
1,2,6-Thiadiazinones as Novel Narrow Spectrum Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) Inhibitors

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Results
2.1. Synthesis
2.2. Initial Kinase Profiling
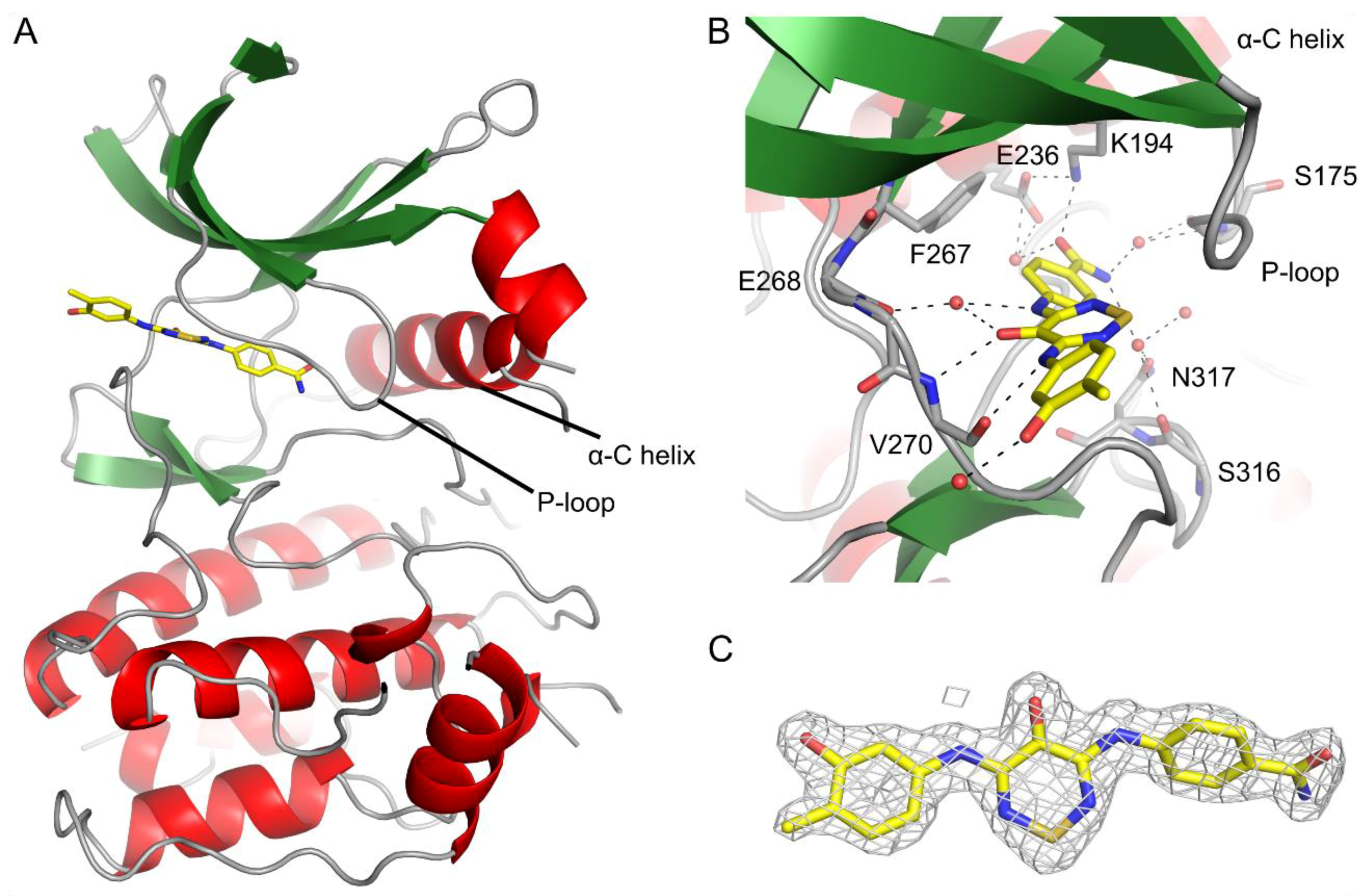
2.3. CaMKK2 Crystallography
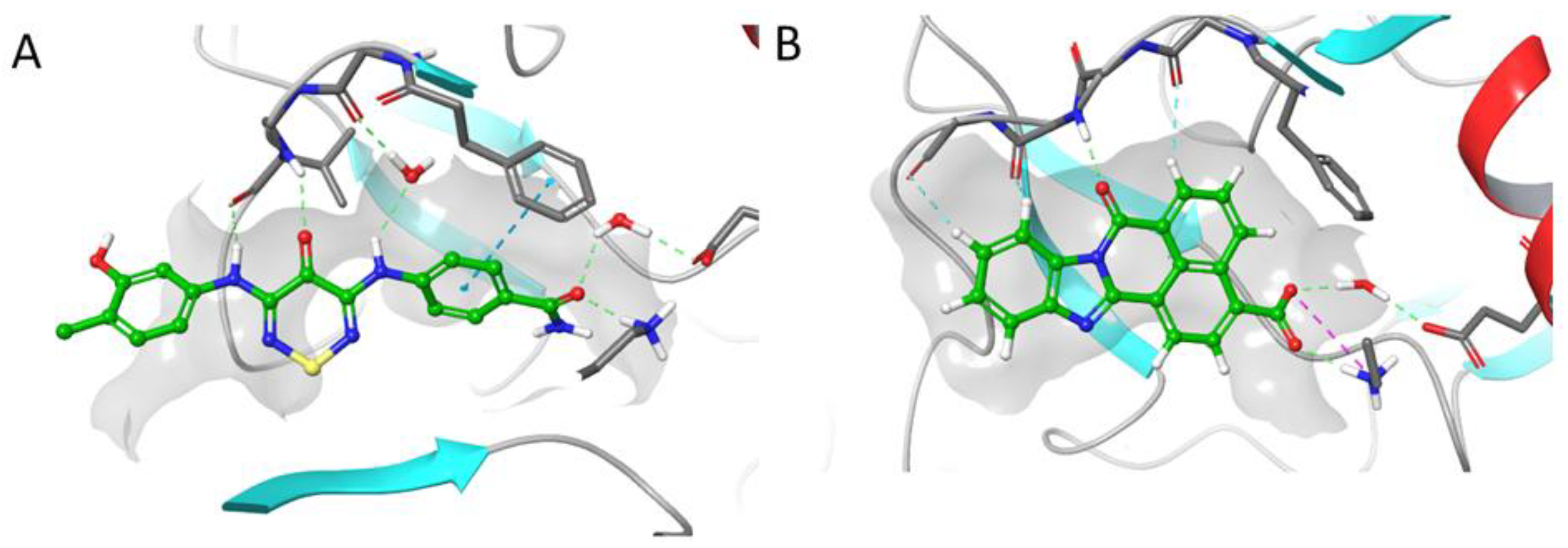
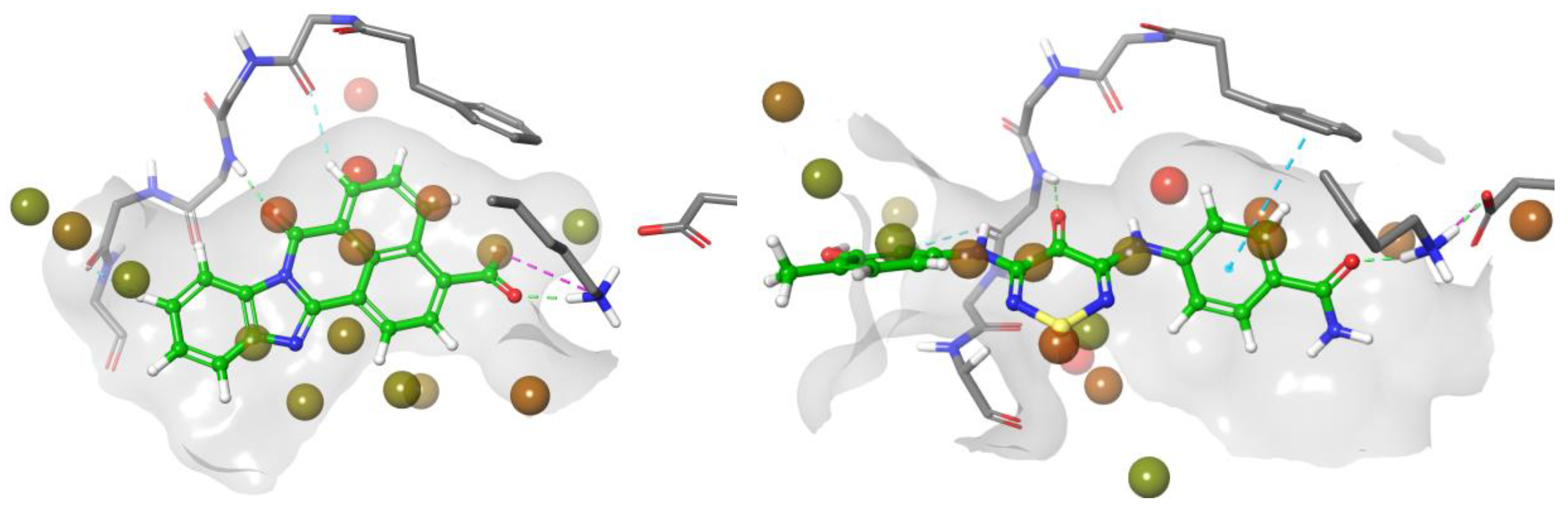
