]
- Maruyama, K.; Naruta, Y. Syntheses of alpha- and beta-lapachones and their homologues by way of photochemical side chain introduction to quinone. Chem. Lett. 1977, 8, 847–850. [Google Scholar] [CrossRef]
- Uno, H.J. Allylation of 2-alkanoyl 1,4-quinones with allylsilanes and allylstannanes. Efficient synthesis of pyranonaphthoquinone antibiotics. J. Org. Chem. 1986, 51, 350–358. [Google Scholar] [CrossRef]
- Brimble, M.A.; Lynds, S.M. A short synthesis of deoxyfrenolicin. J. Chem. Soc. Perkin Trans. 1 1994, 5, 493–496. [Google Scholar] [CrossRef]
- Kraus, G.A.; Maeda, H. A direct preparation of 1,4-benzodiazepines. The synthesis of medazepam and related compounds via a common intermediate. Tetrahedron Lett. 1994, 35, 9189–9190. [Google Scholar] [CrossRef]
- Waske, P.A.; Mattay, J.; Oelgemöller, M. Photoacylations of 2-substituted 1,4-naphthoquinones: A concise access to biologically active quinonoid compounds. Tetrahedron Lett. 2006, 47, 1329–1332. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Pessoa-Mahana, D.; Tapia, R.A.; Rojas de Arias, A.; Nakayama, H.; Torres, S.; Miret, J.; Ferreira, M.E. Studies on quinones. Part 34: The reaction of styrene with activated 1,4-benzoquinones: Access to potential antiprotozoal pyranobenzoquinones. Tetrahedron 2001, 57, 8653–8658. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Benites, J.; Cortés, M.; Pessoa-Mahana, D.; Prina, E.; Fournet, A. Studies on quinones. Part 35: Access to antiprotozoal active euryfurylquinones and hydroquinones. Tetrahedron 2002, 58, 881–886. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Zamorano, C.; González, M.F.; Prina, E.; Fournet, A. Studies on quinones. Part 39: Synthesis and leishmanicidal activity of acylchloroquinones and hydroquinones. Bioorg. Med. Chem. 2005, 13, 4153–4159. [Google Scholar] [CrossRef] [PubMed]
