Design, Synthesis and Evaluation of N-pyrazinylbenzamides as Potential Antimycobacterial Agents

 ,
,  , , and
, , and 
Abstract
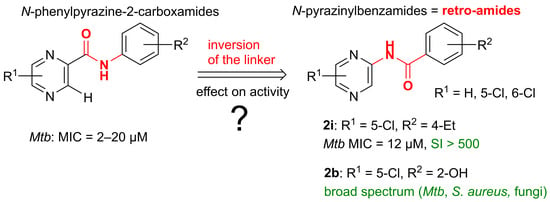
:
1. Introduction
2. Results and Discussion
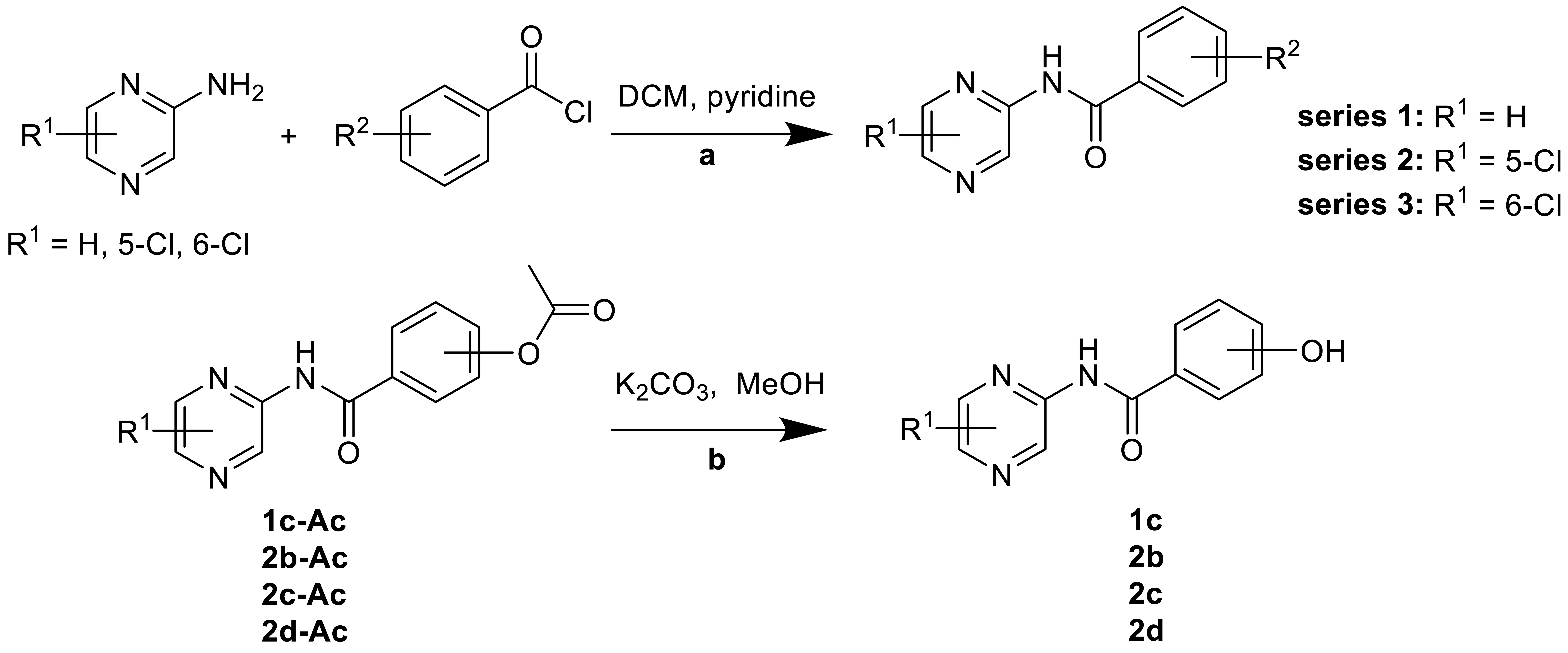
2.1. Chemistry
2.2. Antimycobacterial Activity
2.3. Antibacterial Activity
2.4. Antifungal Activity
2.5. In Vitro Cytotoxicity
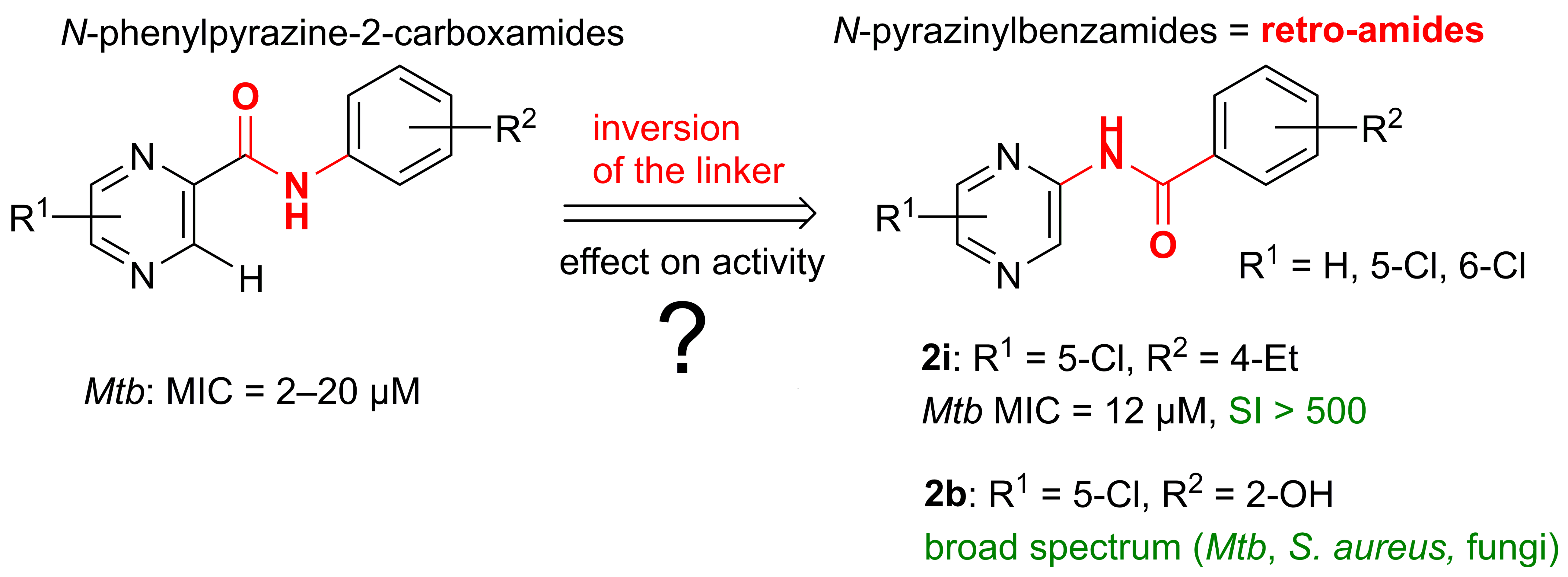
