
Green Methodologies for Copper(I)-Catalyzed Azide-Alkyne Cycloadditions: A Comparative Study

 , and
, and
Abstract
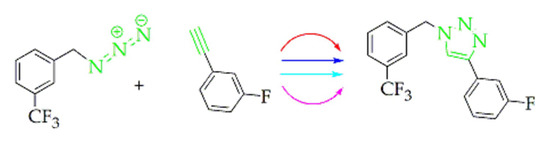
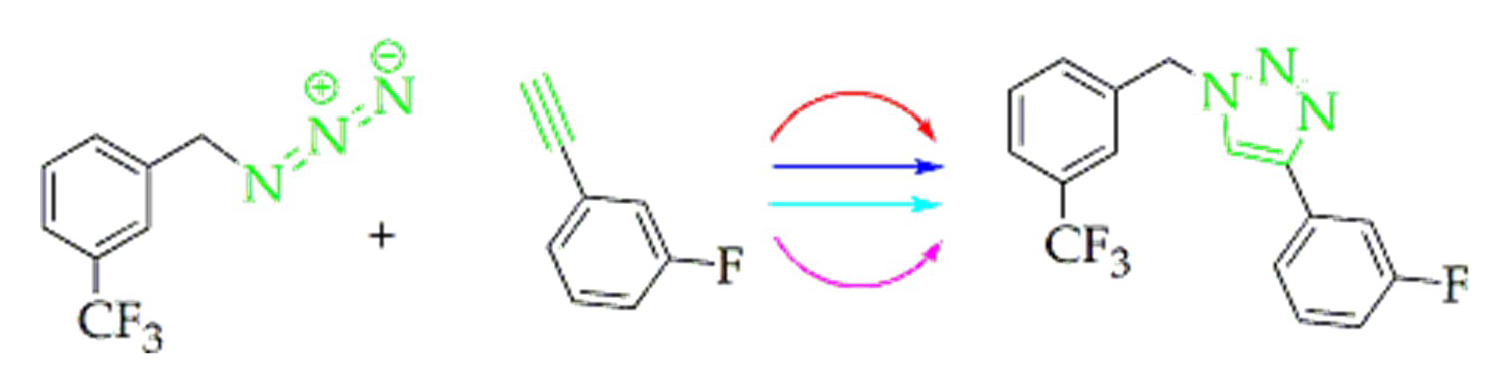
:
1. Introduction
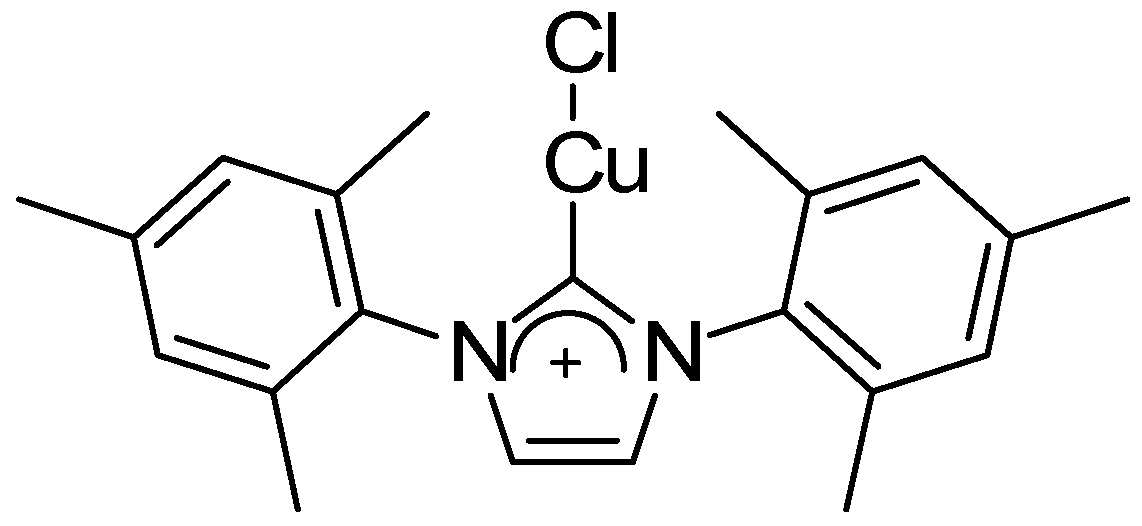
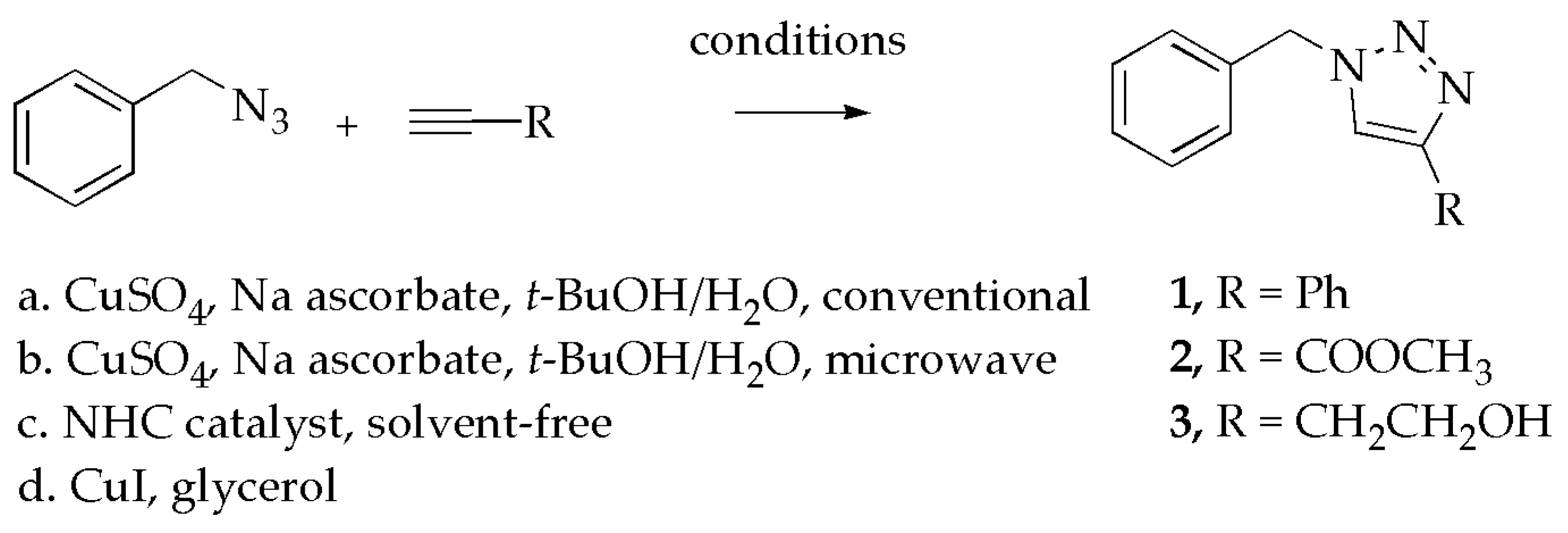
2. Results and Discussion
2.1. Non-Fluorinated Triazoles
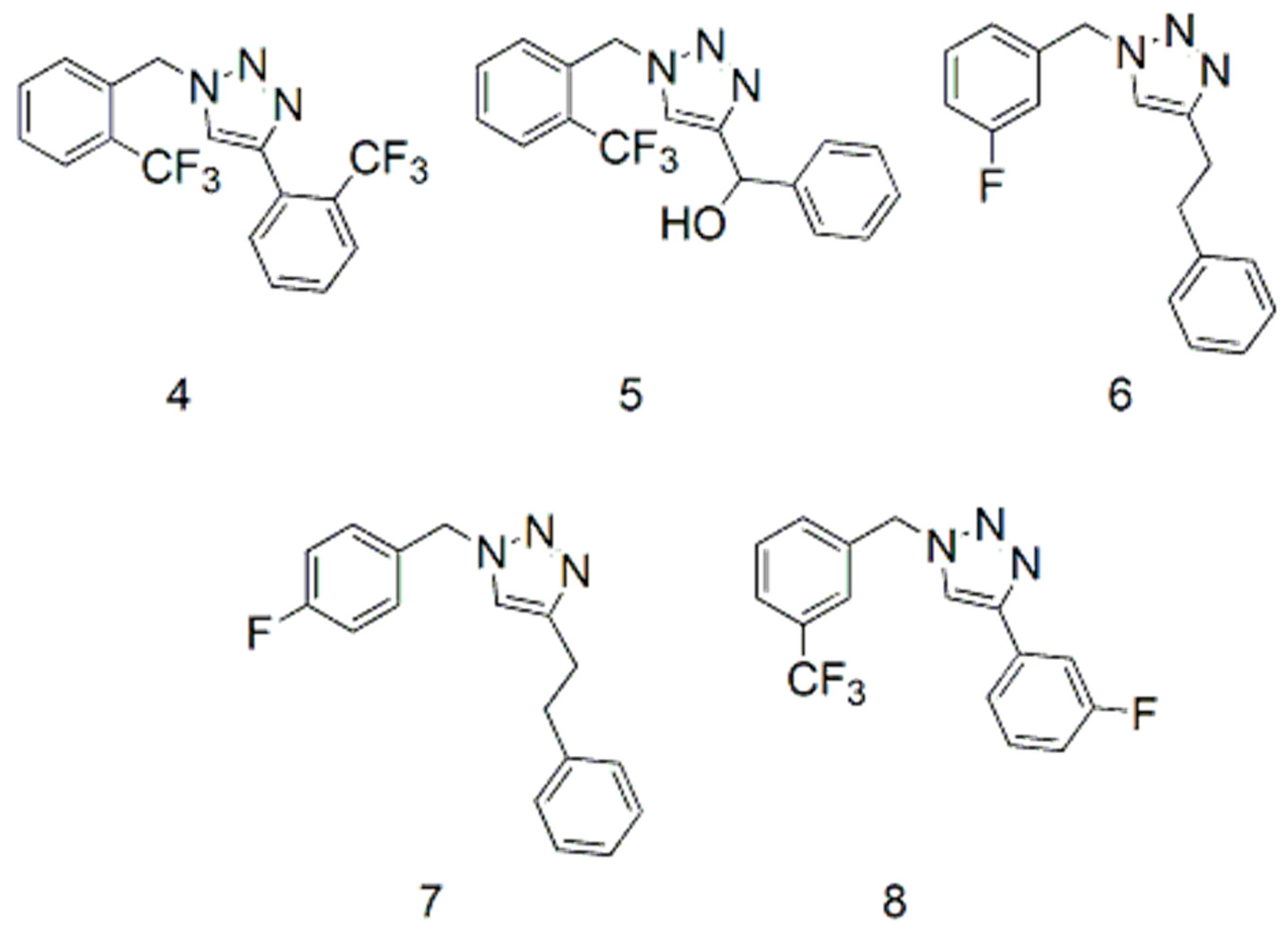
2.2. Fluorinated Triazoles
3. Conclusions
4. Materials and Methods
4.1. General Procedure for the Synthesis of Organic Azides
4.1.1. Procedure for the Conventional Synthesis of 4-Phenyl-1-(phenylmethyl)-1H-1,2,3-triazole (1). Data from One of the Runs
4.1.2. Procedure for the Conventional Synthesis of Methyl 1-(Phenylmethyl)-1H-1,2,3-triazole-4-carboxylate (2). Data from One of the Runs
4.1.3. Procedure for the Conventional Synthesis of 1-(Phenylmethyl)-1H-1,2,3-triazolyl-4-ethanol (3) [40] Data from One of the Runs
4.1.4. Procedure for the Microwave Preparation of Triazoles 1–3
4.1.5. Procedure for the NHC Preparation of Triazoles 1–3
4.1.6. Procedure for the CuI/Glycerol Preparation of Triazoles 1–3
4.1.7. Procedures for the 20 mmol Versions of the Preparation of 1
4.1.8. Procedures for the Preparation of 4-(3-Fluorophenyl)-1-(3-trifluoromethylbenzyl)-1H-1,2,3-triazole (8) (Similar Procedures were Used to Prepare 4–7)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, Z.; Yao, Z.; Xu, X. Recent Advances in Cu-catalyzed Click Reaction and Its Applications. Curr. Org. Chem. 2017, 21, 2240–2248. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Z. Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction. Molecules 2016, 21, 1393. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Brittain, W.D.G.; Buckley, B.R.; Fossey, J.S. Asymmetric copper-catalyzed azide-alkyne cycloadditions. ACS Catal. 2016, 6, 3629–3636. [Google Scholar] [CrossRef]
- Mandoli, A. Recent advances in recoverable systems for the copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC). Molecules 2016, 21, 1174. [Google Scholar] [CrossRef] [PubMed]