- Valderrama, J.A.; González, M.F.; Pessoa-Mahana, D.; Tapia, R.A.; Fillion, H.; Pautet, F.; Rodríguez, J.A.; Theoduloz, C.; Schmeda-Hishmann, G. Studies on quinones. Part 41: Synthesis and cytotoxicity of isoquinoline-containing polycyclic quinones. Bioorg. Med. Chem. 2006, 14, 5003–5011. [Google Scholar] [CrossRef] [PubMed]
- Valderrama, J.A.; Vásquez, D. Design and synthesis of angucyclinone AB-pyrido[2,3-d] pyrimidine analogues. Tetrahedron Lett. 2008, 49, 703–706. [Google Scholar] [CrossRef]
- Vásquez, D.; Rodríguez, J.A.; Theoduloz, C.; Buc Calderon, P.; Valderrama, J.A. Studies on quinones. Part 46. Synthesis and in vitro antitumor evaluation of aminopyrimidoisoquinolinequinones. Eur. J. Med. Chem. 2010, 45, 5234–5242. [Google Scholar] [CrossRef]
- Delgado, V.; Ibacache, A.; Theoduloz, C.; Valderrama, J.A. Synthesis and in vitro cytotoxic evaluation of aminoquinones structurally related to marine isoquinolinequinones. Molecules 2012, 17, 7042–7056. [Google Scholar] [CrossRef] [PubMed]
- Pardo, M.; Joos, K.; Schäfer, W. Über die oxidative Aminierung von 1′,4′-Dihydroxy-2′-acetonaphthon. Liebigs Ann. Chem. 1979, 4, 503–521. [Google Scholar] [CrossRef]
- Schäfer, W.; Aguado, A.; Sezer, U. A new method of preparing heterocyclic quinones. Angew. Chem. Int. Ed. 1971, 10, 406–407. [Google Scholar] [CrossRef]
- Ríos, D.; Benites, J.; Valderrama, J.A.; Farias, M.; Pedrosa, R.C.; Verrax, J.; Buc Calderon, P. Biological evaluation of 3-acyl-2-arylamino-1,4-naphthoquinones as inhibitors of Hsp90 chaperoning function. Curr. Top. Med. Chem. 2012, 12, 2094–2102. [Google Scholar] [CrossRef] [PubMed]
- Buff, H.; Kuckländer, U. Reaction of N-methyl-hydrazones as azaenamines with quinones. Tetrahedron 2000, 56, 5137–5145. [Google Scholar] [CrossRef]
