
Microwave-Assisted Extraction of Multiple Trace Levels of Intermediate Metabolites for Camptothecin Biosynthesis in Camptotheca acuminata and Their Simultaneous Determination by HPLC-LTQ-Orbitrap-MS/MS and HPLC-TSQ-MS

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Plant Materials
2.3. Sample Preparation and MAE Procedure
2.4. Optimization of the Extraction Parameters Based on the Taguchi Design
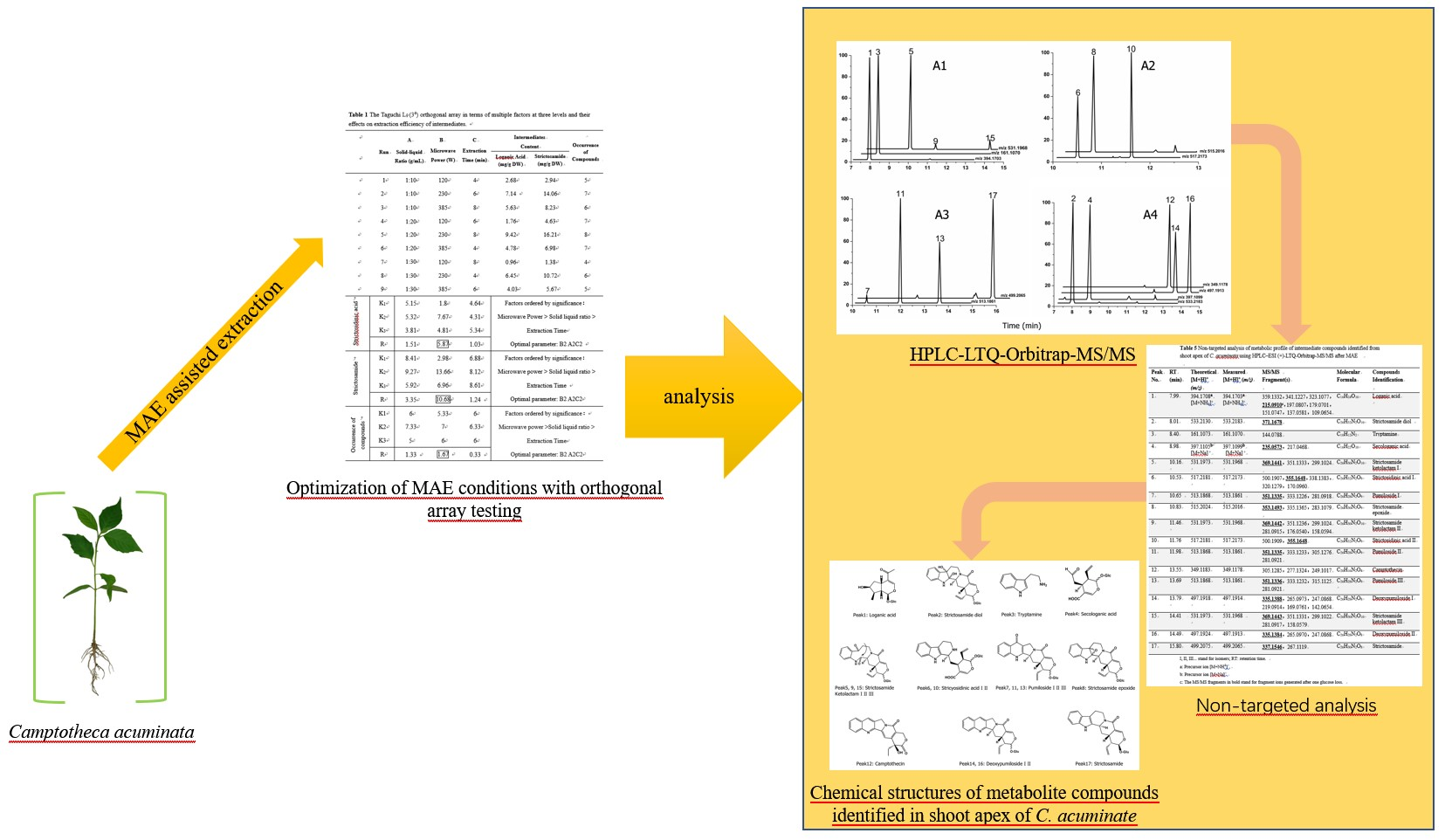
2.5. Determination of Metabolites in C. acuminata by HPLC-LTQ-Orbitrap-MS/MS and HPLC-TSQ-MS
2.6. Scanning Electron Microscopy (SEM) Analysis
3. Results and Discussion
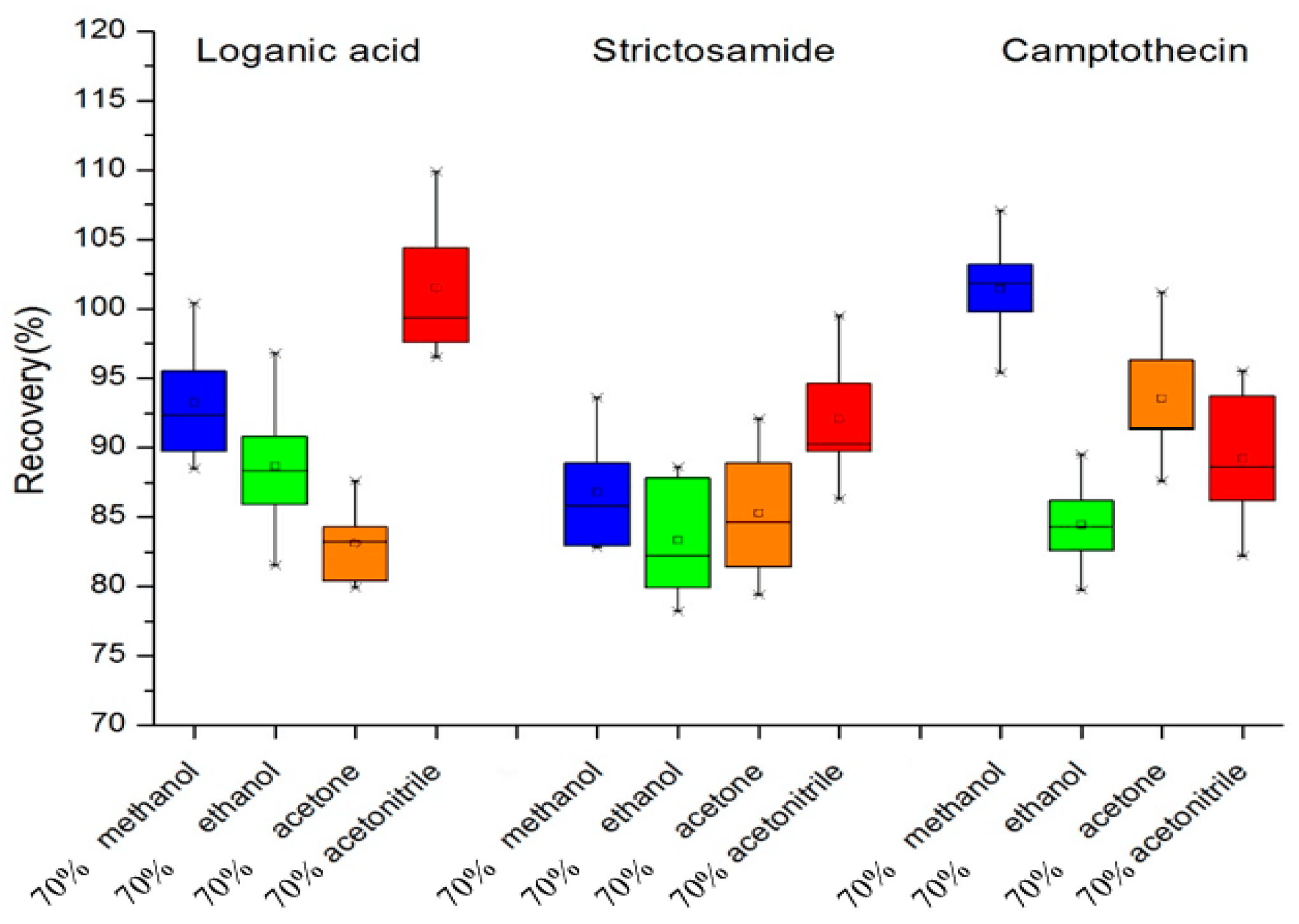
3.1. Selection of Extraction Solvents
3.2. Optimization of MAE Conditions with Orthogonal Array Testing Influential Factors
3.3. SEM Analysis
3.4. Identification of CPT Biosynthetic Intermediates in C. acuminata
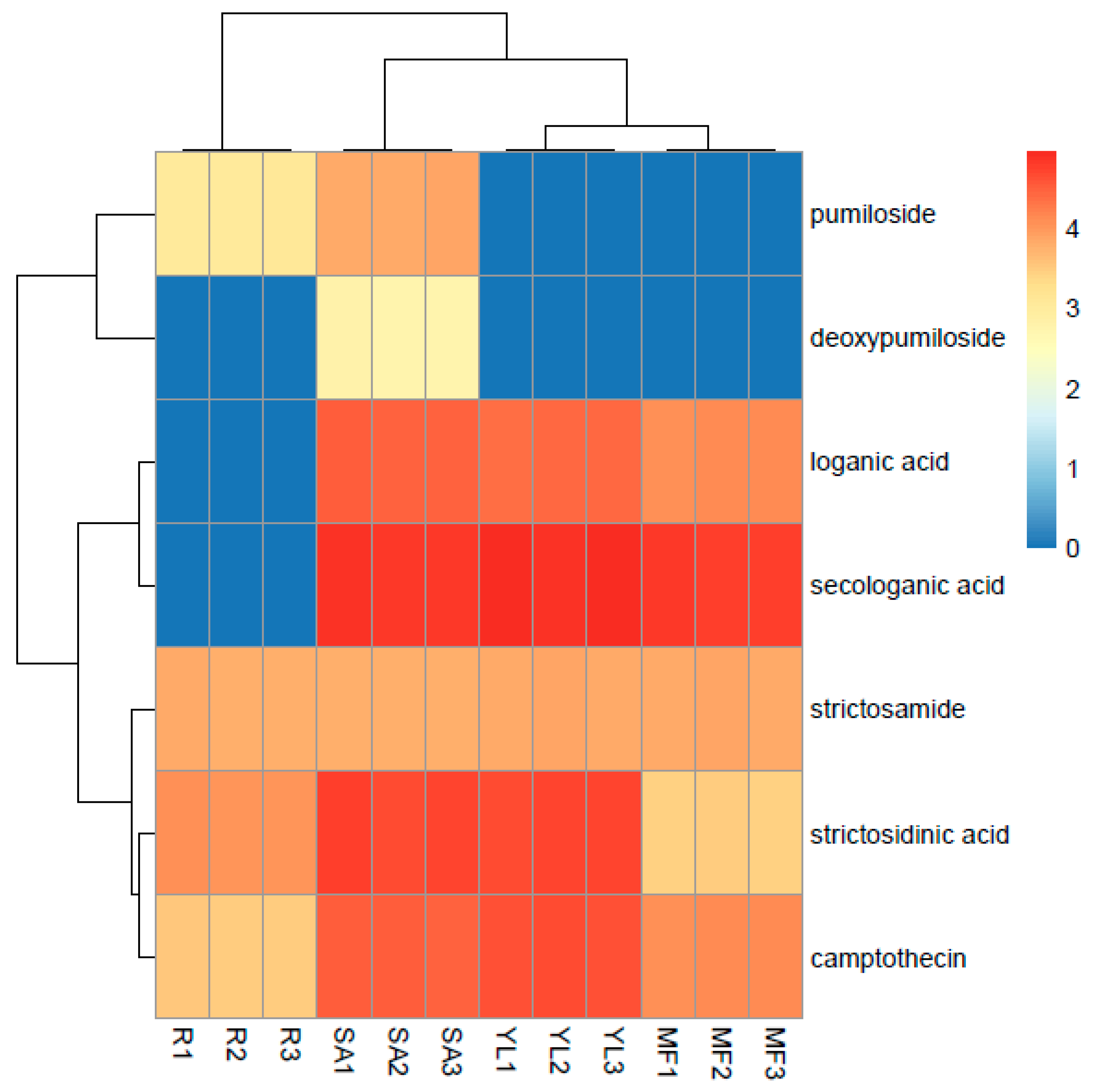
3.5. Comparative Analysis of Metabolites in Plant Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wall, M.E.; Wani, M.C.; Cook, C.E.; Palmer, K.H.; McPhail, A.T.; Sim, G.A. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J. Am. Chem. Soc. 1966, 88, 3887–3890. [Google Scholar] [CrossRef]
- Demain, A.L.; Vaishnav, P. Natural products for cancer chemotherapy. Microb. Biotechnol. 2011, 4, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Uluka, H.; Swaan, P.W. Camptothecins: A review of their chemotherapeutic potential. Drugs 2002, 62, 2039–2057. [Google Scholar] [CrossRef] [PubMed]
- Lorence, A.; Nessler, C.L. Camptothecin, over four decades of surprising findings. Phytochemistry 2004, 65, 2735–2749. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Sudo, H.; Yamazaki, M.; Koseki-Nakamura, M.; Kitajima, M.; Takayama, H.; Aimi, N. Feasible production of camptothecin by hairy root culture of Ophiorrhiza pumila. Plant Cell Rep. 2001, 20, 267–271. [Google Scholar] [CrossRef]