- Alonso, F.; Moglie, Y.; Radivoy, G. Copper nanoparticles in click chemistry. Acc. Chem. Res. 2015, 48, 2516–2528. [Google Scholar] [CrossRef] [PubMed]
- Haldon, E.; Nicasio, M.C.; Perez, P.J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef] [PubMed]
- Castro, V.; Rodriguez, H.; Albericio, F. CuAAC: An efficient click chemistry reaction on solid phase. ACS Comb. Sci. 2016, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Monguchi, Y.; Sawama, Y.; Sajiki, H. Synthesis of triazole, indole, and five or six-membered saturated heterocyclic compounds. Heterocycles 2015, 91, 239–264. [Google Scholar]
- Lima, C.G.S.; Ali, A.; van Berkel, S.S.; Westermann, B.; Paixao, M.W. Emerging approaches for the synthesis of triazoles: Beyond metal-catalyzed and strain-promoted azide-alkyne cycloaddition. Chem. Commun. 2015, 51, 10784–10796. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O.; Van der Eycken, E. Click chemistry under non-classical reaction conditions. Chem. Soc. Rev. 2010, 39, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Totobenazara, J.; Burke, A.J. New click-chemistry methods for 1,2,3-triazoles synthesis: Recent advances and applications. Tetrahedron Lett. 2015, 56, 2853–2859. [Google Scholar] [CrossRef]
- Kappe, C.O.; Dallinger, D.; Murphree, S.S. Practical Microwave Synthesis for Organic Chemists; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Hayes, B.L. Microwave Synthesis: Chemistry at the Speed of Light; CEM Publishing: Mathews, NC, USA, 2002. [Google Scholar]
- Loupy, A. Microwaves in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Elgemeie, G.H.; Mohamed, R.A. Microwave synthesis of fluorescent and luminescent dyes (1990–2017). J. Mol. Struct. 2018, 1173, 707–742. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Banik, B.K. Synthesis of Medicinally Privileged Heterocycles through Dielectric Heating. Curr. Med. Chem. 2017, 24, 4596–4626. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Zhang, Z.; Zhang, G.; Ding, Y.; Li, Q.H. Suzuki-Miyaura cross-coupling reaction catalyzed by supported palladium under microwave irradiation. Curr. Org. Synth. 2017, 14, 462–476. [Google Scholar] [CrossRef]