2.6. Comparison of the Antimycobacterial Activity and HepG2 Cytotoxicity of N-Pyrazinylbenzamides (Retro-Amides) with the Corresponding N-Phenylpyrazine-2-carboxamides (Amides)
2.7. In Silico Prediction of Molecular Structure and Properties
2.7.1. Geometry Optimization
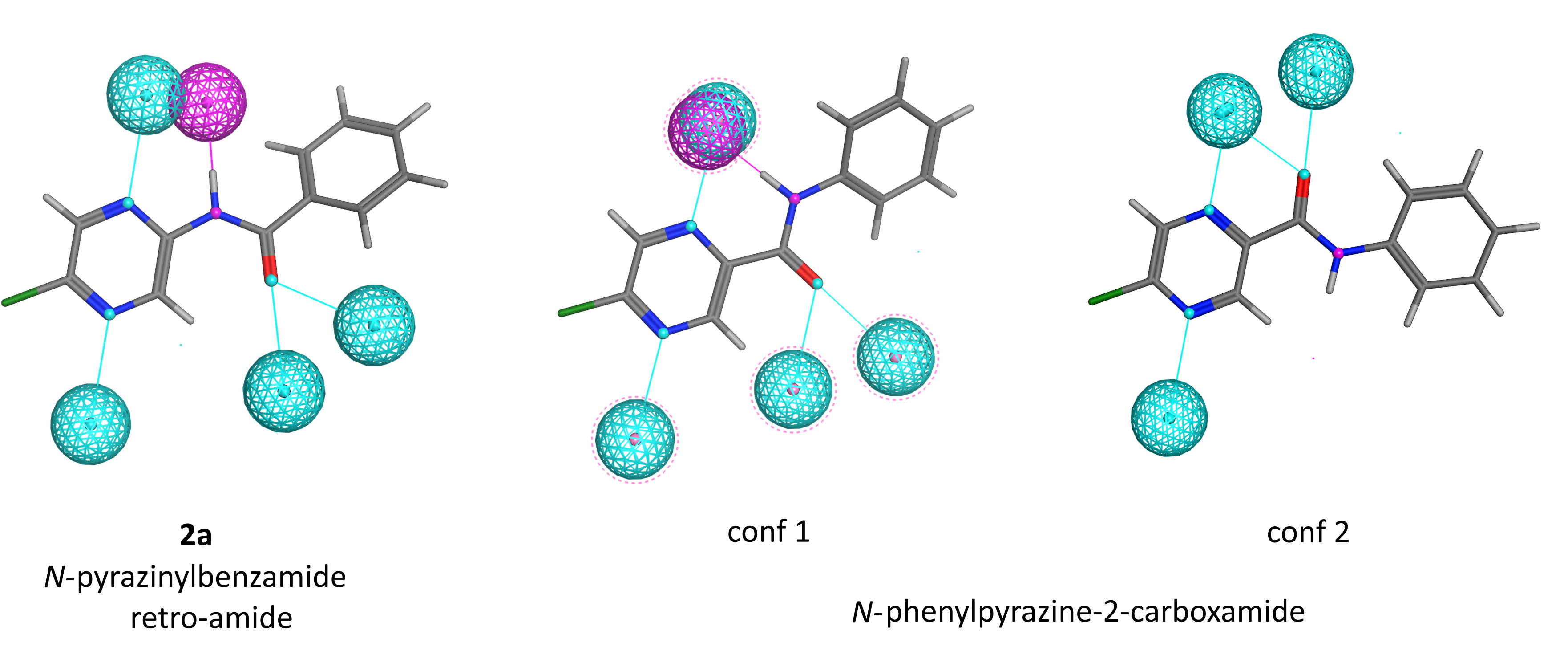
2.7.2. Analysis of Pharmacophore Features
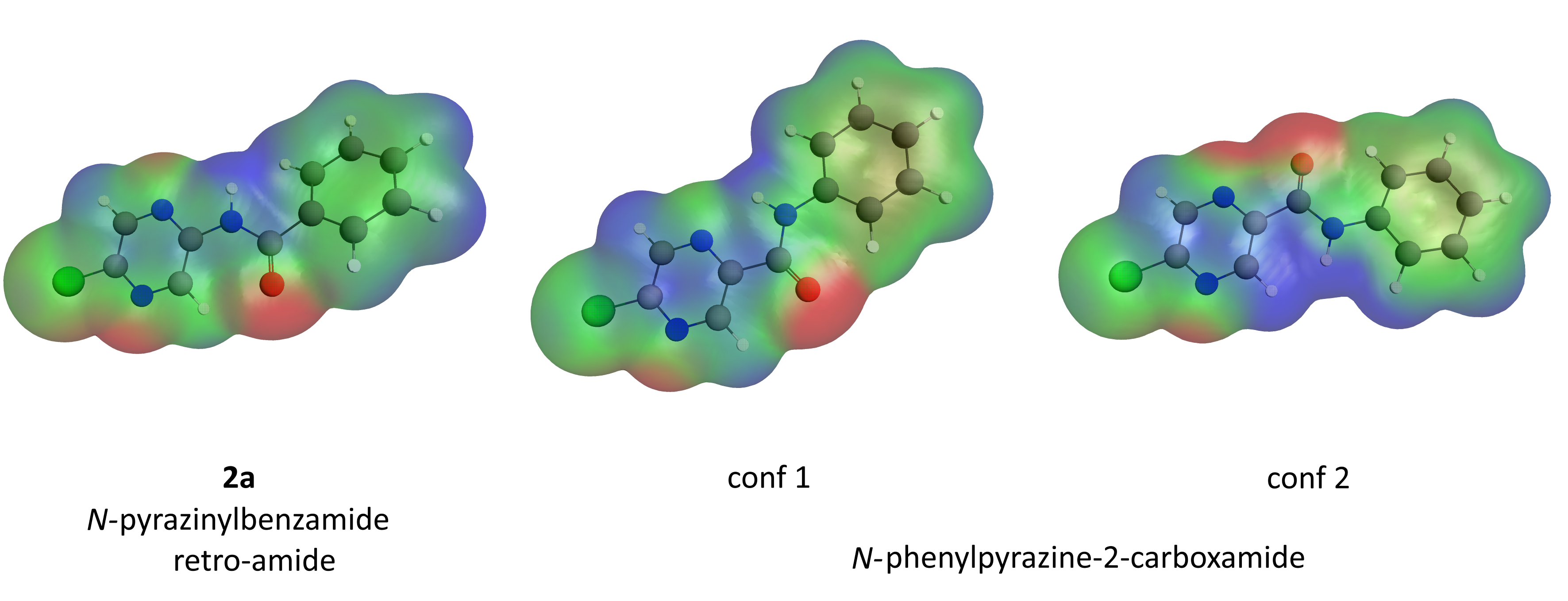
2.7.3. Molecular Electrostatic Potential (MEP)
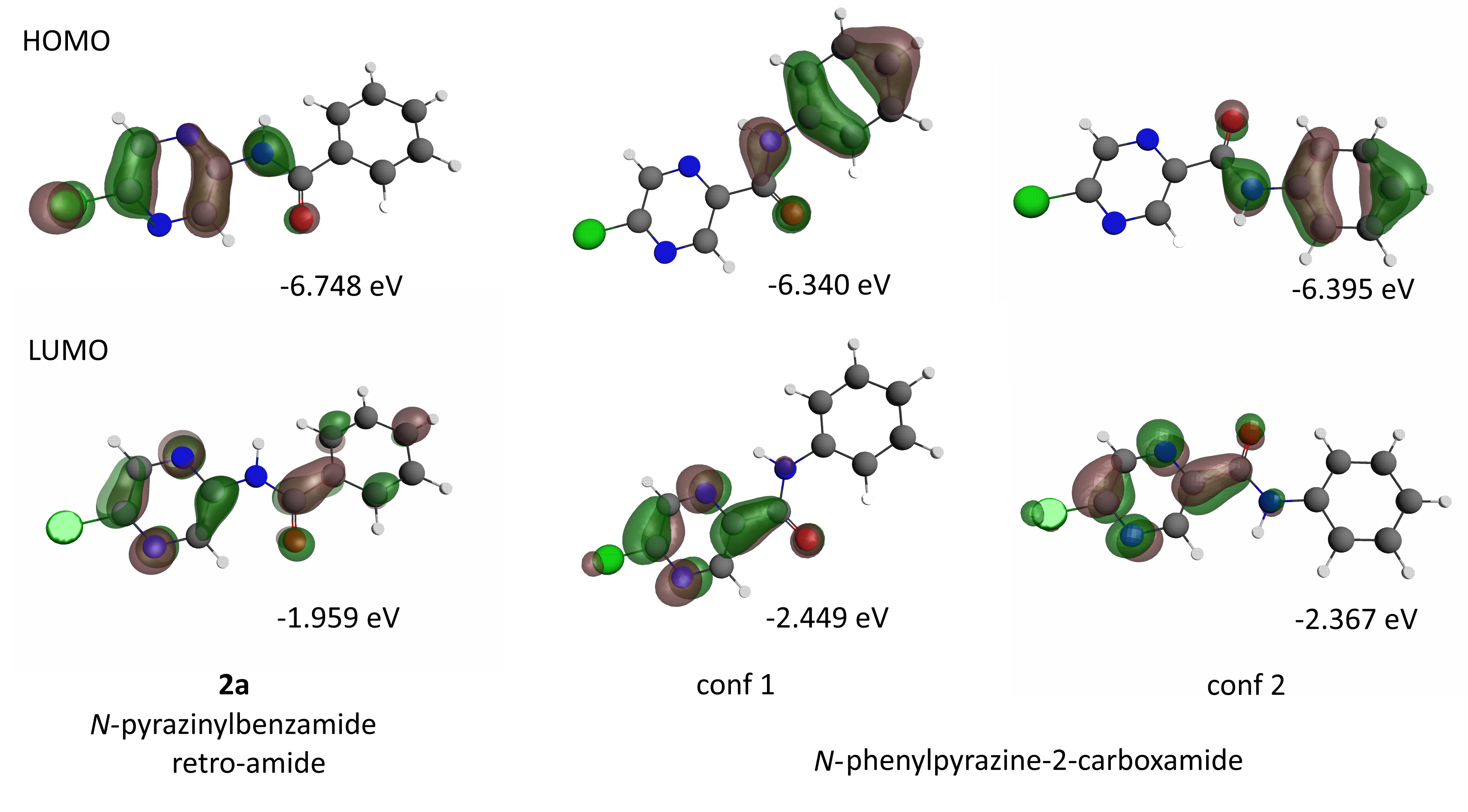
2.7.4. HOMO and LUMO Orbitals
3. Materials and Methods
3.1. General
3.2. Chemistry
3.2.1. Method A (Used in the Synthesis of Final Products from Series 1 and 3)
3.2.2. Method B (Used in the Synthesis of Final Products from Series 2)
3.2.3. Hydrolysis of Acetates
3.3. Analytical Data of Prepared Compounds
3.4. Biological Methods
3.5. In Silico Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2017; WHO/HTM/TB/2017.23; WHO: Geneva, Switzerland, 2017; Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 10 July 2018).
- Houben, R.M.; Dodd, P.J. The Global Burden of Latent Tuberculosis Infection: A Re-estimation Using Mathematical Modelling. PLoS Med. 2016, 13, e1002152. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Treatment Guidelines for Drug-Resistant Tuberculosis (2016 Update). WHO/HTM/TB/2016.04. Available online: http://www.who.int/tb/areas-of-work/drug-resistant-tb/treatment/resources/en/ (accessed on 15 August 2018).