- Geu-Flores, F.; Sherden, N.H.; Courdavault, V.; Burlat, V.; Glenn, W.S.; Wu, C.; Nims, E.; Cui, Y.; O’Connor, S.E. An alternative route to cyclic terpenes by reductive cyclization in iridoid biosynthesis. Nature 2012, 492, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Stöckigt, J.; Barleben, L.; Panjikar, S.; Loris, E.A. 3D-Structure and function of strictosidine synthase—The key enzyme of monoterpenoid indole alkaloid biosynthesis. Plant Physiol. Biochem. 2008, 46, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.X.; Yan, T.S.; Chang, C.H.; Liu, Z.W.; Wang, Y.Y.; Tang, Z.H.; Yu, F. Application of virus-induced gene silencing approach in Camptotheca acuminata. Plant Cell Tissue Org. 2016, 126, 533–540. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Sudo, H.; Yamazaki, M.; Aimi, N.; Saito, K. Camptothecin biosynthetic genes in hairy roots of Ophiorrhizapumila, cloning, characterization and differential expression in tissues and by stress compounds. Plant Cell Physiol. 2003, 44, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Collu, G.; Unver, N.; Peltenburg-Looman, A.M.; van der Heijden, R.; Verpoorte, R.; Memelink, J. Geraniol 10-hydroxylase, a cytochrome P450 enzyme involved in terpenoid indole alkaloid biosynthesis. FEBS Lett. 2001, 508, 215–220. [Google Scholar] [CrossRef]
- Eisenreich, W.; Rohdich, F.; Bacher, A. Deoxyxylulose phosphate pathway to terpenoids. Trends Plant Sci. 2001, 6, 78–84. [Google Scholar] [CrossRef]
- Chen, M.; Li, Y.; Xu, D.; Luo, J.; Kong, L. One-step targeted accumulation and detection of camptothecin analogues from fruits of Camptotheca acuminata Decne using bilayer solid-phase extraction coupled with ultra-high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2017, 1524, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.H.; Wang, S.Y.; Yang, L.; Zu, Y.G.; Yang, F.J.; Zhao, C.J.; Zhang, L.; Zhang, Z.H. Ionic liquid-aqueous solution ultrasonic-assisted extraction of camptothecin and 10-hydroxycamptothecin from Camptothe caacuminata, samara. Chem. Eng. Process 2012, 57, 59–64. [Google Scholar] [CrossRef]
- Nde, D.B.; Boldor, D.; Astete, C. Optimization of microwave assisted extraction parameters of neem (Azadirachtaindica A. Juss) oil using the Doehlert’s experimental design. Ind. Crop. Prod. 2015, 65, 233–240. [Google Scholar] [CrossRef]
