
The Influence of Dimerization on the Pharmacokinetics and Activity of an Antibacterial Enzyme Lysostaphin

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Creation of a Dimerized Version of Lysostaphin
2.2. Lst-HDD Predominantly Exists as A Dimer
2.3. Bacteriolytic But Not the Catalytic Activity of Lysostaphin Is Affected by Dimerization
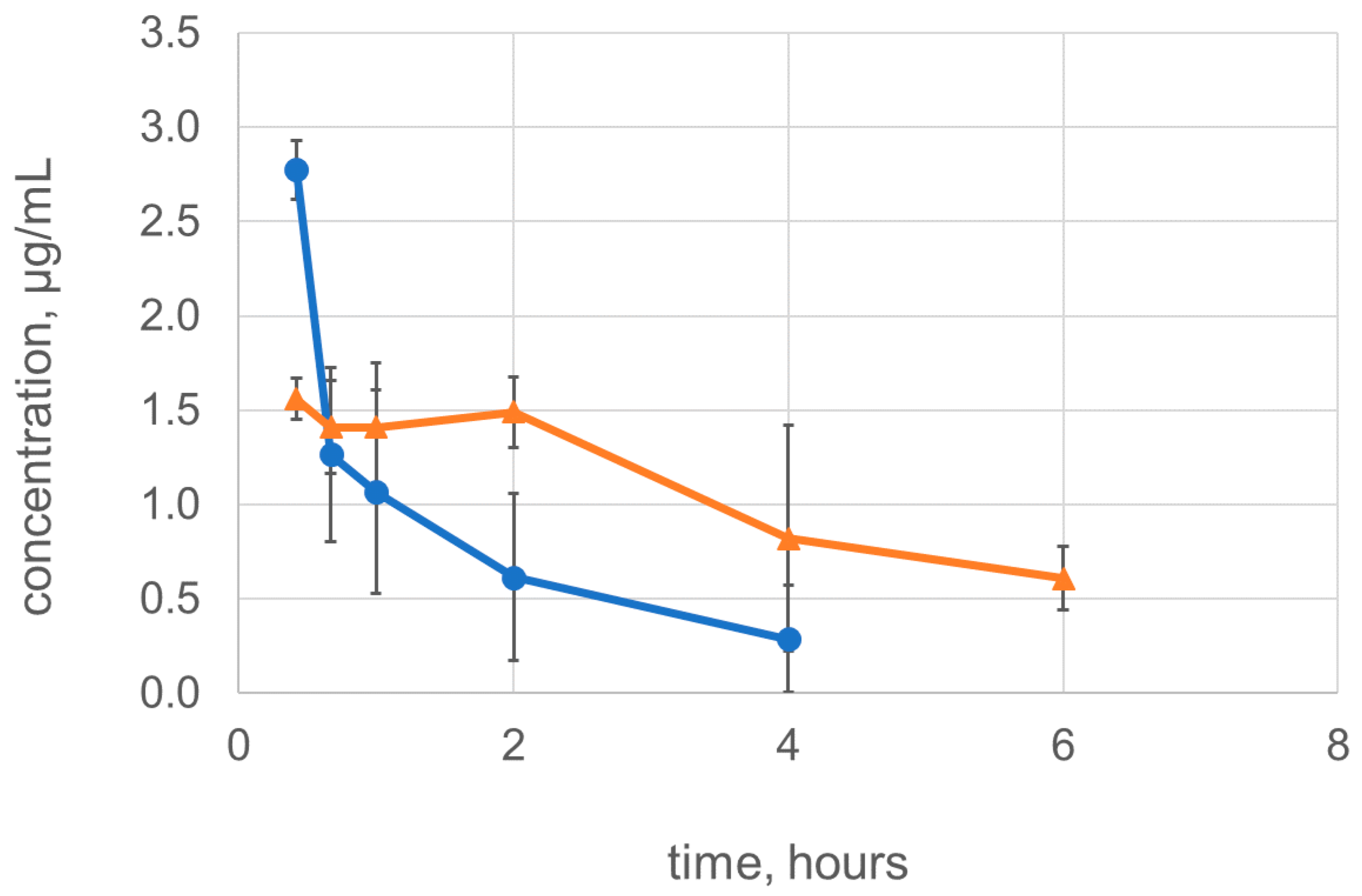
2.4. Dimerization Improves the Pharmacokinetic Characteristics of Lysostaphin
3. Materials and Methods
3.1. Cloning, Expression, and Purification
3.2. Size Exclusion Chromatography
3.3. Catalytic Activity
3.4. Minimum Inhibitory Concentration
3.5. Staphylolytic Activity
3.6. Anti-lysostaphin Antibody Production
3.7. Pharmacokinetics
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| BCA | Bicinchoninic acid |
| BHI | Brain heart infusion |
| CFU | Colony forming units |
| DTT | Dithiothreitol |
| ELISA | Enzyme-linked immunosorbent assay |
| HRP | Horseradish peroxidase |
| IL-1 | Interleukin 1 |
| IPTG | Isopropyl thiogalactoside |
| MALDI | Matrix-assisted laser desorption/ionisation |
| MIC | Minimum inhibitory concentration |
| PEG | Polyethylene glycol |
| SEC | Size exclusion chromatography |
| TBS | Tris-buffered saline |
| TBST | Tris-buffered saline with 0.05% Tween 20 |
| TMB | 3,3′-5,5′-Tetramethylbenzidine |
| VEGF | Vascular endothelial growth factor |
References
- Smith, R.A.; M’Ikanatha, N.M.; Read, A.F. Antibiotic Resistance: A Primer and Call to Action. Health Commun. 2015, 30, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Sabtu, N.; Enoch, D.A.; Brown, N.M. Antibiotic resistance: What, why, where, when and how? Br. Med. Bull. 2015, 116, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [PubMed]
- Cetinkaya, Y.; Falk, P.; Mayhall, C.G. Vancomycin-Resistant Enterococci. Clin. Microbiol. Rev. 2000, 13, 686–707. [Google Scholar] [CrossRef] [PubMed]
- Gajdács, M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics 2019, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Sheu, C.C.; Lin, S.Y.; Chang, Y.T.; Lee, C.Y.; Chen, Y.H.; Hsueh, P.R. Management of infections caused by extended-spectrum β–lactamase-producing Enterobacteriaceae: current evidence and future prospects. Expert Rev. Anti. Infect. Ther. 2018, 16, 205–218. [Google Scholar] [CrossRef]
- El Chakhtoura, N.G.; Saade, E.; Iovleva, A.; Yasmin, M.; Wilson, B.; Perez, F.; Bonomo, R.A. Therapies for multidrug resistant and extensively drug-resistant non-fermenting gram-negative bacteria causing nosocomial infections: a perilous journey toward ‘molecularly targeted’ therapy. Expert Rev. Anti-Infect. Ther. 2018, 16, 89–110. [Google Scholar] [CrossRef]
- Gajdács, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef]
