Effects of In Vitro Digestion on the Content and Biological Activity of Polyphenols from Acacia mearnsii Bark


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Changes in the Total Polyphenol Content (TPC) and Stability of PAs
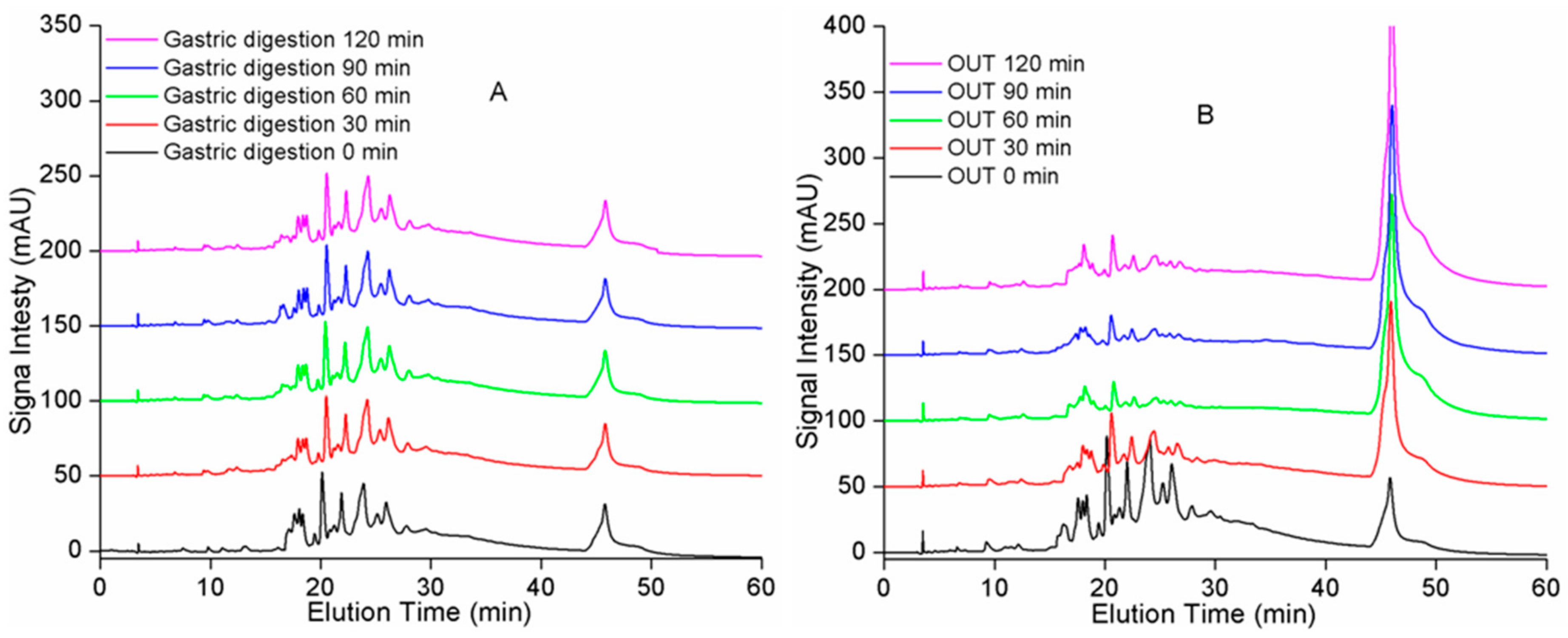
2.2. Gel Permeation Chromatography (GPC) Analysis
2.3. Changes in PA Bioaccessibility during Digestion
2.4. Changes in Antioxidant Activities of Digesta
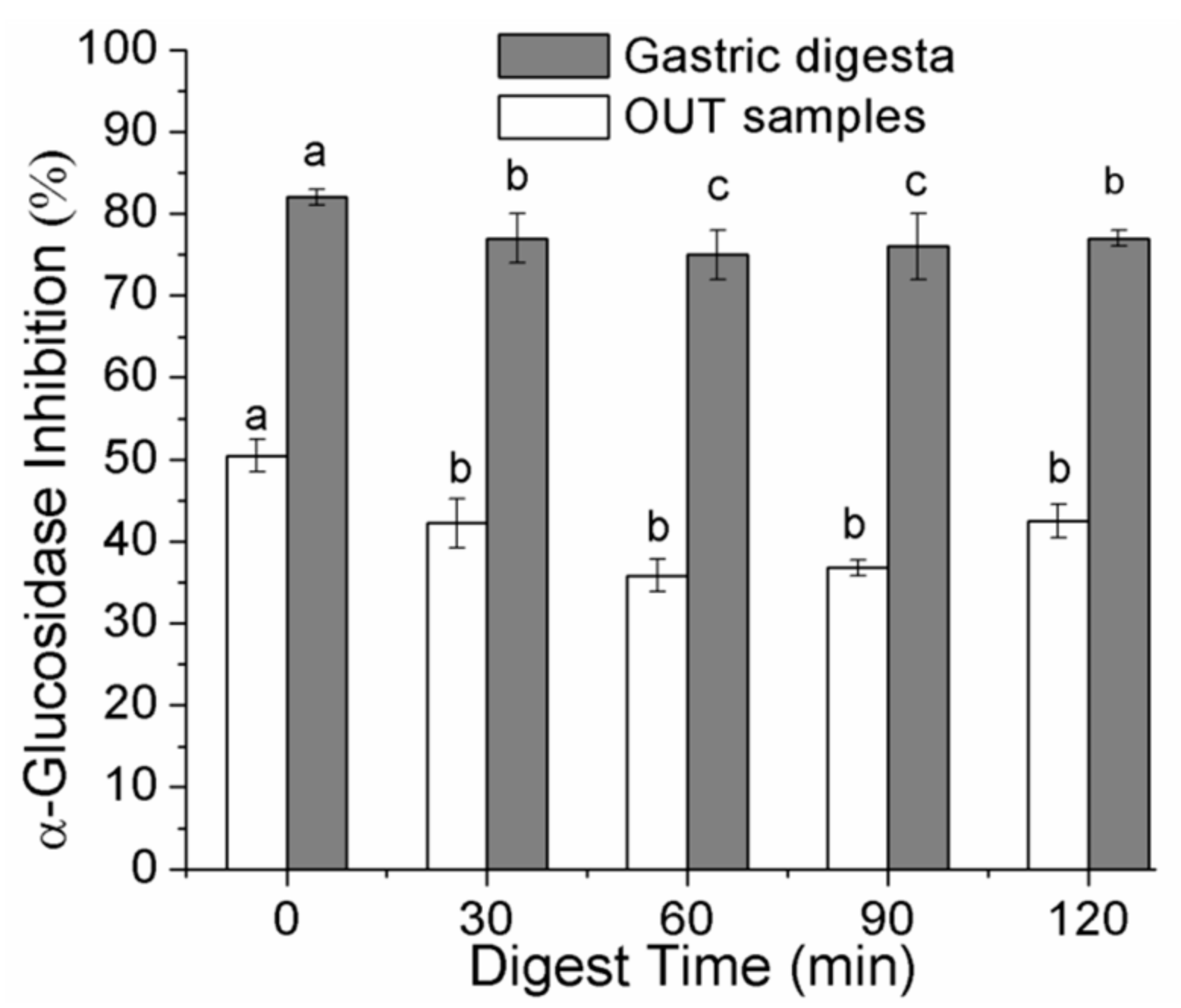
2.5. Changes in α-Glucosidase–Inhibitory Activities of Digesta
3. Materials and Methods
3.1. Materials and Reagents
3.2. Extraction of PAs
3.3. Simulated In Vitro Gastric and Intestinal Digestion
3.4. Analytical Assays
3.4.1. Clean-Up of the IN and OUT Samples
3.4.2. Determination of TPC and NP-HPLC/VWD
3.4.3. GPC
3.4.4. PA Identification: UFLC-QTOF-MS2
3.4.5. Antioxidant-Activity Measurements
3.4.6. α-Glucosidase Inhibition
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aron, P.M.; Kennedy, J.A. Flavan-3-ols: Nature, occurrence and biological activity. Mol. Nutr. Food Res. 2008, 52, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Venter, P.B.; Senekal, N.D.; Kemp, G.; Amra-Jordaan, M.; Khan, P.; Bonnet, S.L.; Westhuizen, J.H.V.D. Analysis of commercial proanthocyanidins. Part 3: The chemical composition of wattle (Acacia mearnsii) bark extract. Phytochemistry 2012, 83, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Graceb, M.H.; Esposito, D.; Wang, F.; Lila, M.A. Phytochemical characterization and anti-inflammatory properties of Acacia mearnsii leaves. Nat. Prod. Commun. 2016, 11, 649–653. [Google Scholar] [PubMed]
- Gey, K.F. The antioxidant hypothesis of cardiovascular disease: Epidemiology and mechanisms. Biochem. Soc. Trans. 1990, 18, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Ness, A.R.; Powles, J.W. Fruit and vegetables, and cardiovascular disease: A review. Int. J. Epidemiol. 1997, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.L.; Doughty, K.; Ali, A. Cocoa and chocolate in human health and disease. Antioxid. Redox Signal. 2011, 15, 2779–2811. [Google Scholar] [CrossRef] [PubMed]
- Roux, D.G.; Ferreira, D.; Hundt, H.K.L.; Malan, E. Structure, stereochemistry, and reactivity of natural condensed tannis as basis for their extended industrial application. Appl. Polym. Symp. 1975, 28, 335–353. [Google Scholar]