- Rokhaya, G.D.; Gregory, D.F.; Shahamat, U.K. Comparison of soxhlet and microwave-assisted extractions for the determination of fenitrothion residues in beans. J. Agric. Food Chem. 2002, 50, 3204–3207. [Google Scholar] [CrossRef]
- Wang, S.Y.; Yang, L.; Zu, Y.G.; Zhao, C.J.; Sun, X.W.; Zhang, L.; Zhang, Z.H. Design and performance evaluation of ionic-liquids-based microwave-assisted environmentally friendly extraction technique for camptothecin and 10-hydroxycamptothecin from samara of Camptotheca acuminata. Ind. Eng. Chem. Res. 2011, 50, 13620–13627. [Google Scholar] [CrossRef]
- Patil, D.M.; Akamanchi, K.G. Microwave assisted process intensification and kinetic modelling: Extraction of camptothecin from Nothapodytes nimmoniana plant. Ind. Crop. Prod. 2017, 98, 60–67. [Google Scholar] [CrossRef]
- Sadre, R.; Magallanes-Lundback, M.; Pradhan, S.; Salim, V.; Mesberg, A.; Jones, A.D.; DellaPenna, D. Metabolite diversity in alkaloid biosynthesis: A multi-lane (diastereomer) highway for camptothecin synthesis in Camptotheca acuminata. Plant Cell 2016, 28, 1926–1944. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.L.; Zu, Y.G.; Yang, L. A novel approach for isolation of essential oil from fresh leaves of Magnolia sieboldii using microwave-assisted simultaneous distillation and extraction. Sep. Purif. Technol. 2015, 154, 271–280. [Google Scholar] [CrossRef]
- Viñas, P.; Campillo, N.; Pastor-Belda, M.; Oller, A.; Hernández-Córdoba, M. Determination of phthalate esters in cleaning and personal care products by dispersive liquid–liquid microextraction and liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2015, 1376, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Li, H.F.; Lin, J.M. Fractional-factorial-design-based microwave-assisted extraction for the determination of organophosphorus and organochlorine residues in tobacco by using GC-MS. J. Sep. Sci. 2016, 40, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Omar, R.; Idris, A.; Yunus, R.; Khalid, K.; Isma, M.A. Characterization of empty fruit bunch for microwave-assisted pyrolysis. Fuel 2011, 90, 1536–1544. [Google Scholar] [CrossRef]