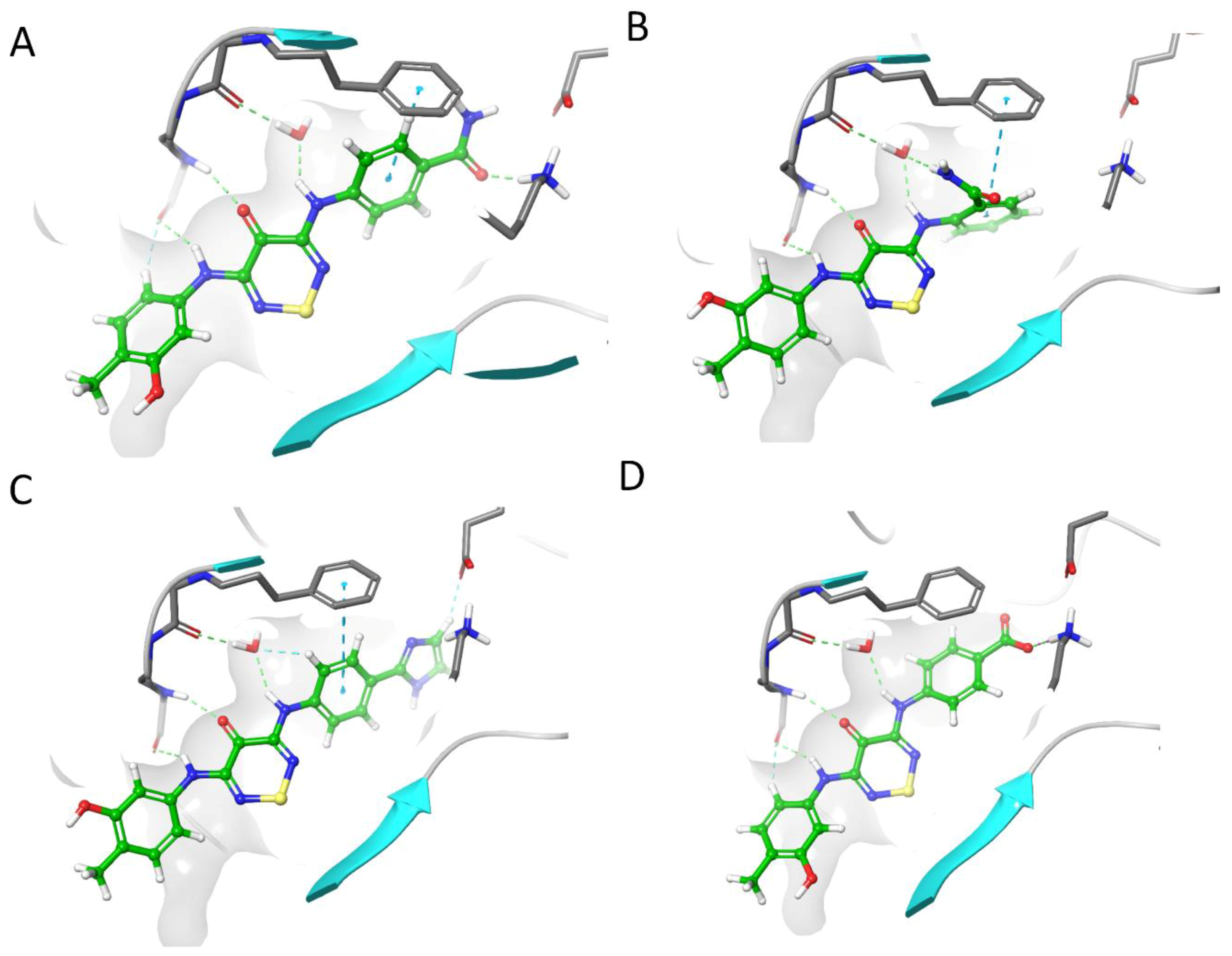
2.4. CaMKK2 Docking and Water Map Simulation
2.5. Optimisation Results on CaMKK2
2.5.1. Outline of Compounds
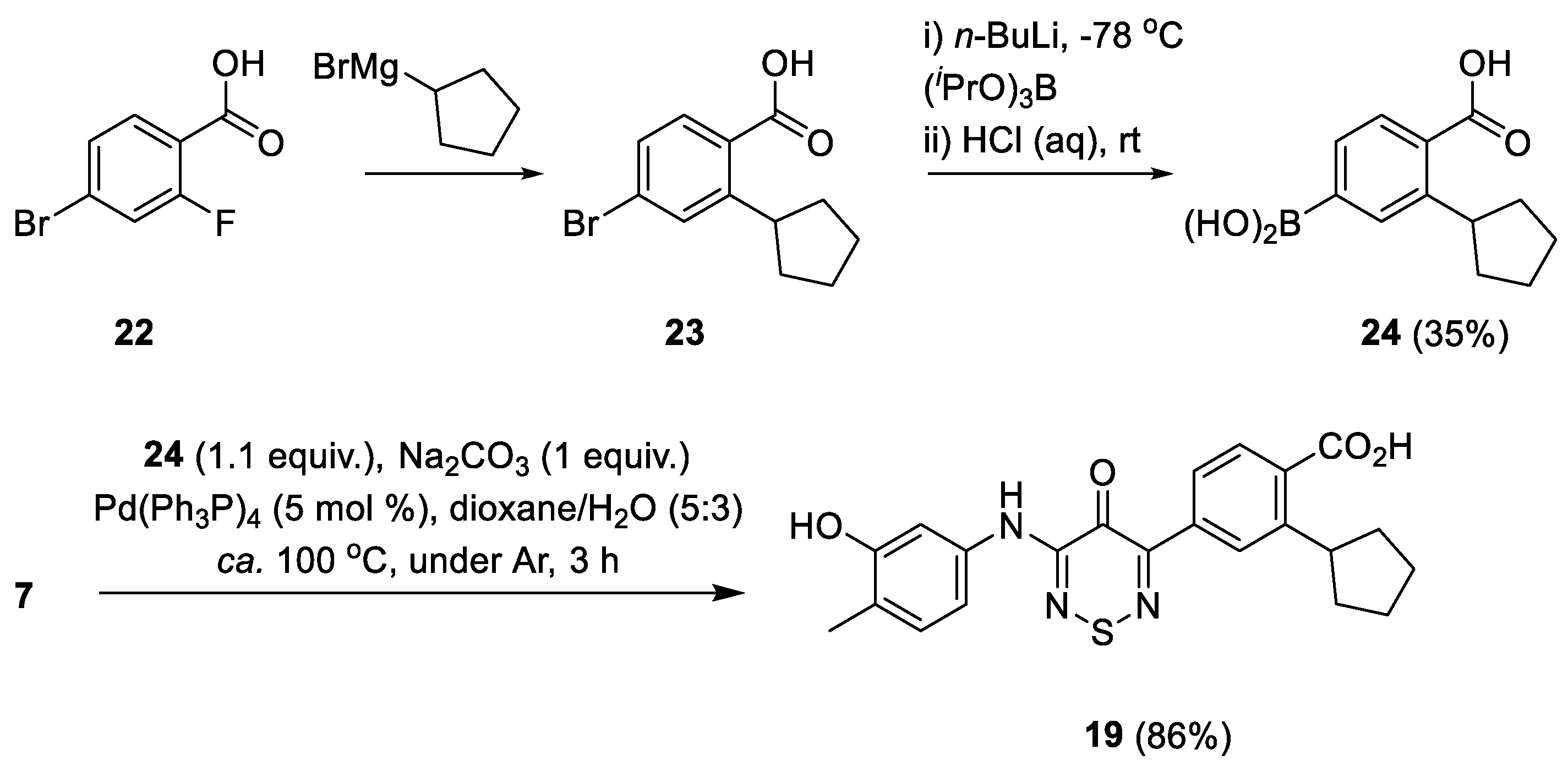
2.5.2. Analogue Synthesis
2.6. Optimization Results on CaMKK2
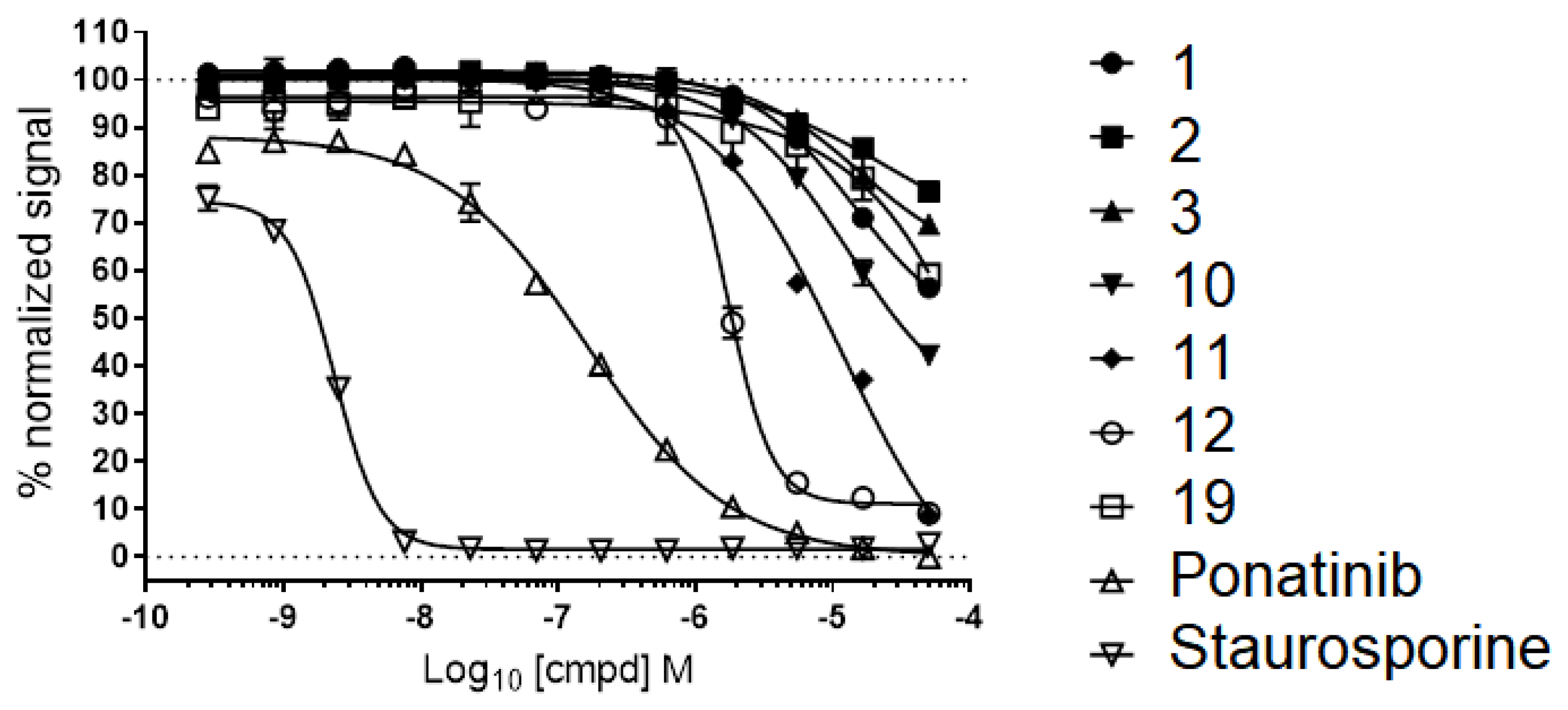
2.7. Advanced Enzyme Assay Results on CaMKK2 Demonstrating Functional Inhibition
3. Discussion
4. Materials and Methods
4.1. Kinase Panel
4.2. CaMKK2 Crystallization
4.2.1. Cloning, Protein Expression and Purification
4.2.2. Protein Crystallization
4.2.3. Structure Solution and Refinement
4.3. Molecular Modelling
4.3.1. Molecular Modelling
4.3.2. Hydration Site Analysis
4.4. Biochemical Assays
4.4.1. CaMKK2B TR-FRET Assay
4.4.2. CaMKK2 Enzyme Assay
4.5. Chemistry Experimental Section
4.5.1. General Methods and Materials
4.5.2. Preparation of Aniline Starting Materials
4.5.3. Preparation of 3-Amino-Substituted-4H-1,2,6-Thiadiazines
4.5.4. Preparation of 3,5-Diaminosubstituted Thiadiazines
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brognard, J.; Hunter, T. Protein Kinase Signalling Networks in Cancer. Curr. Opin. Genet. Dev. 2011, 21, 4–11. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. New drugs at FDA: CDER’s new molecular entities and new therapeutic biological products. Available online: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/ DrugInnovation/default.htm (accessed on 23 April 2018).