- Zimhony, O.; Cox, J.S.; Welch, J.T.; Vilcheze, C.; Jacobs, W.R., Jr. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat. Med. 2000, 6, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, H.I.; Mizrahi, V.; Barry, C.E., 3rd. Effects of pyrazinamide on fatty acid synthesis by whole mycobacterial cells and purified fatty acid synthase I. J. Bacteriol. 2002, 184, 2167–2172. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.C.; Zimhony, O.; Chung, W.J.; Sayahi, H.; Jacobs, W.R., Jr.; Welch, J.T. Inhibition of isolated Mycobacterium tuberculosis fatty acid synthase I by pyrazinamide analogs. Antimicrob. Agents Chemother. 2007, 51, 2430–2435. [Google Scholar] [CrossRef] [PubMed]
- Zimhony, O.; Vilcheze, C.; Arai, M.; Welch, J.T.; Jacobs, W.R., Jr. Pyrazinoic acid and its n-propyl ester inhibit fatty acid synthase type I in replicating tubercle bacilli. Antimicrob. Agents Chemother. 2007, 51, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Sayahi, H.; Zimhony, O.; Jacobs, W.R., Jr.; Shekhtman, A.; Welch, J.T. Pyrazinamide, but not pyrazinoic acid, is a competitive inhibitor of NADPH binding to Mycobacterium tuberculosis fatty acid synthase I. Bioorg. Med. Chem. Lett. 2011, 21, 4804–4807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Chen, J.; Shi, W.; Liu, W.; Zhang, W.; Zhang, Y. Mutations in panD encoding aspartate decarboxylase are associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg. Microbes Infect. 2013, 2, e34. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Chen, J.; Feng, J.; Cui, P.; Zhang, S.; Weng, X.; Zhang, W.; Zhang, Y. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg. Microbes Infect. 2014, 3, e58. [Google Scholar] [CrossRef] [PubMed]
- Pandey, B.; Grover, S.; Tyagi, C.; Goyal, S.; Jamal, S.; Singh, A.; Kaur, J.; Grover, A. Molecular principles behind pyrazinamide resistance due to mutations in panD gene in Mycobacterium tuberculosis. Gene 2016, 581, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Shibayama, K.; Rimbara, E.; Mori, S. Biochemical characterization of quinolinic acid phosphoribosyltransferase from Mycobacterium tuberculosis H37Rv and inhibition of its activity by pyrazinamide. PLoS ONE 2014, 9, e100062. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhang, X.; Jiang, X.; Yuan, H.; Lee, J.S.; Barry, C.E., 3rd; Wang, H.; Zhang, W.; Zhang, Y. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 2011, 333, 1630–1632. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Y.; Bi, J.; Cai, Q.; Liao, X.; Li, W.; Guo, C.; Zhang, Q.; Lin, T.; Zhao, Y.; et al. Structural basis for targeting the ribosomal protein S1 of Mycobacterium tuberculosis by pyrazinamide. Mol. Microbiol. 2015, 95, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Dillon, N.A.; Peterson, N.D.; Feaga, H.A.; Keiler, K.C.; Baughn, A.D. Anti-tubercular Activity of Pyrazinamide is Independent of trans-Translation and RpsA. Sci. Rep. 2017, 7, 6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehr, M.; Elamin, A.A.; Singh, M. Pyrazinamide: The importance of uncovering the mechanisms of action in mycobacteria. Expert Rev. Anti-Infect. Ther. 2015, 13, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, M.; Kesetovic, D.; Zitko, J. Antimycobacterial evaluation of pyrazinoic acid reversible derivatives. Curr. Pharm. Des. 2011, 17, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Servusova, B.; Paterova, P.; Mandikova, J.; Kubicek, V.; Kucera, R.; Hrabcova, V.; Kunes, J.; Soukup, O.; Dolezal, M. Synthesis, antimycobacterial activity and in vitro cytotoxicity of 5-chloro-N-phenylpyrazine-2-carboxamides. Molecules 2013, 18, 14807–14825. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Servusova-Vanaskova, B.; Paterova, P.; Navratilova, L.; Trejtnar, F.; Kunes, J.; Dolezal, M. Design, synthesis and anti-mycobacterial evaluation of some new N-phenylpyrazine-2-carboxamides. Chem. Pap. 2016, 70, 649–657. [Google Scholar] [CrossRef]