- Zhang, H.F.; Yang, X.H.; Wang, Y. Microwave assisted extraction of secondary metabolites from plants: Current status and future directions. Trends Food Sci. Technol. 2015, 22, 672–688. [Google Scholar] [CrossRef]
- Buttress, A.J.; Binner, E.; Yi, C.; Palade, P.; Robinson, J.P.; Kingman, S.W. Development and evaluation of a continuous microwave processing system for hydrocarbon removal from solids. Chem. Eng. J. 2016, 283, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Hernándeza, A.; Aparicio-Saguilána, A.; Reynoso-Mezab, G.; Carrillo-Ahumada, J. Multi-objective optimization of process conditions in the manufacturing of banana (Musa paradisiaca L.) starch/natural rubber films. Carbohyd. Polym. 2017, 157, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.G.; Gao, Y.X.; Li, D.Q.; Liu, C.F.; Jin, M.; Bian, J.; Lv, M.C.; Sun, Y.Q.; Zhang, L.X.; Gao, P.Y. The neuroprotective and antioxidant profiles of selenium-containing polysaccharides from the fruit of Rosa laevigata. Food Funct. 2018, 9, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Pimentel-Moral, S.; Borrás-linares, I.; Lozano-Sánchez, J.; Arráez-Román, D.; Martínez-Férez, A.; Segura-Carretero, A. Microwave-assisted extraction for Hibiscus sabdariffa bioactive compounds. J. Pharm. Biomed. Anal. 2018, 156, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Montoro, P.; Maldini, M.; Piacente, S.; Macchia, M.; Pizza, C. Metabolite fingerprinting of Camptotheca acuminata and the HPLC-ESI-MS/MS analysis of camptothecin and related alkaloids. J. Pharm. Biomed. 2010, 51, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.G.; Zheng, Z.Q.; Deng, X.Q.; Ma, X.J.; Wang, S.N.; Liu, J.; Liu, Y.; Shi, J.L. Flexible and powerful strategy for qualitative and quantitative analysis of valepotriates in Valeriana jatamansi Jones using high performance liquid chromatography with linear ion trap Orbitrap mass spectrometry. J. Sep. Sci. 2017, 40, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.F.; Wu, W.J.; Huang, M.W.; Long, F.; Liu, X.H.; Zhu, Y.Z. Application of high-performance liquid chromatography coupled with linear ion trap quadrupole orbitrap mass spectrometry for qualitative and quantitative assessment of Shejin-Liyan granule supplements. Molecules 2018, 23, 884. [Google Scholar] [CrossRef] [PubMed]
- Rather, G.A.; Sharma, A.; Pandith, S.A.; Kaul, V.; Nandi, U.; Misra, P.; Lattoo, S.K. De novo transcriptome analyses reveals putative pathway genes involved in biosynthesis and regulation of camptothecin in Nothapodytes nimmoniana (Graham) Mabb. Plant Mol. Biol. 2018, 96, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Carpenter, S.B.; Bourgeois, W.J.; Yu, Y.; Constantin, R.J.; Falcon, M.J.; Adams, J.C. Variations in the secondary metabolite camptothecin in relation to tissue age and season in Camptotheca acuminata. Tree Physiol. 1998, 18, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, G.; Xu, S.; Wu, J.; Yin, Y. Provenance variation in camptothecin concentrations of Camptotheca acuminata grown in China. New For. 2002, 24, 215–224. [Google Scholar] [CrossRef]
- Sankar-Thomas, Y.D.; Lieberei, R. Camptothecin accumulation in various organ cultures of Camptotheca acuminata Decne grown in different culture systems. Plant Cell Tissue. Org. 2011, 106, 445–454. [Google Scholar] [CrossRef]
Sample Availability: Samples of the analyzed plant compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | A Solid-Liquid Ratio (g/mL) | B Microwave Power (W) | C Extraction Time (min) | Intermediates Content | Total Number of Intermediate Compounds | ||
|---|---|---|---|---|---|---|---|
| Loganic Acid (mg/g DW) | Strictosamide (mg/g DW) | ||||||
| 1 | 1:10 | 120 | 4 | 2.68 | 2.94 | 5 | |
| 2 | 1:10 | 230 | 6 | 7.14 | 14.06 | 7 | |
| 3 | 1:10 | 385 | 8 | 5.63 | 8.23 | 6 | |
| 4 | 1:20 | 120 | 6 | 1.76 | 4.63 | 7 | |
| 5 | 1:20 | 230 | 8 | 9.42 | 16.21 | 8 | |
| 6 | 1:20 | 385 | 4 | 4.78 | 6.98 | 7 | |
| 7 | 1:30 | 120 | 8 | 0.96 | 1.38 | 4 | |
| 8 | 1:30 | 230 | 4 | 6.45 | 10.72 | 6 | |