- Cohen, P.; Alessi, D.R. Kinase drug discovery—What’S next in the field? ACS Chem. Biol. 2013, 8, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.A.; Biggs, W.H.; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Müller, S.; Knapp, S. The (un)targeted cancer kinome. Nat. Chem. Biol. 2010, 6, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Arruda, P.; Blagg, J.; Burley, S.; Drewry, D.H.; Edwards, A.; Fabbro, D.; Gillespie, P.; Gray, N.S.; Kuster, B.; et al. A public-private partnership to unlock the untargeted kinome. Nat. Chem. Biol. 2013, 9, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Geevers, J.; Trompen, W.P. Synthesis and reactions of 3,5-dichloro-4H-1,2,6-thiadiazin-4-one. Recl. Trav. Chim. Pays-Bas 1974, 93, 270–272. [Google Scholar] [CrossRef]
- Ioannidou, H.A.; Kizas, C.; Koutentis, P.A. Palladium Catalyzed C–C Coupling Reactions of 3,5-Dichloro-4H-1,2,6-thiadiazin-4-one. Org. Lett. 2011, 13, 3466–3469. [Google Scholar] [CrossRef] [PubMed]
- Kalogirou, A.S.; Koutentis, P.A. A Qualitative Comparison of the Reactivities of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine and 4,5-Dichloro-1,2,3-dithiazolium Chloride. Molecules 2015, 20, 14576–14594. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Elkins, J.M.; Fedele, V.; Szklarz, M.; Abdul Azeez, K.R.; Salah, E.; Mikolajczyk, J.; Romanov, S.; Sepetov, N.; Huang, X.P.; Roth, B.L.; et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016, 34, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Drewry, D.H.; Wells, C.I.; Andrews, D.M.; Angell, R.; Al-Ali, H.; Axtman, A.D.; Capuzzi, S.J.; Elkins, J.M.; Ettmayer, P.; Frederiksen, M.; et al. Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS ONE 2017, 12, e0181585. [Google Scholar] [CrossRef] [PubMed]
- Koutentis, P.A.; Rees, C.W. Cyclisation chemistry of 4H-1,2,6-thiadiazines. J. Chem. Soc. Perkin Trans. 1 2000, 2601–2607. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A. Pd-catalyzed C-N Coupling of Primary (Het)arylamines with 5-Substituted 3-Chloro-4H-1,2,6-thiadiazin-4-ones. Tetrahedron Lett. 2018. submitted. [Google Scholar]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Madureira, P.A.; Ferreira, B.; Baptista, I.; Machado, S.; Colaço, L.; dos Santos, M.; Liu, N.; Dopazo, A.; Ugurel, S.; et al. TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat. Commun. 2017, 8, 14687. [Google Scholar] [CrossRef] [PubMed]
- Kukimoto-Niino, M.; Yoshikawa, S.; Takagi, T.; Ohsawa, N.; Tomabechi, Y.; Terada, T.; Shirouzu, M.; Suzuki, A.; Lee, S.; Yamauchi, T.; et al. Crystal structure of the Ca²⁺/calmodulin-dependent protein kinase kinase in complex with the inhibitor STO-609. J. Biol. Chem. 2011, 286, 22570–22579. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.; Washburn, D.G.; Hoang, H.T.; Manns, S.; Frazee, J.S.; Nakamura, H.; Patterson, J.R.; Trizna, W.; Wu, C.; Azzarano, L.M.; et al. Design and synthesis of orally bioavailable serum and glucocorticoid-regulated kinase 1 (SGK1) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4441–4445. [Google Scholar] [CrossRef] [PubMed]
- LanthaScreen® Kinase Binding Assay User Guide. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/LanthaScreen_KinaseBinding_Assay_man.pdf (accessed on 23 April 2018).