- Nursten, H.E. Practical polyphenolics: From structure to molecular recognition and physiological action. Trends Food Sci. Technol. 1999, 10, 339. [Google Scholar] [CrossRef]
- Kusano, R.; Ogawa, S.; Matsuo, Y.; Tanaka, T.; Yazaki, Y.; Kouno, I. Α-amylase and lipase inhibitory activity and structural characterization of Acacia bark proanthocyanidins. J. Nat. Prod. 2011, 74, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Grace, M.H.; Esposito, D.; Komarnytsky, S.; Wang, F.; Lila, M.A. Polyphenols isolated from Acacia mearnsii bark with anti-inflammatory and carbolytic enzyme inhibitory activities. Chin. J. Nat. Med. 2017, 15, 816–824. [Google Scholar] [CrossRef]
- Makino, R.; Ohara, S.; Hashida, K. Radical scavenging characteristics of condensed tannins from barks of various tree species compared with quebracho wood tannin. Holzforschung 2011, 65, 651–657. [Google Scholar] [CrossRef]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Chen, J.; Li, T.; Liu, C.; Wang, X.; Dai, T.; Mcclements, D.J.; Liu, J. Impact of in vitro simulated digestion on the potential health benefits of proanthocyanidins from Choerospondias axillaris peels. Food Res. Int. 2015, 78, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Fernández, K.; Labra, J. Simulated digestion of proanthocyanidins in grape skin and seed extracts and the effects of digestion on the angiotensin I-converting enzyme (ACE) inhibitory activity. Food Chem. 2013, 139, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.E.; Chaudry, F.; Pannala, A.S.; Srai, S.K.; Debnam, E.; Rice-Evans, C. Decomposition of cocoa procyanidins in the gastric milieu. Biochem. Biophys. Res. Commun. 2000, 272, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Woottonbeard, P.C.; Moran, A.; Ryan, L. Stability of the total antioxidant capacity and total polyphenol content of 23 commercially available vegetable juices before and after in vitro digestion measured by FRAP, DPPH, ABTS and Folin–Ciocalteu methods. Food Res. Int. 2011, 44, 217–224. [Google Scholar] [CrossRef]
- Davide, T.; Elena, V.; Davide, B.; Angela, C. In vitro bio-accessibility and antioxidant activity of grape polyphenols. Food Chem. 2010, 120, 599–606. [Google Scholar]