| 9 | 1:30 | 385 | 6 | 4.03 | 5.67 | 5 | |
| Strictosidinic acid | K1 | 5.15 | 1.8 | 4.64 | Factors ordered by significance: Microwave power > Solid liquid ratio > Extraction time Optimal parameter: B2 A2C2 | ||
| K2 | 5.32 | 7.67 | 4.31 | ||||
| K3 | 3.81 | 4.81 | 5.34 | ||||
| R | 1.51 | 1.03 | |||||
| Strictosamide | K1 | 8.41 | 2.98 | 6.88 | Factors ordered by significance: Microwave power > Solid liquid ratio > Extraction time Optimal parameter: B2 A2C2 | ||
| K2 | 9.27 | 13.66 | 8.12 | ||||
| K3 | 5.92 | 6.96 | 8.61 | ||||
| R | 3.35 | 1.24 | |||||
| Occurrence of compounds | K1 | 6 | 5.33 | 6 | Factors ordered by significance: Microwave power > Solid liquid ratio > Extraction time Optimal parameter: B2 A2C2 | ||
| K2 | 7.33 | 7 | 6.33 | ||||
| K3 | 5 | 6 | 6 | ||||
| R | 1.33 | 0.33 | |||||
| Peak No. | RT (min) | Theoretical [M + H]+ (m/z) | Measured [M + H]+ (m/z) | MS/MS Fragment(s) | Molecular Formula | Compound Identification |
|---|---|---|---|---|---|---|
| 1 | 7.99 | 394.1708 a [M + NH4]+ | 394.1703 a [M + NH4]+ | 359.1332, 341.1227, 323.1077, 215.0910 c, 197.0807, 179.0701, 151.0747, 137.0581, 109.0654 | C16H24O10 | Loganic acid |
| 2 | 8.01 | 533.2130 | 533.2183 | 371.1678 | C26H32N2O10 | Strictosamide diol |
| 3 | 8.40 | 161.1073 | 161.1070 | 144.0788 | C10H12N2 | Tryptamine |
| 4 | 8.98 | 397.1105 b [M + Na]+ | 397.1099 b [M + Na]+ | 235.0573, 217.0468 | C16H22O10 | Secologanic acid |
| 5 | 10.16 | 531.1973 | 531.1968 | 369.1441, 351.1333, 299.1024 369.1442, 351.1236, 299.1024 281.0915, 176.0540, 158.0594 369.1443, 351.1331, 299.1022 281.0917, 158.0579 | C26H30N2O10 | Strictosamide ketolactam I |
| 6 | 10.53 | 517.2181 | 517.2173 | 500.1907, 355.1648, 338.1383, 320.1279, 170.0960 500.1909, 355.1648 | C26H32N2O9 | Strictosidinic acid I |
| 7 | 10.65 | 513.1868 | 513.1861 | 351.1335, 333.1226, 281.0918 | C26H28N2O9 | Pumiloside I |
| 8 | 10.83 | 515.2024 | 515.2016 | 353.1493, 335.1365, 283.1079 | C26H30N2O9 | Strictosamide epoxide |
| 9 | 11.46 | 531.1973 | 531.1968 | 369.1442, 351.1236, 299.1024 281.0915, 176.0540, 158.0594 | C26H30N2O10 | Strictosamide ketolactam II |
