Novel Topologically Complex Scaffold Derived from Alkaloid Haemanthamine

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
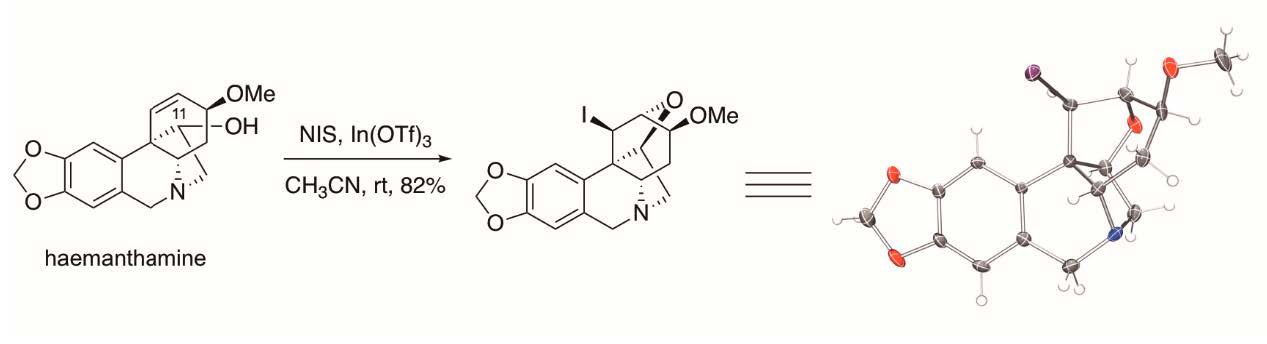
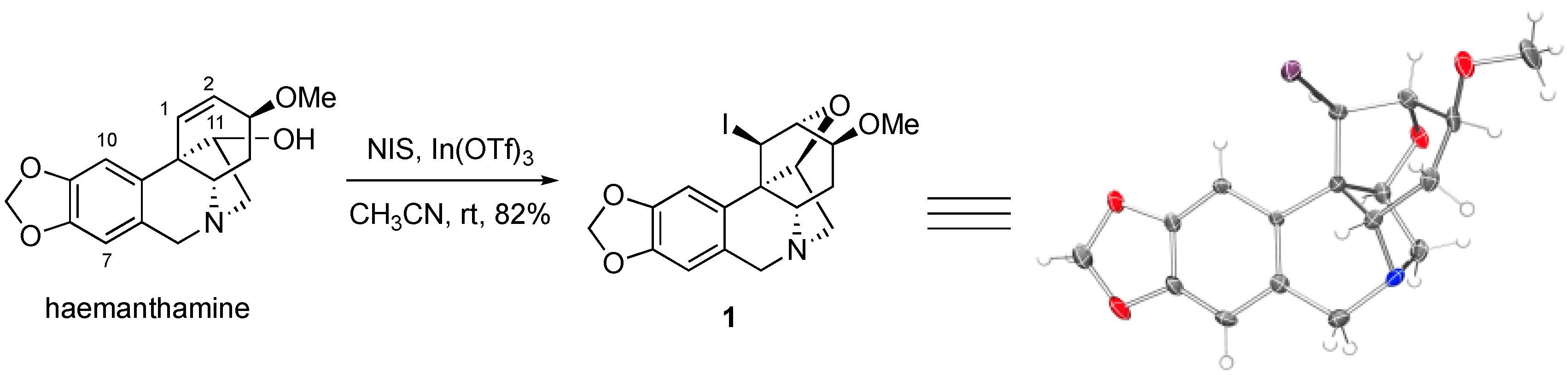
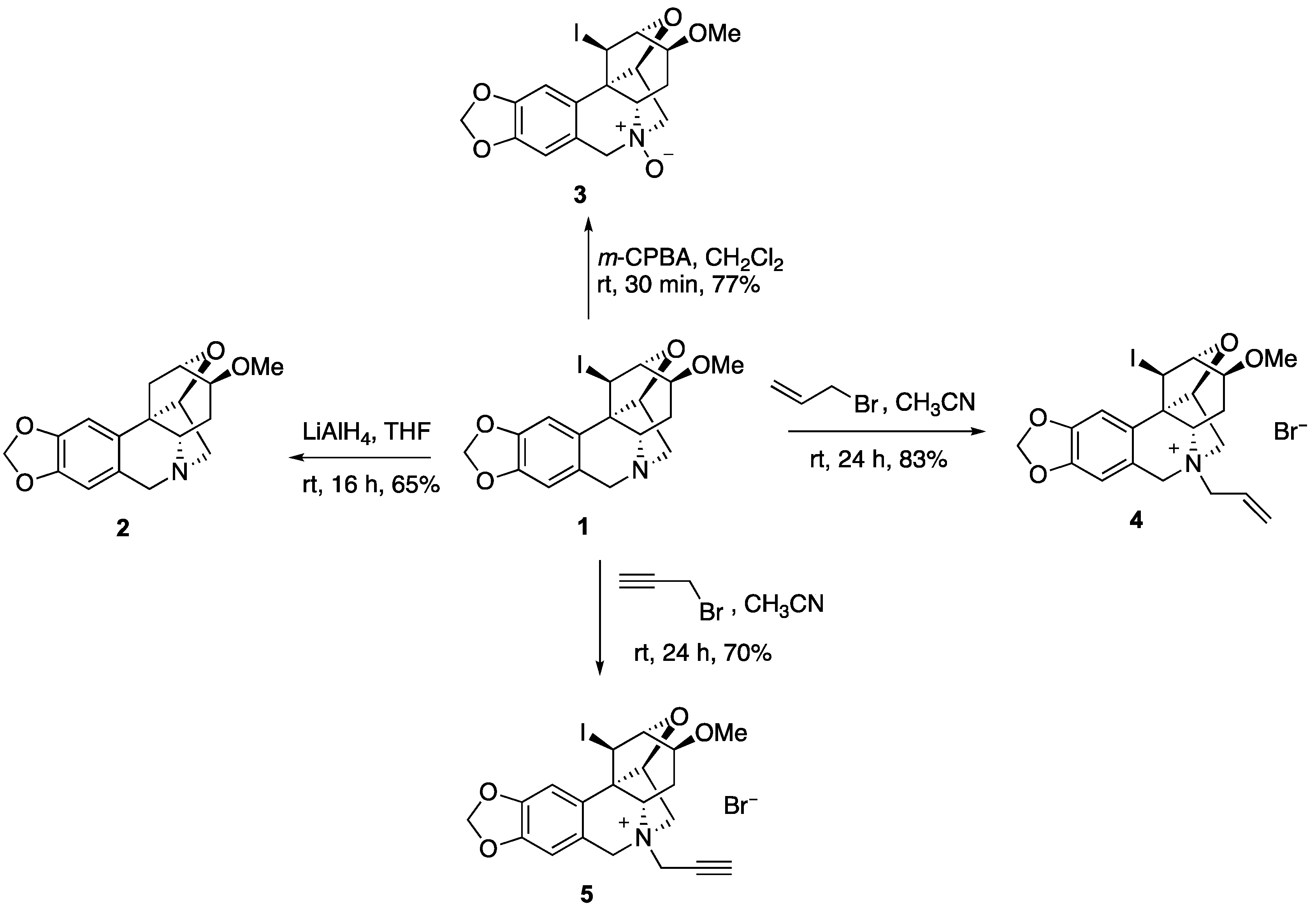
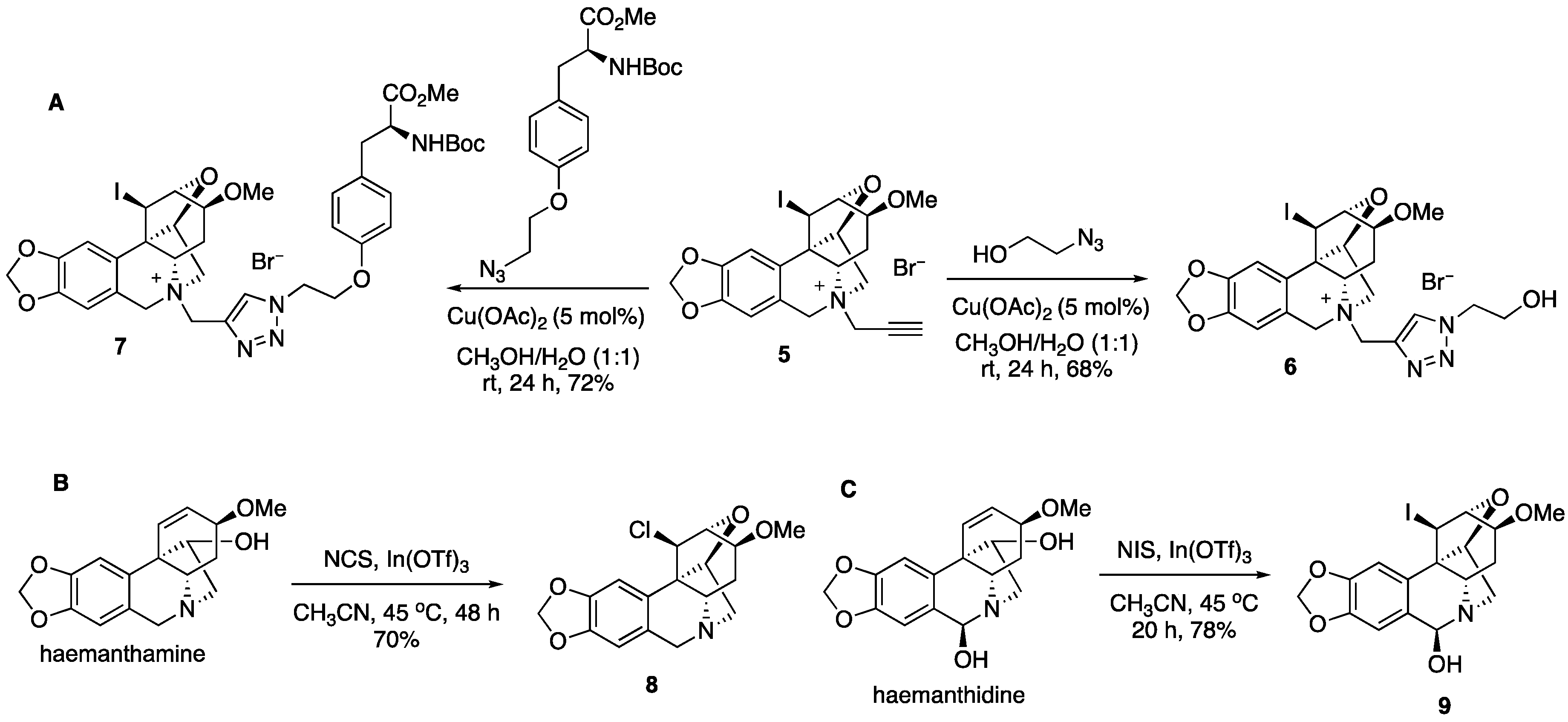
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Crystallographic Information
3.3. Experimental Procedures
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Galloway, W.R.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Castro, M.; Zimmerman, S.; Sankar, M.G.; Kumar, K. Scaffold diversity synthesis and its application in probe and drug discovery. Angew. Chem. Int. Ed. 2016, 55, 7586–7605. [Google Scholar] [CrossRef] [PubMed]
- McLeod, M.C.; Singh, G.; Plampin, J.N., III; Rane, D.; Wang, J.L.; Day, V.W.; Aube, J. Probing chemical space with alkaloid-inspired libraries. Nat. Chem. 2014, 6, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Hao, J.; Ulanovkaya, O.A.; Dundas, J.; Liang, J.; Kozmin, S.A. Creation and manipulation of common functional groups en route to a skeletally diverse chemical library. PNAS 2011, 108, 6763–6768. [Google Scholar] [CrossRef] [PubMed]
- Borzilleri, R.M.; Vite, G.D. Case history: Discovery of ixabepilone (IXEMPRA™), a first-in-class epothilone analogue for treatment of metastatic breast cancer. Annu. Rep. Med. Chem. 2009, 44, 301–322. [Google Scholar]