- Mcdougall, G.J.; Fyffe, S.; Dobson, P.; Stewart, D. Anthocyanins from red wine—Their stability under simulated gastrointestinal digestion. Phytochemistry 2005, 66, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, F.; Gil-Izquierdo, A.; Pérez-Vicente, A.; García-Viguera, C. In vitro gastrointestinal digestion study of broccoli inflorescence phenolic compounds, glucosinolates, and vitamin C. J. Agric. Food Chem. 2004, 52, 7432–7433. [Google Scholar] [CrossRef] [PubMed]
- Rubilar, M.; Jara, C.; Poo, Y.; Acevedo, F.; Gutierrez, C.; Sineiro, J.; Shene, C. Extracts of maqui (Aristotelia chilensis) and murta (Ugni molinae turcz.): Sources of antioxidant compounds and α-glucosidase/α-amylase inhibitors. J. Agric. Food Chem. 2011, 59, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.A.; Noble, A.C.; Singleton, V.L. Bitterness and astringency of phenolic fractions in wine. J. Agric. Food Chem. 1980, 28, 675–678. [Google Scholar] [CrossRef]
- Porter, L.J.; Woodruffe, J. Haemanalysis: The relative astringency of proanthocyanidin polymers. Phytochemistry 1984, 23, 1255–1256. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; Scalbert, A. Proanthocyanidins and tannin-like compounds–nature, occurrence, dietary intake and effects on nutrition and health. J. Sci. Food Agric. 2000, 80, 1094–1117. [Google Scholar] [CrossRef]
- Bermudezsoto, M.J.; TomáSbarberáN, F.A.; GarcíAconesa, M.T. Stability of polyphenols in chokeberry (Aronia melanocarpa) subjected to in vitro gastric and pancreatic digestion. Food Chem. 2007, 102, 865–874. [Google Scholar] [CrossRef]
- Ortega, N.; Reguant, J.; Romero, M.P.; Macià, A.; Motilva, M.J. Effect of fat content on the digestibility and bioaccessibility of cocoa polyphenol by an in vitro digestion model. J. Agric. Food Chem. 2009, 57, 5743–5749. [Google Scholar] [CrossRef] [PubMed]
- Rodríguezroque, M.J.; Rojasgraü, M.A.; Elezmartínez, P.; Martínbelloso, O. Soymilk phenolic compounds, isoflavones and antioxidant activity as affected by in vitro gastrointestinal digestion. Food Chem. 2013, 136, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Ou, K.; Gu, L. Absorption and metabolism of proanthocyanidins. J. Funct. Foods 2014, 7, 43–53. [Google Scholar] [CrossRef]
- Parkar, S.G.; Trower, T.M.; Stevenson, D.E. Fecal microbial metabolism of polyphenols and its effects on human gut microbiota. Anaerobe 2013, 23, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Parkar, S.G.; Stevenson, D.E.; Skinner, M.A. The potential influence of fruit polyphenols on colonic microflora and human gut health. Int. J. Food Microbiol. 2008, 124, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Dudonné, S.; Vitrac, X.; Coutière, P.; Woillez, M.; Mérillon, J.M. Comparative study of antioxidant properties and total phenolic content of 30 plant extracts of industrial interest using DPPH, ABTS, FRAP, SOD, and ORAC assays. J. Agric. Food Chem. 2009, 57, 1768–1774. [Google Scholar] [CrossRef] [PubMed]