- Cai, Z.; Ding, Z.; Hao, Y.; Ni, T.; Xie, F.; Zhao, J.; Li, R.; Yu, S.; Wang, T.; Chai, X.; et al. Design, synthesis, and SAR study of 3-(benzo[d][1,3]dioxol-5-yl)-N-benzylpropanamide as novel potent synergists against fluconazole-resistant Candida albicans. Bioorg. Med. Chem. Lett. 2017, 27, 4571–4575. [Google Scholar] [CrossRef] [PubMed]
- Cabaret, D.; Adediran, S.A.; Pratt, R.F.; Wakselman, M. New substrates for beta-lactam-recognizing enzymes: Aryl malonamates. Biochemistry 2003, 42, 6719–6725. [Google Scholar] [CrossRef] [PubMed]
- Adediran, S.A.; Cabaret, D.; Lohier, J.F.; Wakselman, M.; Pratt, R.F. Benzopyranones with retro-amide side chains as (inhibitory) beta-lactamase substrates. Bioorg. Med. Chem. Lett. 2004, 14, 5117–5120. [Google Scholar] [CrossRef] [PubMed]
- Adediran, S.A.; Lohier, J.F.; Cabaret, D.; Wakselman, M.; Pratt, R.F. Synthesis and reactivity with beta-lactamases of a monobactam bearing a retro-amide side chain. Bioorg. Med. Chem. Lett. 2006, 16, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Adediran, S.A.; Cabaret, D.; Lohier, J.F.; Wakselman, M.; Pratt, R.F. Substituted aryl malonamates as new serine beta-lactamase substrates: Structure-activity studies. Bioorg. Med. Chem. 2010, 18, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, V.; Gogou, M.; Giannoussi, A.; Skobridis, K. Insights into the N,N-diacylation reaction of 2-aminopyrimidines and deactivated anilines: An alternative N-monoacylation reaction. Arkivoc 2014, 11–23. [Google Scholar] [CrossRef]
- Collins, L.; Franzblau, S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [Google Scholar] [PubMed]
- Bispo, M.D.F.; Goncalves, R.S.B.; Lima, C.H.D.; Cardoso, L.N.D.; Lourenco, M.C.S.; de Souza, M.V.N. Synthesis and Antitubercular Evaluation of N-Arylpyrazine and N,N′-Alkyl-diylpyrazine-2-carboxamide Derivatives. J. Heterocycl. Chem. 2012, 49, 1317–1322. [Google Scholar] [CrossRef]
- Ananthan, S.; Faaleolea, E.R.; Goldman, R.C.; Hobrath, J.V.; Kwong, C.D.; Laughon, B.E.; Maddry, J.A.; Mehta, A.; Rasmussen, L.; Reynolds, R.C.; et al. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis 2009, 89, 334–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, H.; Sun, Z. Susceptibility of Mycobacterium tuberculosis to weak acids. J. Antimicrob. Chemother. 2003, 52, 56–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, M.M.; Zhang, Y. Effects of weak acids, UV and proton motive force inhibitors on pyrazinamide activity against Mycobacterium tuberculosis in vitro. J. Antimicrob. Chemother. 2006, 58, 936–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolezal, M.; Palek, L.; Vinsova, J.; Buchta, V.; Jampilek, J.; Kralova, K. Substituted pyrazinecarboxamides: Synthesis and biological evaluation. Molecules 2006, 11, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, M.; Cmedlova, P.; Palek, L.; Vinsova, J.; Kunes, J.; Buchta, V.; Jampilek, J.; Kralova, K. Synthesis and antimycobacterial evaluation of substituted pyrazinecarboxamides. Eur. J. Med. Chem. 2008, 43, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, M.; Zitko, J.; Osicka, Z.; Kunes, J.; Vejsova, M.; Buchta, V.; Dohnal, J.; Jampilek, J.; Kralova, K. Synthesis, antimycobacterial, antifungal and photosynthesis-inhibiting activity of chlorinated N-phenylpyrazine-2-carboxamides. Molecules 2010, 15, 8567–8581. [Google Scholar] [CrossRef] [PubMed]