- Schillaci, D.; Spanò, V.; Parrino, B.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G.; Cascioferro, S. Pharmaceutical Approaches to Target Antibiotic Resistance Mechanisms. J. Med. Chem. 2017, 60, 8268–8297. [Google Scholar] [CrossRef]
- Spengler, G.; Kincses, A.; Gajdács, M.; Amaral, L. New Roads Leading to Old Destinations: Efflux Pumps as Targets to Reverse Multidrug Resistance in Bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef]
- Assis, L.M.; Nedeljković, M.; Dessen, A. New strategies for targeting and treatment of multi-drug resistant Staphylococcus aureus. Drug Resist. Updat. 2017, 31, 1–14. [Google Scholar] [CrossRef]
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Pastagia, M.; Schuch, R.; Fischetti, V.A.; Huang, D.B. Lysins: the arrival of pathogen-directed anti-infectives. J. Med. Microbiol. 2013, 62, 1506–1516. [Google Scholar] [CrossRef] [Green Version]
- Sharma, U.; Vipra, A.; Channabasappa, S. Phage-derived lysins as potential agents for eradicating biofilms and persisters. Drug Discov. Today 2018, 00, 1–10. [Google Scholar] [CrossRef]
- São-José, C. Engineering of Phage-Derived Lytic Enzymes: Improving Their Potential as Antimicrobials. Antibiotics 2018, 7, 29. [Google Scholar] [CrossRef]
- Jun, S.Y.; Jang, I.J.; Yoon, S.; Jang, K.; Yu, K.-S.; Cho, J.Y.; Seong, M.-W.; Jung, G.M.; Yoon, S.J.; Kang, S.H. Pharmacokinetics and Tolerance of the Phage Endolysin-Based Candidate Drug SAL200 after a Single Intravenous Administration among Healthy Volunteers. Antimicrob. Agents Chemother. 2017, 61, 1–11. [Google Scholar] [CrossRef]
- Cassino, C.; Murphy, M.G.; Boyle, J.; Rotolo, J.; Wittekind, M. Results of the First in Human Study of Lysin CF-301 Evaluating the Safety, Tolerability and Pharmacokinetic Profile in Healthy Volunteers. In Proceedings of the 26th European Congress of Clinical Microbiology and Infectious Diseases, Amsterdam, The Netherlands, 9–12 April 2016. [Google Scholar]
- Tang, L.; Meibohm, B. Pharmacokinetics of Peptides and Proteins. In Pharmacokinetics and Pharmacodynamics of Biotech Drugs: Principles and Case Studies in Drug Development; Wiley-VCH: Weinheim, Germany, 2006; ISBN 3527314083. [Google Scholar]
- Walsh, S.; Shah, A.; Mond, J. Improved Pharmacokinetics and Reduced Antibody Reactivity of Lysostaphin Conjugated to Polyethylene Glycol. Antimicrob. Agents Chemother. 2003, 47, 554–558. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, J.M.; Djurkovic, S.; Fischetti, V.A. Phage Lytic Enzyme Cpl-1 as a Novel Antimicrobial for Pneumococcal Bacteremia. Infect. Immun. 2003, 71, 6199–6204. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Cerrato, V.; García, P.; Huelves, L.; García, E.; Del Prado, G.; Gracia, M.; Ponte, C.; López, R.; Soriano, F. Pneumococcal LytA autolysin, a potent therapeutic agent in experimental peritonitis-sepsis caused by highly β-lactam-resistant Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2007, 51, 3371–3373. [Google Scholar] [CrossRef]