- Fasolo, A.; Sessa, C. Current and future directions in mammalian target of rapamycin inhibitor development. Expert Opin. Investig. Drugs. 2011, 20, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.C.; Hergenrother, P.J. Natural products as starting points for the synthesis of complex and diverse compounds. Nat. Prod. Rep. 2014, 31, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Pelish, H.E.; Westwood, N.J.; Feng, Y.; Kirchhausen, T.; Shair, M.D. Use of biomimetic diversity-oriented synthesis to discover galanthamine-like molecules with biological properties beyond those of natural product. J. Am. Chem. Soc. 2001, 123, 6740–6741. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kiuchi, M.; Tallarico, J.A.; Schreiber, S.L. Small-molecule diversity using a skeletal transformation strategy. Org. Lett. 2005, 7, 2535–2538. [Google Scholar] [CrossRef] [PubMed]
- Balthaser, B.R.; Maloney, M.C.; Beeler, A.B.; Porco, J.A., Jr.; Snyder, J.K. Remodeling of the natural product fumagillol employing a reaction discovery approach. Nat. Chem. 2011, 3, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Appendino, G.C.; Tron, G.C.; Jarevang, T.; Sterner, O. Unnatural natural products from the transannular cyclization of lathyrane diterpenes. Org. Lett. 2001, 3, 1609–1612. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, M.C.; Garcia, I.; Sierra, M.A. Diversity oriented synthesis of hispanane-like terpene derivatives from (R)-(+)-scareolide. Chem. Eur. J. 2005, 11, 3659–3667. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, T.; Iwabuchi, R.; Minagawa, S.; Shiomi, F.; Cappiello, J.; Sawada, S.; Utsumi, R.; Okamoto, T. Unprecedented olefin-dependent histidine-kinase inhibitory of zerumbone ring-opening material. Bioorg. Med. Chem. Lett. 2004, 14, 5943–5946. [Google Scholar] [CrossRef] [PubMed]
