Rippled β-Sheet Formation by an Amyloid-β Fragment Indicates Expanded Scope of Sequence Space for Enantiomeric β-Sheet Peptide Coassembly


Abstract
:
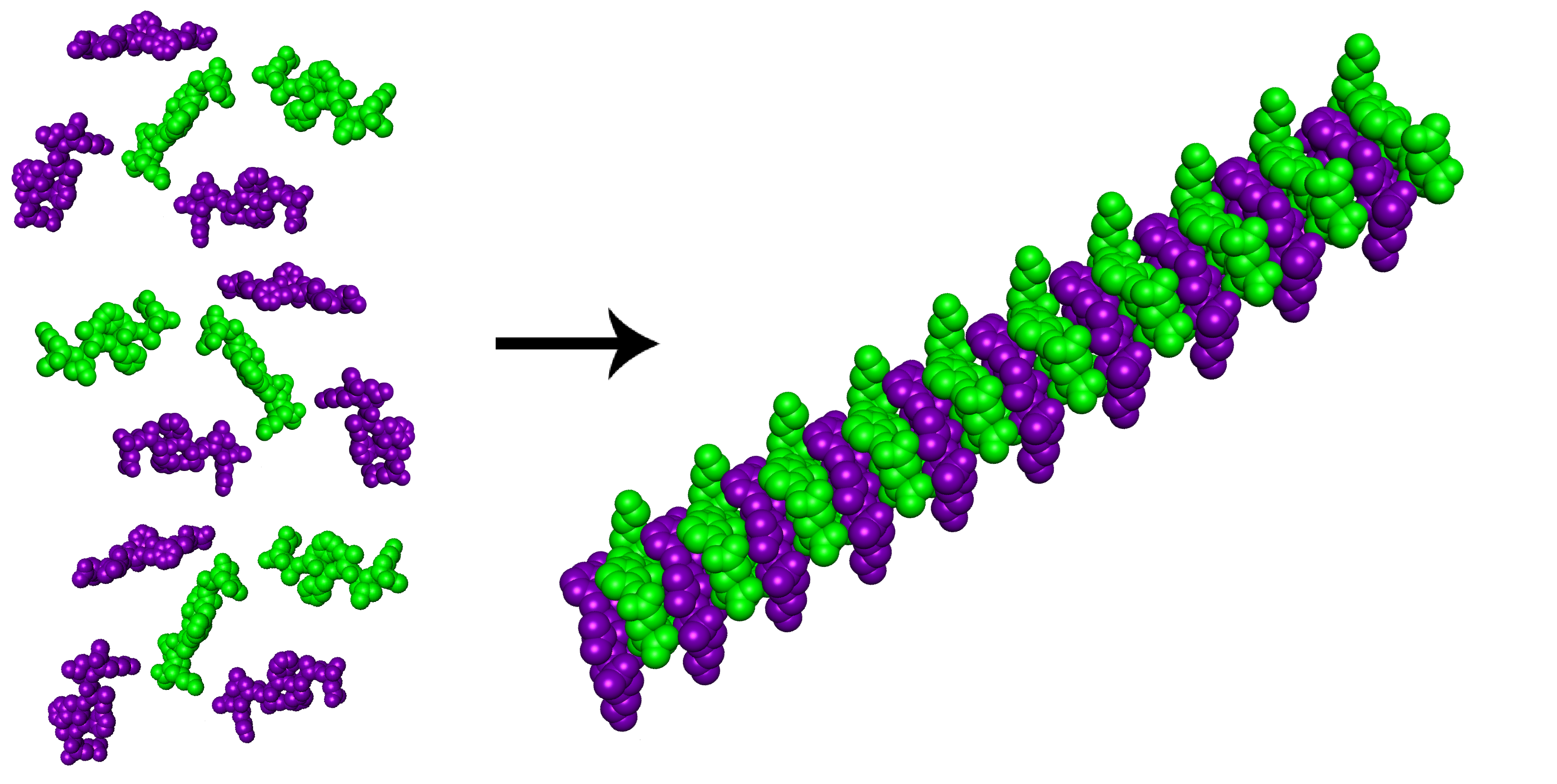{kind=link}