- Abdelkader, K.; Gerstmans, H.; Saafan, A.; Dishisha, T.; Briers, Y. The Preclinical and Clinical Progress of Bacteriophages and Their Lytic Enzymes: The Parts are Easier than the Whole. Viruses 2019, 11, 96. [Google Scholar] [CrossRef]
- Entenza, J.M.; Loeffler, J.M.; Grandgirard, D.; Fischetti, V.A.; Moreillon, P. Therapeutic Effects of Bacteriophage Cpl-1 Lysin against Streptococcus pneumoniae Endocarditis in Rats. Antimicrob. Agents Chemother. 2005, 49, 4789–4792. [Google Scholar] [CrossRef] [Green Version]
- Kokai-Kun, J.F.; Chanturiya, T.; Mond, J.J. Lysostaphin as a treatment for systemic Staphylococcus aureus infection in a mouse model. J. Antimicrob. Chemother. 2007, 60, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Swierczewska, M.; Lee, K.C.; Lee, S. What is the future of PEGylated therapies? Expert Opin. Emerg. Drugs 2015, 20, 531–536. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Liu, M.; Wan, H. Discussion about Several Potential Drawbacks of PEGylated Therapeutic Proteins. Biol. Pharm. Bull. 2013, 37, 335–339. [Google Scholar] [CrossRef]
- Resch, G.; Moreillon, P.; Fischetti, V.A. PEGylating a bacteriophage endolysin inhibits its bactericidal activity. AMB Express 2011, 1, 29. [Google Scholar] [CrossRef]
- Cuesta, A.M.; Sainz-Pastor, N.; Bonet, J.; Oliva, B.; Alvarez-Vallina, L. Multivalent antibodies: when design surpasses evolution. Trends Biotechnol. 2010, 28, 355–362. [Google Scholar] [CrossRef]
- Sytkowski, A.; Lunn, E. Human erythropoietin dimers with markedly enhanced in vivo activity. Proc. Nat. Acad. Sci. USA 1998, 95, 1184–1188. [Google Scholar] [CrossRef] [Green Version]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Resch, G.; Moreillon, P.; Fischetti, V.A. A stable phage lysin (Cpl-1) dimer with increased antipneumococcal activity and decreased plasma clearance. Int. J. Antimicrob. Agents 2011, 38, 516–521. [Google Scholar] [CrossRef]
- Kokai-kun, J.F. Lysostaphin: A silver bullet for staph. In Antimicrobial Drug Discovery: Emerging Strategies; CAB International: Wallingford, UK, 2012; pp. 147–165. ISBN 978-1-84593-943-4. [Google Scholar]
- Gurnon, D.G.; Whitaker, J.A.; Oakley, M.G. Design and Characterization of a Homodimeric Antiparallel Coiled Coil. J. Am. Chem. Soc. 2003, 125, 7518–7519. [Google Scholar] [CrossRef]
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. Version 9.0, 2019. Available online: http://www.eucast.org (accessed on 11 April 2019).