- Allen, G.R., Jr.; Weiss, M.J. Behavior of 2-carbomethoxy- and 2-acetyl-1,4-benzoquinone in the Nenitzescu indole syntheses. J. Org. Chem. 1968, 33, 198–200. [Google Scholar] [CrossRef]
- Valderrama, J.A.; González, M.F.; Colonelli, P.; Vásquez, D. Design and synthesis of angucyclinone 5-Aza analogues. Synlett 2006, 17, 2777–2780. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Ríos, D.; Muccioli, G.G.; Buc Calderon, P.; Brito, I.; Benites, J. Hetero-annulation reaction between 2-acylnaphthoquinones and 2-aminobenzothiazoles. A new synthetic route to antiproliferative benzo[g]benzothiazolo[2,3-b]quinazoline-7,12-quinones. Tetrahedron Lett. 2015, 56, 5103–5105. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Cabrera, M.; Benites, J.; Ríos, D.; Inostroza-Rivera, R.; Muccioli, G.G. Synthetic approaches and in vitro cytotoxic evaluation of 2-acyl-3-(3,4,5-trimethoxyanilino)-1,4-naphthoquinones. RSC Adv. 2017, 7, 24813–24821. [Google Scholar] [CrossRef]
- Vasquez, D.; Rodriguez, J.A.; Theoduloz, C.; Verrax, J.; Buc Calderon, P.; Valderrama, J.A. Synthesis and antitumor evaluation of 8-phenylaminopyrimido[4,5-c]isoquinolinequinones. Bioorg. Med. Chem. 2009, 19, 5060–5062. [Google Scholar] [CrossRef] [PubMed]
- Janáky, T.; Juhász, A.; Bajusz, S.; Csernus, V.; Srkalovic, G.; Bokser, L.; Milovanovic, S.; Redding, T.W.; Rékási, Z.; Nagy, A. Analogues of luteinizing hormone-releasing hormone containing cytotoxic groups. Proc. Natl. Acad. Sci. USA 1992, 89, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Rahimipour, S.; Weiner, L.; Fridkin, M.; Bade Shrestha-Dawadi, P.; Bittner, S. Novel naphthoquinonyl derivatives: Potential structural components for the synthesis of cytotoxic peptides. Lett. Peptide. Sci. 1996, 3, 263–274. [Google Scholar] [CrossRef]