- Ben Nejma, A.; Znati, M.; Daich, A.; Othman, M.; Lawson, A.M.; Ben Jannet, H. Design and semisynthesis of new herbicide as 1,2,3-triazole derivatives of the natural maslinic acid. Steroids 2018, 138, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Kaoukabi, H.; Kabri, Y.; Curti, C.; Taourirte, M.; Rodriguez-Ubis, J.C.; Snoeck, R.; Andrei, G.; Vanelle, P.; Lazrek, H.B. Dihydropyrimidinone/1,2,3-triazole hybrid molecules: Synthesis and anti-varicella-zoster virus (VZV) evaluation. Eur. J. Med. Chem. 2018, 155, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Basu, B. Microwave-induced triazole synthesis via 1,3-dipolar azide-alkyne cycloaddition: Recent advances. Curr. Green Chem. 2016, 3, 195–213. [Google Scholar] [CrossRef]
- Sarmiento-Sanchez, J.I.; Ochoa-Teran, A.; Rivero, I.A. Conventional and microwave assisted synthesis of 1,4-disubstituted 1,2,3-triazoles from Huisgen cycloaddition. Arkivoc 2011, 9, 177–188. [Google Scholar]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Beletskaya, I.P.; Kustov, L.M. Catalysis as an important tool of green chemistry. Russ. Chem. Rev. 2010, 79, 441–461. [Google Scholar] [CrossRef]
- Padmaja, R.D.; Chanda, K. A short review on synthetic advances toward the synthesis of Rufinamide, an antiepileptic drug. Org. Proc. Res. Dev. 2018, 22, 457–466. [Google Scholar] [CrossRef]
- Feroci, M.; Chiarotto, I.; Inesi, A. Electrolysis of ionic liquids. A possible keystone for the achievement of green solvent-catalyst systems. Curr. Org. Chem. 2013, 17, 204–219. [Google Scholar] [CrossRef]
- Sebest, F.; Dunsford, J.J.; Adams, M.; Pivot, J.; Newman, P.D.; Diez-Gonzalez, S. Ring-expanded N-heterocyclic carbenes for copper-mediated azide-alkyne click cycloaddition reactions. ChemCatChem 2018, 10, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Misztalewska-Turkowitz, I.; Markiewicz, K.H.; Michalak, M.; Wilczewska, A.Z. NHC-copper complexes immobilized on magnetic nanoparticles: Synthesis and catalytic activity in the CuAAC reactions. J. Catal. 2018, 362, 46–54. [Google Scholar] [CrossRef]