- Neilson, A.P.; O’Keefe, S.F.; Bolling, B.W. High-molecular-weight proanthocyanidins in foods: Overcoming analytical challenges in pursuit of novel dietary bioactive components. Annu. Rev. Food Sci. Technol. 2015, 7, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Gilizquierdo, A.; Zafrilla, P.; Tomasbarberan, F.A. An in vitro method to simulate phenolic compound release from the food matrix in the gastrointestinal tract. Eur. Food Res. Technol. 2002, 214, 155–159. [Google Scholar] [CrossRef]
- Damiano, S.; Forino, M.; De, A.; Vitali, L.A.; Lupidi, G.; Taglialatela-Scafati, O. Antioxidant and antibiofilm activities of secondary metabolites from Ziziphus jujuba leaves used for infusion preparation. Food Chem. 2017, 230, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, J.; Escribano-Bailon, M.T.; Prieur, C.; Souquet, J.M.; Cheynier, V. Normal-phase high-performance liquid chromatographic separation of procyanidins from cacao beans and grape seeds. J. Chromatogr. 1993, 654, 255–260. [Google Scholar] [CrossRef]
- Lerma-Herrera, M.A.; Núñez-Gastélum, J.A.; Ascacio-Valdés, J.; Aguilar, C.N.; Rodrigo-García, J.; Díaz-Sánchez, A.G.; Alvarez-Parrilla, E.; Rosa, L.A.D.L. Estimation of the mean degree of polymerization of condensed tannins from the kernel and shell of Carya illinoinensis by HPLC/MS and spectrophotometric methods. Food Anal. Methods 2017, 10, 3023–3031. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rive-Evans, C. Antioxidant activity applying a improved ABTS radical cation assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Boue, S.M.; Daigle, K.W.; Chen, M.H.; Cao, H.; Heiman, M.L. Antidiabetic potential of purple and red rice (Oryza sativa L.) bran extracts. J. Agric. Food Chem. 2016, 64, 5345–5353. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Digestion Time (min) | TPC of Digestion Samples (mg/mL) | ||
|---|---|---|---|
| Gastric Digestion | OUT Sample | IN Sample | |
| 0 | 5.97 ± 0.05a | 4.69 ± 0.03a | 0 |
| 30 | 5.47 ± 0.04b | 3.86 ± 0.05b | 0.19 ± 0.01a |