- Servusova-Vanaskova, B.; Paterova, P.; Garaj, V.; Mandikova, J.; Kunes, J.; Naesens, L.; Jilek, P.; Dolezal, M.; Zitko, J. Synthesis and Antimicrobial Evaluation of 6-Alkylamino-N-phenylpyrazine-2-carboxamides. Chem. Biol. Drug Des. 2015, 86, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, M.; Miletin, M.; Kunes, J.; Kralova, K. Substituted amides of pyrazine-2-carboxylic acids: Synthesis and biological activity. Molecules 2002, 7, 363–373. [Google Scholar] [CrossRef]
- Sebastian, S.H.R.; Al-Alshaikh, M.A.; El-Emam, A.A.; Panicker, C.Y.; Zitko, J.; Dolezal, M.; VanAlsenoy, C. Spectroscopic, quantum chemical studies, Fukui functions, in vitro antiviral activity and molecular docking of 5-chloro-N-(3-nitrophenyl) pyrazine-2-carboxamide. J. Mol. Struct. 2016, 1119, 188–199. [Google Scholar] [CrossRef]
- Bode, B.M.; Gordon, M.S. Macmolplt: A graphical user interface for GAMESS. J. Mol. Graph. Model. 1998, 16, 133–138. [Google Scholar] [CrossRef]
- Matyk, J.; Waisser, K.; Drazkova, K.; Kunes, J.; Klimesova, V.; Palat, K., Jr.; Kaustova, J. Heterocyclic isosters of antimycobacterial salicylanilides. Farmaco 2005, 60, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Kratky, M.; Vinsova, J. Salicylanilide Ester Prodrugs as Potential Antimicrobial Agents—A Review. Curr. Pharm. Des. 2011, 17, 3494–3505. [Google Scholar] [CrossRef] [PubMed]
- Baranyai, Z.; Kratky, M.; Vinsova, J.; Szabo, N.; Senoner, Z.; Horvati, K.; Stolarikova, J.; David, S.; Bosze, S. Combating highly resistant emerging pathogen Mycobacterium abscessus and Mycobacterium tuberculosis with novel salicylanilide esters and carbamates. Eur. J. Med. Chem. 2015, 101, 692–704. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds from series 1, 2, and 3 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
| R1 | |||
| R2 | H | 5-Cl | 6-Cl |
| H | 1a | 2a | 3a |
| 2-OH | 1b | 2b | n.a. |
| 3-OH | 1c | 2c | n.a. |
| 4-OH | n.a. | 2d | n.a. |
| 2-OCH3 | 1e | 2e | 3e |
| 3-OCH3 | 1f | 2f | 3f |
| 4-OCH3 | 1g | 2g | 3g |
| 4-CH3 | 1h | 2h | 3h |
| 4-Et | 1i | 2i | 3i |
| 2-Cl | 1j | 2j | 3j |
| 3-Cl | 1k | 2k | 3k |
| 4-Cl | 1l | 2l | 3l |
| 4-Br | n.a. | 2m | n.a. |
| 3-CF3 | 1n | 2n | 3n |
| 2-OAc | n.a. | 2b-Ac | n.a. |
| 3-OAc | 1c-Ac | 2c-Ac | n.a. |
| 4-OAc | 1d-Ac | 2d-Ac | n.a. |
| Cpd | R1 | R2 | MW | Mtb H37Rv ATCC 27294 MIC (µg/mL) | M. kans. ATCC 12478 MIC (µg/mL) | M. avium ATCC 15769 MIC (µg/mL) | M. smeg. ATCC 607 MIC (µg/mL) |
|---|---|---|---|---|---|---|---|
| 1a | H | H | 199.21 | 100 | >100 | >100 | ≥500 |
| 1e | H | 2-OCH3 | 229.24 | >100 | 100 | >100 | 125 |
| 1g | H | 4-OCH3 | 229.24 | 25 | >100 | >100 | ≥500 |
| 1j | H | 2-Cl | 233.66 | >100 | >100 | >100 | 250 |
| 1k | H | 3-Cl | 233.66 | 50 | 50 | >100 | ≥500 |
| 1l | H | 4-Cl | 233.66 | 50 | 50 | >100 | ≥500 |
| 1l-SP b | H | 4-Cl b | 372.21 | 50 | >100 | >100 | ≥500 |
| 2b-Ac | 5-Cl | 2-OAc | 291.69 | 12.5 | 12.5 | 50 | 31.25 |
| 2b | 5-Cl | 2-OH | 249.65 | 12.5 | 50 | 50 | 15.625 |
| 2c-Ac | 5-Cl | 3-OAc | 291.69 | >100 | >100 | >100 | 250 |
| 2d-Ac | 5-Cl | 4-OAc | 291.69 | >100 | >100 | >100 | 250 |
| 2h | 5-Cl | 4-CH3 | 247.68 | 6.25 | >100 | >100 | ≥500 |
| 2i | 5-Cl | 4-Et | 261.71 | 3.13 | >100 | >100 | ≥500 |
| 2n | 5-Cl | 3-CF3 | 301.65 | 25 | >100 | >100 | ≥500 |
| 3f | 6-Cl | 3-OCH3 | 263.68 | 50 c | 100 c | 100 c | ≥500 c |
| 3n | 6-Cl | 3-CF3 | 301.65 | 25 | 25 | 50 | 125 |
| INH | - | - | 137.14 | 0.1–0.39 | 6.25–12.5 | 6.25–12.5 | 15.625 |
| RFM | - | - | 822.95 | - | - | - | 1.56 |
| CPX | - | - | 331.35 | - | - | - | 0.195 |