- Scott, J.W.; Park, E.; Rodriguiz, R.M.; Oakhill, J.S.; Issa, S.M.A.; O’Brien, M.T.; Dite, T.A.; Langendorf, C.G.; Wetsel, W.C.; Means, A.R.; et al. Autophosphorylation of CaMKK2 generates autonomous activity that is disrupted by a T85S mutation linked to anxiety and bipolar disorder. Sci. Rep. 2015, 5, 14436. [Google Scholar] [CrossRef] [PubMed]
- Hermerschmidt, F.; Kalogirou, A.S.; Min, J.; Zissimou, G.A.; Tuladhar, S.M.; Ameri, T.; Faber, H.; Itskos, G.; Choulis, S.A.; Anthopoulos, T.D.; et al. 4H-1,2,6-Thiadiazin-4-one-containing small molecule donors and additive effects on their performance in solution-processed organic solar cells. J. Mater. Chem. C 2015, 3, 2358–2365. [Google Scholar] [CrossRef]
- Racioppi, L.; Means, A.R. Calcium/Calmodulin-dependent Protein Kinase Kinase 2: Roles in Signaling and Pathophysiology. J. Biol. Chem. 2012, 287, 31658–31665. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Marcelo, K.L.; Rajapakshe, K.; Coarfa, C.; Dean, A.; Wilganowski, N.; Robinson, H.; Sevick, E.; Bissig, K.-D.; Goldie, L.C.; et al. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology 2015, 62, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Drewry, D.H.; Schaller, L.T.; Thompson, B.D.; Reid, P.R.; Maloney, P.R.; Liang, X.; Banker, P.; Buckholz, R.G.; Selley, P.K.; et al. Bioorg. Med. Chem. Lett. 2018. [CrossRef]
- Racioppi, L. CaMKK2: A novel target for shaping the androgen-regulated tumor ecosystem. Trends Mol Med. 2013, 19, 83–88. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.T.; Oakhill, J.S.; Ling, N.X.Y.; Langendorf, C.G.; Hoque, A.; Dite, T.A.; Means, A.R.; Kemp, B.E.; Scott, J.W. Impact of Genetic Variation on Human CaMKK2 Regulation by Ca2+-Calmodulin and Multisite Phosphorylation. Sci. Rep. 2017, 7, 43264. [Google Scholar] [CrossRef] [PubMed]
- Levine, Y.C.; Li, G.K.; Michel, T. Agonist-modulated Regulation of AMP-activated Protein Kinase (AMPK) in Endothelial Cells. Evidence for an AMPK→Rac1→Akt→Endothelial Nitric-oxide synthase pathway. J. Biol. Chem. 2007, 282, 20351–20364. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Inuzuka, H.; Ishikawa, Y.; Kobayashi, R. A Single Amino Acid Difference between α and β Ca2+/Calmodulin-dependent Protein Kinase Kinase Dictates Sensitivity to the Specific Inhibitor, STO-609. J. Biol. Chem. 2003, 278, 10908–10913. [Google Scholar] [CrossRef] [PubMed]
- Kinase Profiling Inhibitor Database. Available online: http://www.kinase-screen.mrc.ac.uk/screening-compounds/348780 (accessed on 23 April 2018).