- Mitkowski, P.; Jagielska, E.; Nowak, E.; Bujnicki, J.M.; Stefaniak, F.; Niedziałek, D.; Bochtler, M.; Sabała, I. Structural bases of peptidoglycan recognition by lysostaphin SH3b domain. Sci. Rep. 2019, 9, 5965. [Google Scholar] [CrossRef]
- Rennke, H.G.; Patel, Y.; Venkatachalam, M.A. Glomerular filtration of proteins: Clearance of anionic, neutral, and cationic horseradish peroxidase in the rat. Kidney Int. 1978, 13, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Boksha, I.S.; Lavrova, N.V.; Grishin, A.V.; Demidenko, A.V.; Lyashchuk, A.M.; Galushkina, Z.M.; Ovchinnikov, R.S.; Umyarov, A.M.; Avetisian, L.R.; Chernukha, M.I.; et al. Staphylococcus simulans recombinant lysostaphin: Production, purification, and determination of antistaphylococcal activity. Biochemistry 2016, 81, 502–510. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havliš, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2007, 1, 2856–2860. [Google Scholar] [CrossRef]
- Vetchinin, S.S.; Pavlov, V.M.; Galkina, E.V.; Vakhrameeva, G.M.; Grishaeva, N.S.; Mokrievich, A.N.; Djatlov, I.A. Strain of Hybrid Cultured Animal Cells Mus Musculus 2f9-Producer of Monoclonal Antibodies Specific for Lysostaphin and Inhibiting Its Lytic Activity. Patent RU 2525663 C1, 20 August 2014. [Google Scholar]
- Gabrielsson, J.; Weiner, D. Non-compartmental Analysis. In Computational Toxicology: Volume I, Methods in Molecular Biology; Humana Press: New York, NY, USA, 2012; Volume 929, pp. 377–389. ISBN 9781627030502. [Google Scholar]
- Diehl, K.H.; Hull, R.; Morton, D.; Pfister, R.; Rabemampianina, Y.; Smith, D.; Vidal, J.M.; Van De Vorstenbosch, C. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J. Appl. Toxicol. 2001, 21, 15–23. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples pL330 and pL473 plasmids are available from the authors. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grishin, A.V.; Lavrova, N.V.; Lyashchuk, A.M.; Strukova, N.V.; Generalova, M.S.; Ryazanova, A.V.; Shestak, N.V.; Boksha, I.S.; Polyakov, N.B.; Galushkina, Z.M.; et al. The Influence of Dimerization on the Pharmacokinetics and Activity of an Antibacterial Enzyme Lysostaphin. Molecules 2019, 24, 1879. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101879
Grishin AV, Lavrova NV, Lyashchuk AM, Strukova NV, Generalova MS, Ryazanova AV, Shestak NV, Boksha IS, Polyakov NB, Galushkina ZM, et al. The Influence of Dimerization on the Pharmacokinetics and Activity of an Antibacterial Enzyme Lysostaphin. Molecules. 2019; 24(10):1879. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101879
Chicago/Turabian StyleGrishin, Alexander V., Natalia V. Lavrova, Alexander M. Lyashchuk, Natalia V. Strukova, Maria S. Generalova, Anna V. Ryazanova, Nikita V. Shestak, Irina S. Boksha, Nikita B. Polyakov, Zoya M. Galushkina, and et al. 2019. "The Influence of Dimerization on the Pharmacokinetics and Activity of an Antibacterial Enzyme Lysostaphin" Molecules 24, no. 10: 1879. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101879