- Vidal, C.; García-Álvarez, J. Glycerol: A biorenewable solvent for base-free Cu(I)-catalyzed 1,3-dipolar cycloaddition of azides with terminal and 1-iodoalkynes. Highly efficient transformations and catalyst recycling. Green Chem. 2014, 16, 3515–3521. [Google Scholar] [CrossRef]
- Vibhute, S.P.; Mhaldar, P.M.; Korade, S.N.; Gaikwad, D.S.; Shejawal, R.V.; Pore, D.M. Synthesis of magnetically separable catalyst Cu-ACP-Am-Fe3O4@SiO2 for Huisgen 1,3-dipolar cycloaddition. Tetrahedron Lett. 2018, 59, 3643–3652. [Google Scholar] [CrossRef]
- Gupta, A.; Jamatia, R.; Dam, B.; Pal, A.K. Development of synergistic, dual Pd-Cu@rGO catalyst for Suzuki, Heck and click reactions: Facile synthesis of triazole or tetrazole containing biaryls and stilbenes. ChemistrySelect 2018, 3, 8212–8220. [Google Scholar] [CrossRef]
- Fu, F.; Ciganda, R.; Wang, Q.; Tabey, A.; Wang, C.; Escobar, A.; Martinez-Villacorta, A.M.; Hernandez, R.; Moya, S.; Fouquet, E. Cobaltocene reduction of Cu and Ag salts and catalytic behavior of the nanoparticles formed. ACS Catal. 2018, 8, 8100–8106. [Google Scholar] [CrossRef]
- Zhou, Z.; Peng, X.; Zhong, L.; Li, X.; Sun, R. Lignin nanosphere-supported cuprous oxide as an efficient catalyst for huisgen [3 + 2] cycloadditions under relatively mild conditions. Polymers 2018, 10, 724. [Google Scholar] [CrossRef]
- Singh, G.; Kalra, P.; Aanchal Arora, S.; Sharma, G.; Singh, A.; Verma, V. Design and synthesis of indole triazole pendant siloxy framework as a chemo sensor for sensing of Cu2+ and Ni2+: A comparison between traditional and microwave method. Inorg. Chim. Acta 2018, 473, 186–193. [Google Scholar] [CrossRef]