- Monteiro, P.; Gilot, D.; Langouet, S.; Fardel, O. Activation of the aryl hydrocarbon receptor by the calcium/calmodulin-dependent protein kinase kinase inhibitor 7-oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid (STO-609). Drug Metab. Dispos. 2008, 36, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.D.; Sherman, W.; Farid, R. Understanding kinase selectivity through energetic analysis of binding site waters. Chem. Med. Chem. 2010, 5, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Kohlmann, A.; Zhu, X.; Dalgarno, D. Application of MM-GB/SA and WaterMap to SRC Kinase Inhibitor Potency Prediction. ACS Med. Chem. Lett. 2012, 3, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Higgs, C.; Beuming, T.; Sherman, W. Hydration Site Thermodynamics Explain SARs for Triazolylpurines Analogues Binding to the A2A Receptor. ACS Med. Chem. Lett. 2010, 1, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Newman, J. Novel buffer systems for macromolecular crystallization. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Small-Molecule Drug Discovery Suite 2018-1; Schrödinger, LLC: New York, NY, USA, 2018.
- Keates, T.; Cooper, C.D.O.; Savitsky, P.; Allerston, C.K.; Phillips, C.; Hammarström, M.; Daga, N.; Berridge, G.; Mahajan, P.; Burgess-Brown, N.A.; Müller, S.; Gräslund, S.; Gileadi, O. Expressing the human proteome for affinity proteomics: Optimising expression of soluble protein domains and in vivo biotinylation. N. Biotechnol. 2012, 29, 515–525. [Google Scholar] [CrossRef] [PubMed]
- LanthaScreen™ Eu kinase binding assay for CAMKK2. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/CAMKK2_LanthaScreen_Binding.pdf (accessed on 23 April 2018).
- Harwood, L.M. “Dry-Column” Flash Chromatography. Aldrichimica Acta 1985, 18, 25. [Google Scholar]
Sample Availability: Samples of the compounds 1–5 and 10–19 are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Ar1 | Ar2 | Number and Kinases Hit DSF > 2 °C a | |
| 1 |  |  | 1 | TRIB2 |
| 2 |  |  | 16 | CAMK1D/G/K1/K2B, CDC42BPA, CDK2 |
| CHEK2, DYRK2, MAP2K7, PHKG2 | ||||
| PIM1, PKMYT1, RPSKA6, STK3, STK17, TTK | ||||
| 3 |  |  | 9 | CAMK1G/2B, CDC42BPA, |
| CLK1, MAP2K7, PIM1, | ||||
| STK10, TRIB2, TTK | ||||
| 4 |  |  | 0 | |
| - | ||||
| 5 |  |  | 0 | |
| - | ||||
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | FRET (IC50) a | Compound | R1 | FRET (IC50) a |
| 1 |  | >50 | 14 |  | >50 |
| 2 |  | >50 | 15 |  | >50 |
| 3 |  | 34 | 16 |  | >50 |
| 10 |  | 7.8 | 17 |  | >50 |
| 11 |  | 3.2 | 18 |  | >50 |
| 12 |  | 10.5 | 19 |  | 38 |
| 13 |  | 43 | - | Ponatinib Staurosporine | 0.4 & 0.0023 |
| Compound | FRET (IC50) a | CaMKK2 Enzyme Assay b |
|---|---|---|
| IC50 (μM) | ||
| 10 | 7.8 | 11.9 |
| 11 | 10.5 | 6.5 |
| 12 | 3.2 | 4.1 |
| STO-609 | - | 0.04 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asquith, C.R.M.; Godoi, P.H.; Couñago, R.M.; Laitinen, T.; Scott, J.W.; Langendorf, C.G.; Oakhill, J.S.; Drewry, D.H.; Zuercher, W.J.; Koutentis, P.A.; et al. 1,2,6-Thiadiazinones as Novel Narrow Spectrum Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) Inhibitors. Molecules 2018, 23, 1221. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23051221
Asquith CRM, Godoi PH, Couñago RM, Laitinen T, Scott JW, Langendorf CG, Oakhill JS, Drewry DH, Zuercher WJ, Koutentis PA, et al. 1,2,6-Thiadiazinones as Novel Narrow Spectrum Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) Inhibitors. Molecules. 2018; 23(5):1221. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23051221
Chicago/Turabian StyleAsquith, Christopher R. M., Paulo H. Godoi, Rafael M. Couñago, Tuomo Laitinen, John W. Scott, Christopher G. Langendorf, Jonathan S. Oakhill, David H. Drewry, William J. Zuercher, Panayiotis A. Koutentis, and et al. 2018. "1,2,6-Thiadiazinones as Novel Narrow Spectrum Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) Inhibitors" Molecules 23, no. 5: 1221. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23051221