| 10 | 11.76 | 517.2181 | 517.2173 | 500.1909, 355.1648 | C26H32N2O9 | Strictosidinic acid II |
| 11 | 11.98 | 513.1868 | 513.1861 | 351.1335, 333.1233, 305.1276 281.0921 | C26H28N2O9 | Pumiloside II |
| 12 | 13.55 | 349.1183 | 349.1178 | 305.1285, 277.1324, 249.1017 | C20H16N2O4 | Camptothecin |
| 13 | 13.69 | 513.1868 | 513.1861 | 351.1336, 333.1232, 315.1125 281.0921 | C26H28N2O9 | Pumiloside III |
| 14 | 13.79 | 497.1918 | 497.1914 | 335.1388, 265.0973, 247.0868 219.0914, 169.0761, 142.0654 | C26H28N2O8 | Deoxypumiloside I |
| 15 | 14.41 | 531.1973 | 531.1968 | 369.1443, 351.1331, 299.1022 281.0917, 158.0579 | C26H30N2O10 | Strictosamide ketolactam III |
| 16 | 14.49 | 497.1924 | 497.1913 | 335.1384, 265.0970, 247.0868 | C26H28N2O8 | Deoxypumiloside II |
| 17 | 15.80 | 499.2075 | 499.2065 | 337.1546, 267.1119 | C26H30N2O8 | Strictosamide |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Z.; Wan, R.; Yan, R.; Su, Y.; Huang, H.; Zi, L.; Yu, F. Microwave-Assisted Extraction of Multiple Trace Levels of Intermediate Metabolites for Camptothecin Biosynthesis in Camptotheca acuminata and Their Simultaneous Determination by HPLC-LTQ-Orbitrap-MS/MS and HPLC-TSQ-MS. Molecules 2019, 24, 815. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24040815
Jin Z, Wan R, Yan R, Su Y, Huang H, Zi L, Yu F. Microwave-Assisted Extraction of Multiple Trace Levels of Intermediate Metabolites for Camptothecin Biosynthesis in Camptotheca acuminata and Their Simultaneous Determination by HPLC-LTQ-Orbitrap-MS/MS and HPLC-TSQ-MS. Molecules. 2019; 24(4):815. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24040815
Chicago/Turabian StyleJin, Zhaoxia, Ruyi Wan, Ruxue Yan, Yingying Su, Honglan Huang, Lihan Zi, and Fang Yu. 2019. "Microwave-Assisted Extraction of Multiple Trace Levels of Intermediate Metabolites for Camptothecin Biosynthesis in Camptotheca acuminata and Their Simultaneous Determination by HPLC-LTQ-Orbitrap-MS/MS and HPLC-TSQ-MS" Molecules 24, no. 4: 815. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24040815