- Kale, S.; Kahandal, S.; Disale, S.; Jayaram, R. Conventional and microwave-assisted multicomponent reaction of alkyne, halide and sodium azide catalyzed by copper apatite as heterogeneous base and catalyst in water. Curr. Chem. Lett. 2012, 1, 69–80. [Google Scholar] [CrossRef]
- Kraljevic, T.G.; Harej, A.; Sedic, M.; Pavelic, S.K.; Stepanic, V.; Drenjancevic, D.; Talapko, J.; Raic-Malic, S. Synthesis, in vitro anticancer and antibacterial activities and in silico studies of new 4-substituted 1,2,3-triazole-coumarin hybrids. Eur. J. Med. Chem. 2016, 124, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Manju, N.; Kalluraya, B.; Kumar, A.; Kumar, M.S. Microwave assisted neat synthesis of 1,2,3-triazoles bearing pyrazole moiety. Heterocycl. Lett. 2018, 8, 619–629. [Google Scholar]
- Kaur, N. Synthesis of five-membered N,N,N- and N,N,N,N-heterocyclic compounds: Applications of microwaves. Synth. Commun. 2015, 45, 1711–1742. [Google Scholar] [CrossRef]
- Appukkuttan, P.; Dehaen, W.; Fokin, V.V.; Van der Eycken, E. A microwave-assisted click chemistry synthesis of 1,4-disubstituted 1,2,3-triazoles via a copper(I)-catalyzed three-component reaction. Org. Lett. 2004, 6, 4223–4225. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.P.; Neuhardt, E.A.; St. John, J.P.; Findlater, M.; Abernethy, C.D. Benchtop synthesis and crystal structure determination of a monomeric N-heterocyclic carbene complex of copper(I) chloride. Transit. Met. Chem. 2010, 35, 415–418. [Google Scholar] [CrossRef]
- Ison, E.A.; Ison, A. Synthesis of well-defined copper N-heterocyclic carbene complexes and their use as catalysts for a “click reaction”: A multistep experiment that emphasizes the role of catalysis in green chemistry. J. Chem. Educ. 2012, 89, 1575–1577. [Google Scholar] [CrossRef]
- Chong, Q.; Zhang, S.; Cheng, F.; Wang, J.; Hong, X.; Meng, F. N-Heterocyclic carbene-Cu-catalyzed enantioselective allenyl conjugate addition. Org. Lett. 2018, 20, 6896–6900. [Google Scholar] [CrossRef] [PubMed]