- Rahimipour, S.; Weiner, L.; Bade Shrestha-Dawadi, P.; Bittner, S.; Koch, Y.; Fridkin, M. Cytotoxic peptides: Naphthoquinonyl derivatives of luteinizing hormone-releasing hormone. Lett. Peptide Sci. 1998, 5, 421–427. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Huang, L.; Sakhuja, R. Efficient syntheses of naphthoquinone-dipeptides. Synthesis 2010, 12, 2011–2016. [Google Scholar] [CrossRef]
- de Moraes, T.A.P.; Filha, M.J.S.; Camara, C.A.; Silva, T.M.S.; Soares, B.M.; Bomfim, I.S.; Pessoa, C.; Ximenes, G.C.; Silva, V.A., Jr. Synthesis and cytotoxic Evaluation of a series of 2-amino-naphthoquinones against human cancer cells. Molecules 2014, 19, 13188–13199. [Google Scholar] [CrossRef] [PubMed]
- Valderrama, J.A.; Delgado, V.; Sepúlveda, S.; Benites, J.; Theoduloz, C.; Buc Calderon, P.; Muccioli, G.G. Synthesis and Cytotoxic Activity on human cancer cells of novel isoquinolinequinone–Amino acid derivatives. Molecules 2016, 21, 1199–2013. [Google Scholar] [CrossRef] [PubMed]
- Alnabari, M.; Bittner, S. New quinone-amino acid conjugates linked via a vinylic spacer. Amino Acids 2001, 20, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds 5a, 7 and 9 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
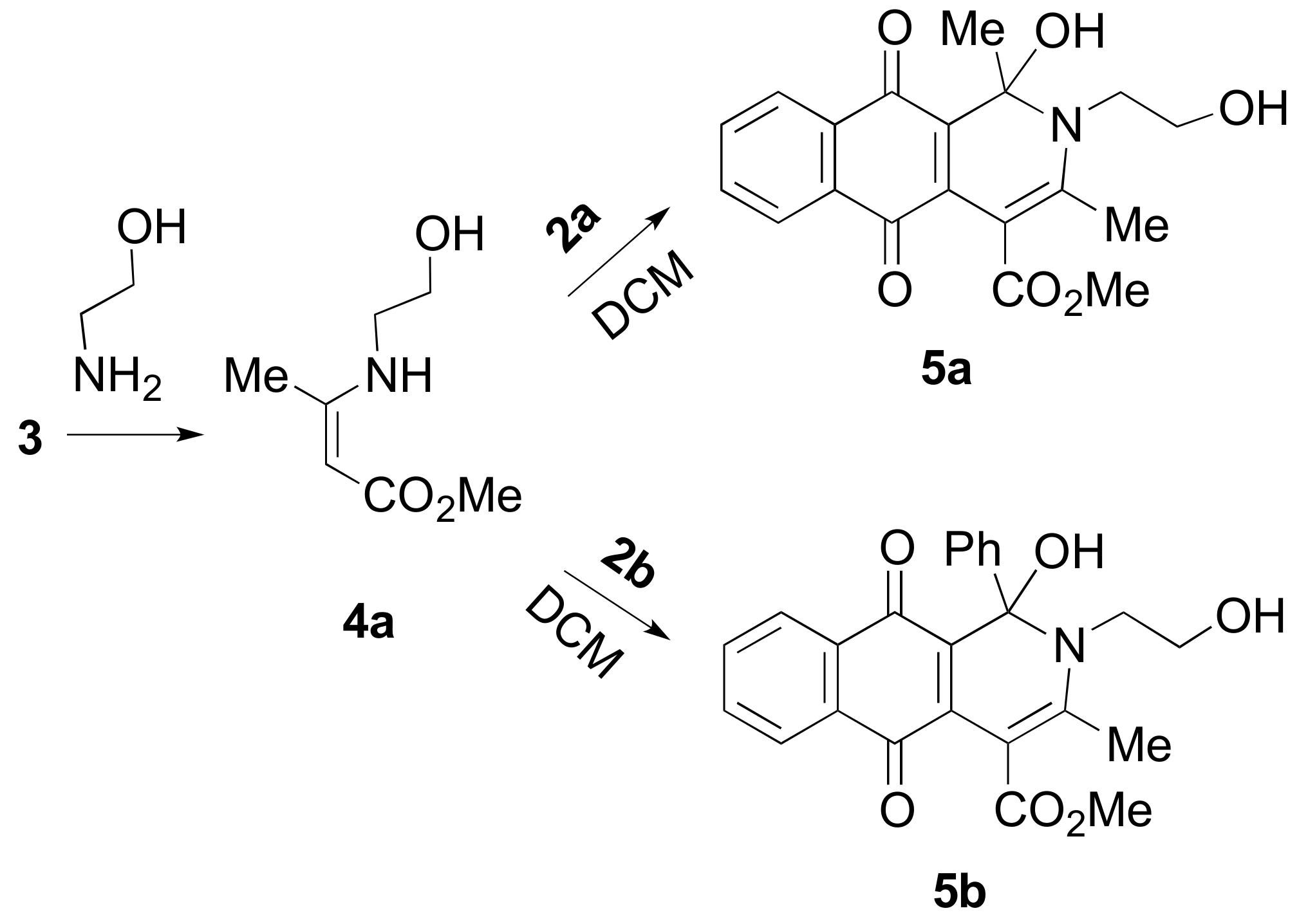{kind=link}
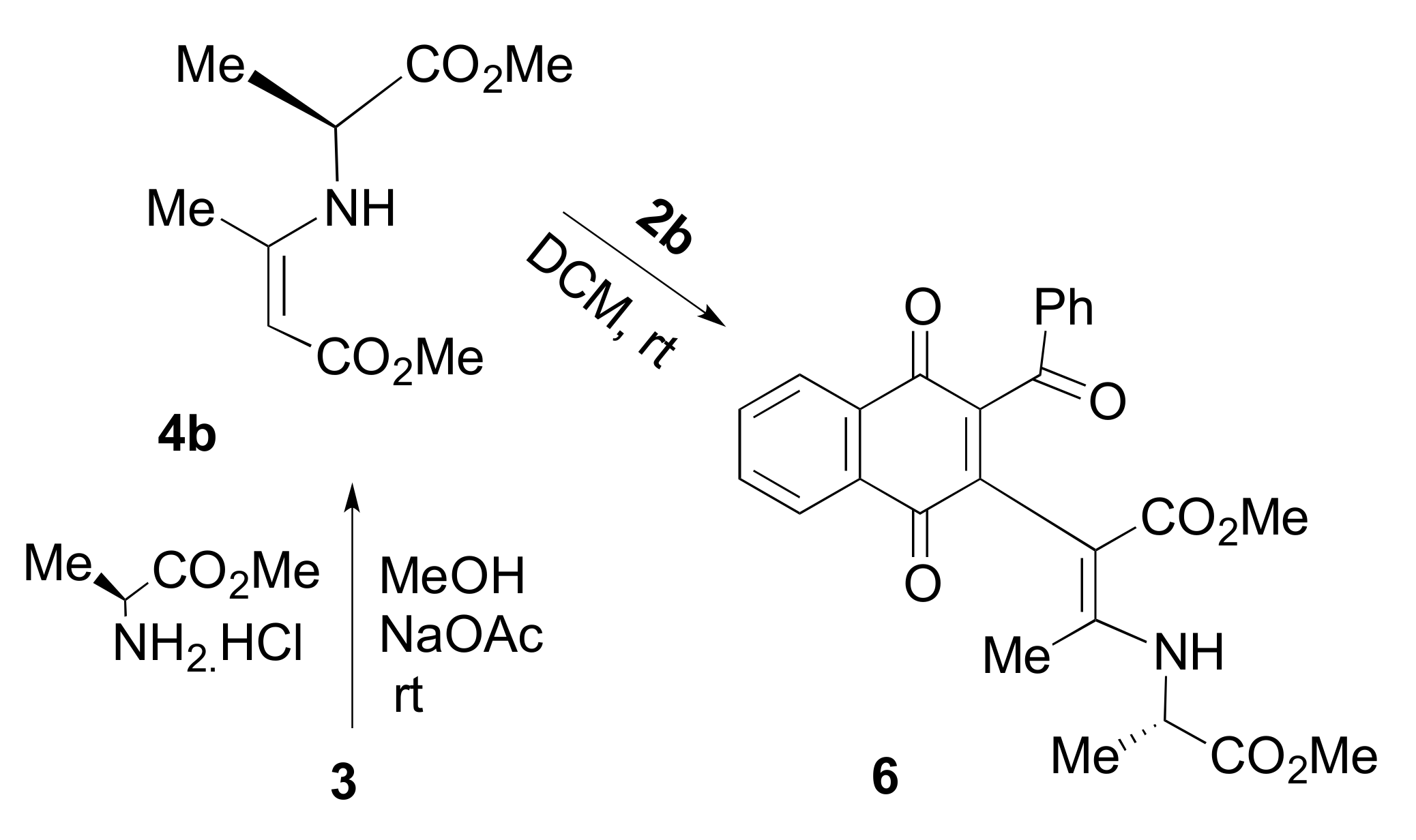{kind=link}

| Compound No. | Structure | Yield (%) * | Compound No. | Structure | Yield (%) * |
|---|---|---|---|---|---|
| 4b |  | 71 | 4e |  | 83 |
| 4c |  | 85 | 4f |  | 76 |
| 4d |  | 80 | 4g |  | 84 |

| Amino Acid | R1 | R2 | Products | Yield * (%) |
|---|---|---|---|---|
| l-Ala | Ph | CH3 | 6 | 58 |
| d-Ala | Ph | CH3 | 7 | 52 |
| l-Leu | Ph | (CH3)2CHCH2 | 8 | 42 |
| l-Phe | Ph | PhCH2 | 9 | 65 |
| d-Phe | Ph | PhCH2 | 10 | 58 |
| l-Trp | Ph | 3-IndolylCH2 | 11 | 71 |