- Huisgens, R.W., III; Morrison, K.C.; Hicklin, R.W.; Flood, T.A., Jr.; Richter, M.F.; Hergenrother, P.J. A ring-distortion strategy to construct stereochemically complex and structurally diverse compounds from natural products. Nat. Chem. 2013, 5, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Van Goietsenoven, G.; Andolfi, A.; Lallemand, B.; Cimmino, A.; Lamoral-Theys, D.; Gras, T.; Abou-Donia, A.; Dubois, J.; Lefranc, F.; Mathieu, V.; et al. Amaryllidaceae alkaloids belonging to different structural subgroups display activity against apoptosis-resistant cancer cells. J. Nat. Prod. 2010, 73, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Cedron, J.C.; Gutierrez, D.; Flores, N.; Ravelo, A.G.; Estevez-Braun, A. Synthesis and antimalarial activity of new haemanthamine-type derivatives. Bioorg. Med. Chem. 2012, 20, 5464–5472. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.-Y.; Li, J.; Peddibholta, S.; Romo, D. Mild arming and derivatization of natural products via an In(OTf)3-catalyzed arene iodination. Org. Lett. 2010, 12, 2104–2107. [Google Scholar] [CrossRef] [PubMed]
- Cedron, J.C.; Estevez-Braun, A.; Ravelo, A.G.; Guiterez, D.; Flores, N.; Bucio, M.A.; Perez-Hernandez, N.; Joseph-Nathan, P. Bioactive montanine derivatives from halide-induced rearrangements of haemanthamine-type Alkaloids. Absolute configuration by VCD. Org. Lett. 2009, 11, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Inubushi, Y.; Fales, H.M.; Warnhoff, E.W.; Wildman, W.C. Structures of montanine, coccinine, and manthine. J. Org. Chem. 1960, 25, 2153–2164. [Google Scholar] [CrossRef]
- Pellegrino, S.; Meyer, M.; Zorbas, C.; Bouchta, S.A.; Saraf, K.; Pelly, S.C.; Yusupova, G.; Evidente, A.; Mathieu, V.; Kornienko, A.; et al. The Amaryllidacae alkaloid haemanthamine binds the eukaryotic ribosome to repress cancer cell growh. Structure 2018, in press. [Google Scholar]
Sample Availability: Alkaloid haemanthamine is available from the authors. |



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govindaraju, K.; Masi, M.; Colin, M.; Mathieu, V.; Evidente, A.; Hudnall, T.W.; Kornienko, A. Novel Topologically Complex Scaffold Derived from Alkaloid Haemanthamine. Molecules 2018, 23, 255. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23020255
Govindaraju K, Masi M, Colin M, Mathieu V, Evidente A, Hudnall TW, Kornienko A. Novel Topologically Complex Scaffold Derived from Alkaloid Haemanthamine. Molecules. 2018; 23(2):255. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23020255
Chicago/Turabian StyleGovindaraju, Karthik, Marco Masi, Margaux Colin, Veronique Mathieu, Antonio Evidente, Todd W. Hudnall, and Alexander Kornienko. 2018. "Novel Topologically Complex Scaffold Derived from Alkaloid Haemanthamine" Molecules 23, no. 2: 255. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23020255