| 60 | 5.24 ± 0.03c | 3.88 ± 0.02b | 0.40 ± 0.02b |
| 90 | 5.47 ± 0.06b | 3.90 ± 0.03b | 0.91 ± 0.02c |
| 120 | 5.45 ± 0.03b | 4.49 ± 0.06c | 1.09 ± 0.03d |
| Retention Time (min) | Compounds | [M − H]− m/z | Molecular Fragments | In 30 min | In 60 min | In 90 min | In 120 min |
|---|---|---|---|---|---|---|---|
| 3.0 | C | 289 | 245, 151, 139, 123 | X | X | ||
| 1.5 | GC | 305 | 219, 203, 167, 125 | X | X | ||
| 5.8 | Dimer (F-C) | 561 | 561, 409, 289, 161 | X | X | X | X |
| 4.2 | Dimer (F-GC/R-C) | 577 | 577, 425, 409, 407, 305, 177, 161 | X | X | X | X |
| 2.6 | Dimer (R-GC) | 593 | 593, 425, 407, 305, 177 | X | X | ||
| 10.9 | Trimer (F-C-F) | 833 | 833, 681, 561, 289, 161 | X | X | X | X |
| 9.6 | Trimer (F-C-R/F-GC-F) | 849 | 849, 697, 561, 409, 305, 289, 177 | X | X | ||
| 7.9 | Trimer (R-C-R) | 865 | 865, 713, 577, 407, 289, 177 | X | X | X | |
| 5.9 | Trimer (R-GC-R) | 881 | 880, 713, 593, 407, 305, 177 | X | X | X |
| Incubation Time (min) | DPPH IC50 (nL) | ABTS IC50 (nL) | ||||
|---|---|---|---|---|---|---|
| Gastric Fraction | OUT Sample | IN Sample | Gastric Fraction | OUT Sample | IN Sample | |
| 0 | 6.9 ± 0.6a | 9.7 ± 0.2a | 0a | 4.2 ± 0.3a | 5.0 ± 0.4a | 0a |
| 30 | 7.2 ± 0.4b | 16.4 ± 0.3b | 120 ± 11a | 4.4 ± 0.4b | 14.8 ± 0.5a | 71.1 ± 6a |
| 60 | 7.8 ± 0.5a | 15.7 ± 0.5c | 85.6 ± 9a | 4.7 ± 0.5c | 11.7 ± 0.2a | 36.9 ± 7a |
| 90 | 7.4 ± 0.6c | 11.5 ± 0.6d | 57.3 ± 7a | 4.5 ± 0.5d | 9.9 ± 0.2a | 18.8 ± 4a |
| 120 | 7.3 ± 0.3d | 10.1 ± 0.4e | 47.4 ± 12a | 4.5 ± 0.3e | 5.9 ± 0.3a | 11.4 ± 6a |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Xiong, J.; He, L.; Zhang, Y.; Li, X.; Zhang, L.; Wang, F. Effects of In Vitro Digestion on the Content and Biological Activity of Polyphenols from Acacia mearnsii Bark. Molecules 2018, 23, 1804. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23071804
Chen X, Xiong J, He L, Zhang Y, Li X, Zhang L, Wang F. Effects of In Vitro Digestion on the Content and Biological Activity of Polyphenols from Acacia mearnsii Bark. Molecules. 2018; 23(7):1804. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23071804
Chicago/Turabian StyleChen, Xiao, Jia Xiong, Lingxiao He, Yu Zhang, Xun Li, Liping Zhang, and Fei Wang. 2018. "Effects of In Vitro Digestion on the Content and Biological Activity of Polyphenols from Acacia mearnsii Bark" Molecules 23, no. 7: 1804. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23071804