- Teyssot, M.-L.; Chevry, A.; Traïkia, M.; El-Ghozzi, M.; Avignant, D.; Gautier, A. Improved copper(I)−NHC catalytic efficiency on Huisgen reaction by addition of aromatic nitrogen donors. Chem.-Eur. J. 2009, 15, 6322–6326. [Google Scholar] [CrossRef] [PubMed]
- Díez-Gonzalez, S.; Correa, A.; Cavallo, L.; Nolan, S.P. (NHC)Copper(I)-catalyzed [3 + 2] cycloaddition of azides and mono- or disubstituted alkynes. Chem.-Eur. J. 2006, 12, 7558–7564. [Google Scholar]
- Bruyat, P.; Gautier, A.; Jean, L.; Renard, P.-Y. Use of an air-stable Cu(I)-NHC catalyst for the synthesis of peptidotriazoles. J. Org. Chem. 2018, 83, 13515–13522. [Google Scholar] [CrossRef] [PubMed]
- Topchiy, M.A.; Ageshina, A.A.; Gribanov, P.S.; Masoud, S.M.; Akmalov, T.R.; Nefedov, S.E.; Osipov, S.N.; Nechaev, M.S.; Asachenko, A.F. Azide-alkyne cycloaddition (CuAAC) in alkane solvents catalyzed by fluorinated NHC copper(I) complex. Eur. J. Org. Chem. 2018. [Google Scholar] [CrossRef]
- Guo, S.; Zhou, Y.; Dai, B.; Huo, C.; Liu, C.; Zhao, Y. CuI/Et2NH-catalyzed one-pot highly efficient synthesis of 1,4-disubstituted 1,2,3-triazoles in green solvent glycerol. Synthesis 2018, 50, 2191–2199. [Google Scholar]
- Wharton, Y. Microwave chemistry-out of the lab and into production. In Institute of Chemical Engineers Symposium Series (2011); (3rd European Process Intensification Conference); Institute of Chemical Engineers: London, UK, 2011; Volume 157, pp. 117–121. [Google Scholar]
- Pokhodylo, N.T. Multicomponent and domino reactions leading to 1,2,3-triazoles. Top. Heterocycl. Chem. 2015, 40, 269–324. [Google Scholar]
- Curtius, T.; Ehrhart, G. Decomposition of benzyl azide in indifferent media and in malonic ester. Ber. Dtsch. Chem. Ges. B 1922, 55B, 1559–1571. [Google Scholar] [CrossRef]
- Pietruszka, J.; Solduga, G. Enantiomerically pure cyclopropylamines from cyclopropylboronic ester. Eur. J. Org. Chem. 2009, 34, 5998–6008. [Google Scholar] [CrossRef]