| d-Phe | C3H7 | PhCH2 | 12 | 45 |
| l-Leu | C5H11 | (CH3)2CHCH2 | 13 | 40 |
| l-Trp | C5H11 | 3-IndolylCH2 | 14 | 58 |
| Product Number | MRC-5 b | AGS c | SK-MES-1 d | J82 e |
|---|---|---|---|---|
| 6 | 58.0 ± 4.1 | 52.7 ± 3.7 | 47.8 ± 3.3 | 37.1 ± 2.2 |
| 7 | 17.4 ± 0.7 | 20.5 ± 1.1 | 53.4 ± 4.3 | 35.6 ± 1.4 |
| 8 | 26.6 ± 1.3 | 26.9 ± 1.5 | 34.5 ± 1.7 | 12.9 ± 0.9 |
| 9 | 33.3 ± 2.2 | 19.3 ± 1.5 | 39.3 ± 1.6 | 22.0 ± 1.1 |
| 10 | 46.7 ± 2.9 | 30.7 ± 1.5 | 34.2 ± 2.3 | 23.0 ± 0.7 |
| 11 | 13.0 ± 0.8 | 10.1 ± 0.1 | 18.9 ± 0.5 | 21.4 ± 0.8 |
| 12 | 21.8 ± 1.5 | 12.8 ± 0.7 | 15.9 ± 0.9 | 14.0 ± 0.6 |
| 13 | 24.9 ± 0.7 | 17.0 ± 1.1 | 21.6 ± 1.3 | 23.5 ± 1.6 |
| 14 | 22.1 ± 1.6 | 9.3 ± 0.6 | 5.5 ± 0.2 | 4.5 ± 0.2 |
| Ref. f | 2.2 ± 0.1 | 0.4 ± 0.0 | 2.9 ± 0.2 | 3.3 ± 0.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valderrama, J.A.; Garrido, J.; Delgado, V.; Benites, J.; Theoduloz, C. Access to New Cytotoxic Quinone-Amino Acid Conjugates Linked through A Vinylic Spacer from 2-Acylnaphthoquinones and Methyl 3-Aminocrotonate. Molecules 2017, 22, 2281. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22122281
Valderrama JA, Garrido J, Delgado V, Benites J, Theoduloz C. Access to New Cytotoxic Quinone-Amino Acid Conjugates Linked through A Vinylic Spacer from 2-Acylnaphthoquinones and Methyl 3-Aminocrotonate. Molecules. 2017; 22(12):2281. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22122281
Chicago/Turabian StyleValderrama, Jaime A., Joel Garrido, Virginia Delgado, Julio Benites, and Cristina Theoduloz. 2017. "Access to New Cytotoxic Quinone-Amino Acid Conjugates Linked through A Vinylic Spacer from 2-Acylnaphthoquinones and Methyl 3-Aminocrotonate" Molecules 22, no. 12: 2281. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22122281