| Strain | Time | Compound (Code, R2) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1n | 2b-Ac | 2b | 2c-Ac | 2c | 2d-Ac | 2d | 2f | 2j | Neom | ||
| 3-CF3 | 2-OAc | 2-OH | 3-OAc | 3-OH | 4-OAc | 4-OH | 3-OCH3 | 2-Cl | - | ||
| SA | 24 h | >125 | 62.5 | 15.62 | >125 | 500 | 0.98 | 31.25 | 31.25 | 62.50 | 3.90 |
| 48 h | >125 | 125 | 15.62 | >125 | 500 | 0.98 | 31.25 | 31.25 | 62.50 | 3.90 | |
| MRSA | 24 h | >125 | 125 | 62.50 | >125 | >500 | >125 | >500 | >500 | >500 | 0.98 |
| 48 h | >125 | 250 | 62.50 | >125 | >500 | >125 | >500 | >500 | >500 | 0.98 | |
| SE | 24 h | 62.50 | 125 | 62.50 | >125 | >500 | >125 | 500 | >500 | >500 | 3.90 |
| 48 h | 62.50 | 250 | 62.50 | >125 | >500 | >125 | 500 | >500 | >500 | 7.81 | |
| Cpd | HepG2 | Mtb | SA | ||
|---|---|---|---|---|---|
| IC50 (μM) | MIC (μM) | SI | MIC (μM) | SI | |
| 2b-Ac | 95.37 | 42.85 | 2 | 65.20 | 1 |
| 2b | 155.30 | 50.07 | 3 | 15.62 | 10 |
| 2d-Ac | >500 * | inactive | n.a. | 0.98 | >510 |
| 2h | >250 * | 25.23 | >10 | >125 | n.a. |
| 2i | >250 * | 11.96 | >21 | >125 | n.a. |
| 3n | >250 * | 82.88 | >3 | n.t. | n.a. |
| Compound | IC50 (µM) after 24 h Exposure | IC50 (µM) after 48 h Exposure | Range of Tested Concentrations (µM) |
|---|---|---|---|
| 2b | 545.3 | 187.6 | 10–2000 |
| 2i | 264.3 | 250.0 | 1–1000 |
| R1 | R2 |  Retro-Amides | Relation |  Amides | ||
|---|---|---|---|---|---|---|
| Code | MIC a (μg/mL) | MIC (μg/mL) | Inhibition (%) at 6.25 μg/mL (TAACF) | |||
| H | H | 1a | 100 | = | >100 b [27] | 0 [31] |
| H | 2-OCH3 | 1e | >100 | = | >100 b [27] | |
| H | 3-OCH3 | 1f | >100 | = | >100 b [27] | |
| H | 4-OCH3 | 1g | 25 | < | >100 b [27] | |
| H | 4-CH3 | 1h | >100 | < | 100 b [27] | 86 [32] |
| H | 2-Cl | 1j | >100 | < | 50 c [19] | |
| H | 3-Cl | 1k | 50 | =? | >100 b [27] | 14 [33] |
| H | 4-Cl | 1l | 50 | >=? | 50 b [27] | 4 [33] |
| H | 3-CF3 | 1n | >100 | < | 6.25 [32], 50 b [27] | 99 [32] |
| 5-Cl | H | 2a | >100 | < | 3.13 (1.56) c [18] | |
| 5-Cl | 2-OH | 2b | 12.5 | < | 3.13 (0.78) c [18] | |
| 5-Cl | 3-OH | 2c | >100 | < | 6.25 c [18] | |
| 5-Cl | 4-OH | 2d | >100 | < | 3.13 (12.5) c [18] | |
| 5-Cl | 4-Et | 2i | 3.13 | = | 1.56 (0.78) c [18] | |
| 5-Cl | 2-Cl | 2j | >100 | < | 3.13 (0.78) c | |
| 5-Cl | 3-Cl | 2k | >100 | < | 6.25 (3.13) c [18] | |
| 5-Cl | 4-Br | 2m | >100 | < | 3.13 c [18] | |
| 5-Cl | 3-CF3 | 2n | 25 | < | 3.13 (6.25) c [18] | |
| 6-Cl | H | 3a | >100 | < | 25 d [34] | 32 [31] |
| 6-Cl | 2-OCH3 | 3e | >100 | <? | 6 [35] | |
| 6-Cl | 3-OCH3 | 3f | 50 | >? | 2 [31] | |
| 6-Cl | 4-CH3 | 3h | >100 | < | 71 [32] | |
| 6-Cl | 2-Cl | 3j | >100 | < | 100 d [34] | |
| 6-Cl | 3-Cl | 3k | >100 | < | 14 [33] | |
| 6-Cl | 4-Cl | 3l | >100 | < | 65 [33] | |
| 6-Cl | 3-CF3 | 3n | 25 | < | 77 [32] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zitko, J.; Mindlová, A.; Valášek, O.; Jand’ourek, O.; Paterová, P.; Janoušek, J.; Konečná, K.; Doležal, M. Design, Synthesis and Evaluation of N-pyrazinylbenzamides as Potential Antimycobacterial Agents. Molecules 2018, 23, 2390. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092390
Zitko J, Mindlová A, Valášek O, Jand’ourek O, Paterová P, Janoušek J, Konečná K, Doležal M. Design, Synthesis and Evaluation of N-pyrazinylbenzamides as Potential Antimycobacterial Agents. Molecules. 2018; 23(9):2390. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092390
Chicago/Turabian StyleZitko, Jan, Alžběta Mindlová, Ondřej Valášek, Ondřej Jand’ourek, Pavla Paterová, Jiří Janoušek, Klára Konečná, and Martin Doležal. 2018. "Design, Synthesis and Evaluation of N-pyrazinylbenzamides as Potential Antimycobacterial Agents" Molecules 23, no. 9: 2390. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092390