- Giorgi, I.; Bianucci, A.M.; Biagi, G.; Livi, O.; Scartoni, V.; Leonardi, M.; Pietra, D.; Coi, A.; Massarelli, I.; Nofal, F.A.; et al. Synthesis, biological activity and molecular modelling of new trisubstituted 8-azaadenines with high affinity for A1 adenosine receptors. Eur. J. Med. Chem. 2007, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zhang, S.; He, X.; Luo, Z.; Wang, X.; Liu, W.; Qin, X. Design and synthesis of novel β-diketo derivatives as HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2012, 20, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, Z.; Liu, J.; Huang, B.; Kang, D.; Zhang, H.; De Clercq, E.; Daelemans, D.; Pannecouque, C.; Lee, K.-H.; et al. Targeting the entrance channel of NNIBP: Discovery of diarylnicotinamide 1,4-disubstituted triazoles as novel HIV-1 NNRTIs with high potency against wild-type and E138K mutant virus. Eur. J. Med. Chem. 2018, 151, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Dey, S.; Sawoo, S.; Adarsh, N.N.; Sarkar, A. Regioselective 1,3-Dipolar Cycloaddition Reaction of Azides with Alkoxy Alkynyl Fischer Carbene Complexes. Organometallics 2010, 29, 6619–6622. [Google Scholar] [CrossRef]
- Gompper, R. Azole series. VI. The course of the quaternation of 1-substituted 4- and 5-phenyl-1,2,3-triazoles. Chem. Berichte 1957, 90, 382–386. [Google Scholar] [CrossRef]
- Luebbe, F.; Grosz, K.P.; Hillebrand, W.; Sucrow, W. Photolysis of a 2-tetrazene from an enehydrazine. Tetrahedron Lett. 1981, 22, 227–228. [Google Scholar] [CrossRef]
Sample Availability: Samples of compounds 1–5 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Maximum Temperature °C | % Yield (Crude) | % Yield (Pure) |
|---|---|---|---|
| 1 | 60 | 72.1 | 53.4 |
| 2 | 70 | 95.0 | 80.3 |
| 3 | 80 | 100.0 | 89.0 |
| 4 | 90 | 96.2 | 80.8 |
| 5 | 100 | 96.2 | 68.2 |
| 6 | 110 | 89.4 | 68.5 |
| 7 | 120 | 92.6 | 78.9 |
| Method | a. Conventional | b. Microwave | ||||
| Compound | Yield % | Time (h) | Mp °C 1 | Yield % | Time (h) | Mp °C 1 |
| 1 | 82.9 | 2.0 | 128.0–129.5 | 91.8 | 0.25 | 128.7–130.3 |
| 2 | 80.9 | 3.0 | 105.0–105.7 | 88.0 | 0.25 | 106.1–106.4 |
| 3 | 71.3 2 | 2.0 | 40.0–40.9 | 92.1 2 | 0.25 | 44.9–45.5 |
| Avg. | 78.4 | 2.3 | 90.6 | 0.25 | ||
| Method | c. Solvent-Free | d. CuI in Glycerol | ||||
| Compound | Yield % | Time (h) | Mp °C 1 | Yield % | Time (h) | Mp °C 1 |
| 1 | 93.7 | 1.0 | 124.4–126.9 | 62.2 4 | 24 | 126.6–128.4 |
| 2 | 67.9 | 0.5 | 101.8–103.9 | 96.9 | 24 | 103.8–104.7 |
| 3 | 68.4 2 | 1.0 | 42.4–44.0 | 52.0 | 24 | oil |
| Avg. | 76.7 3 | 0.8 | 70.6 | 24 | ||
| Method | Conventional (3 h) | Microwave (5 min) | Solvent-Free (0.5 h) | CuI/Glycerol (1 day) | ||||
|---|---|---|---|---|---|---|---|---|
| Compound | Yield % | Mp, °C | Yield % | Mp, °C | Yield % | Mp, °C | Yield % | Mp, °C |
| 4 | 76.3 | 72.2–72.7 | 96.9 | 71.4–72.0 | 96.4 | 68.5–70.9 | ||
| 5 | 96.1 | 93.2–94.4 | 92.9 | 92.8–94.4 | 100.0 | 92.9–94.2 | 84.0 | 88.1–92.5 |
| 6 | 74.7 | 76.7–77.3 | 78.2 | 76.7–77.3 | 94.5 | 76.3–77.1 | 67.9 | 74.8–75.8 |
| 7 | 91.9 | 94.2–95.5 | 87.8 | 94.2–95.6 | 71.5 | 94.8–95.6 | 85.6 | 93.9–94.5 |
| 8 | 85.7 | 62.5–63.2 | 96.7 | 62.6–63.2 | 88.9 | 59.4–63.4 | 83.7 | 61.1–61.7 |
| Avg. | 84.9 | 90.5 | 90.3 | 80.3 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trujillo, M.; Hull-Crew, C.; Outlaw, A.; Stewart, K.; Taylor, L.; George, L.; Duensing, A.; Tracey, B.; Schoffstall, A. Green Methodologies for Copper(I)-Catalyzed Azide-Alkyne Cycloadditions: A Comparative Study. Molecules 2019, 24, 973. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050973
Trujillo M, Hull-Crew C, Outlaw A, Stewart K, Taylor L, George L, Duensing A, Tracey B, Schoffstall A. Green Methodologies for Copper(I)-Catalyzed Azide-Alkyne Cycloadditions: A Comparative Study. Molecules. 2019; 24(5):973. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050973
Chicago/Turabian StyleTrujillo, Marissa, Clayton Hull-Crew, Andrew Outlaw, Kevin Stewart, Loren Taylor, Laura George, Allison Duensing, Breanna Tracey, and Allen Schoffstall. 2019. "Green Methodologies for Copper(I)-Catalyzed Azide-Alkyne Cycloadditions: A Comparative Study" Molecules 24, no. 5: 973. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050973