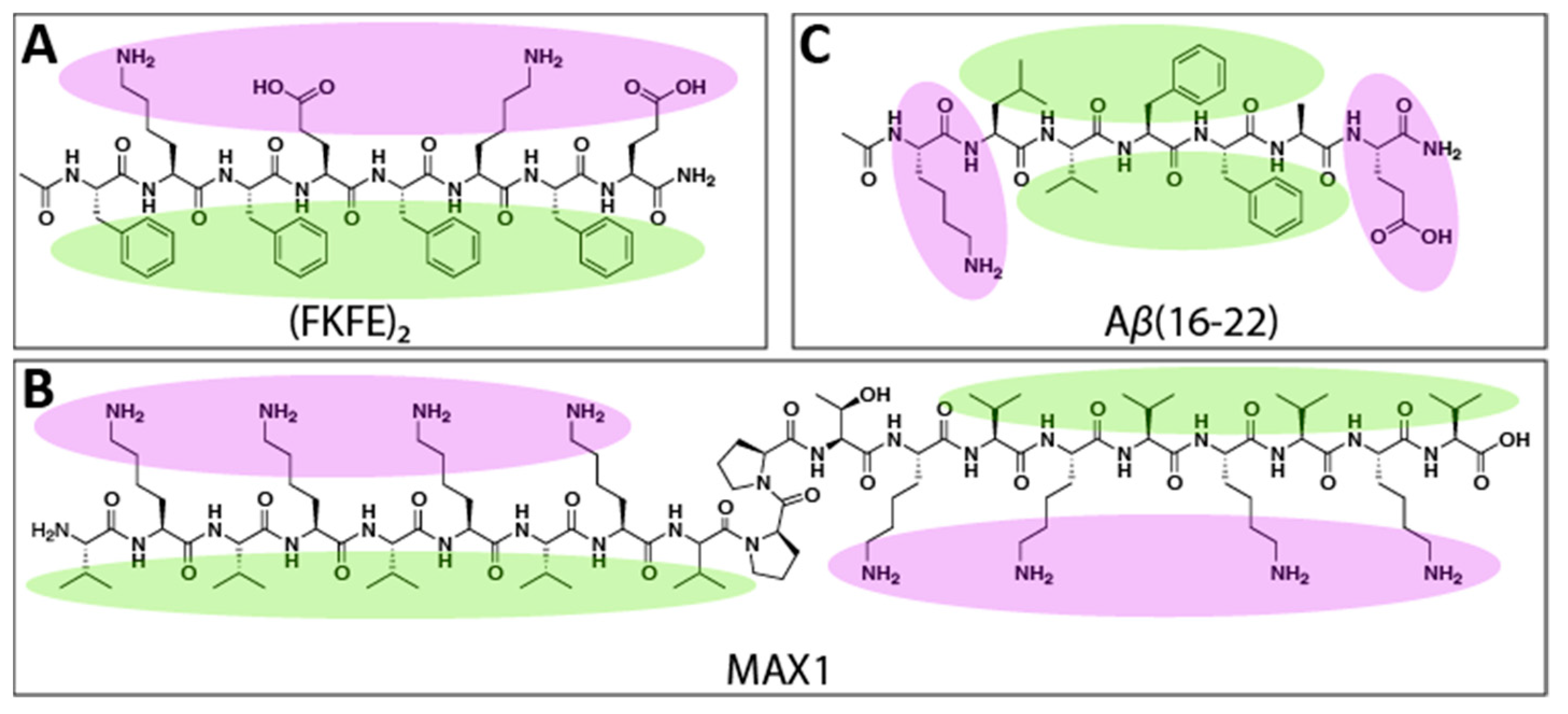{kind=link}
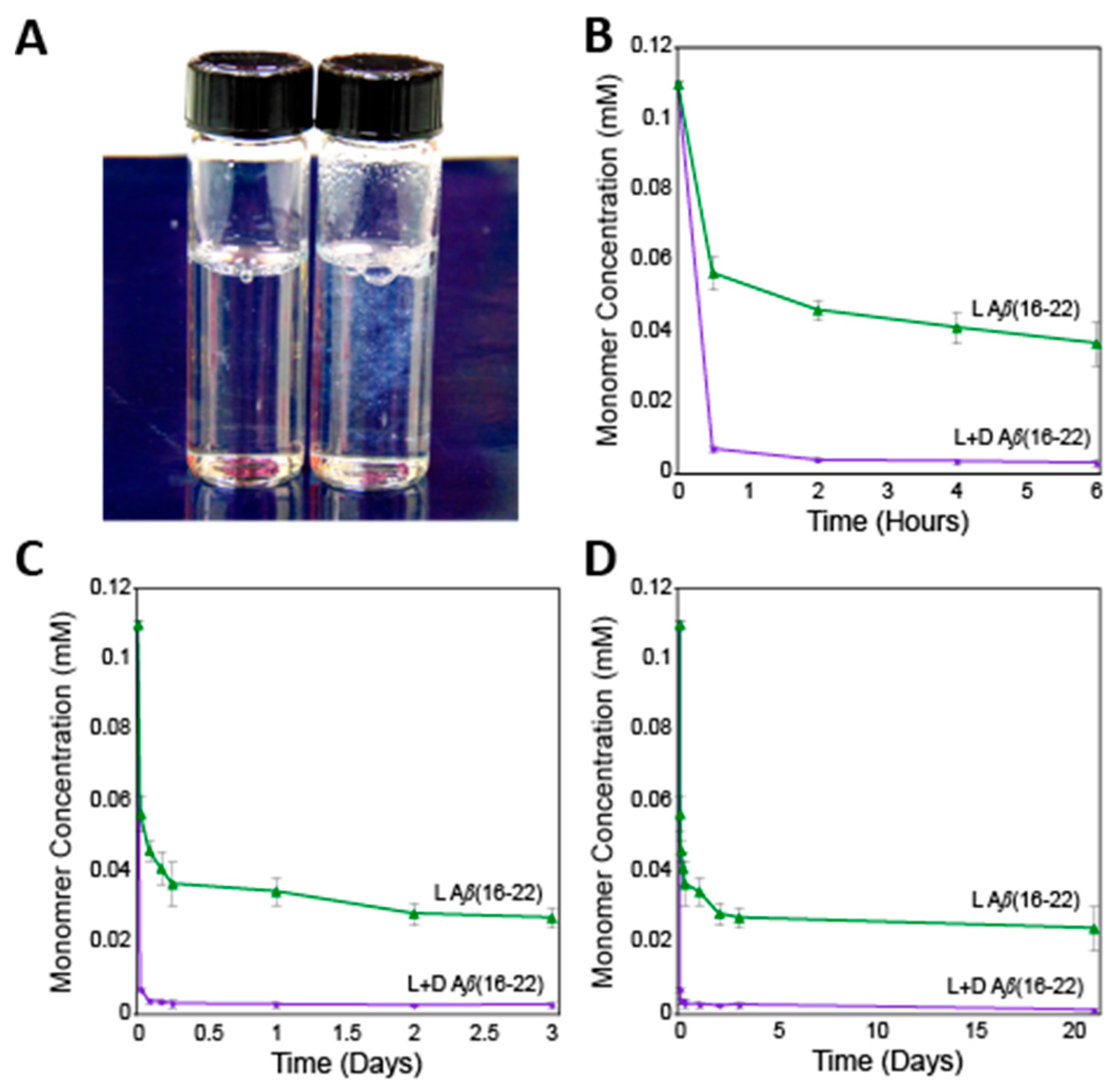{kind=link}
{kind=link}
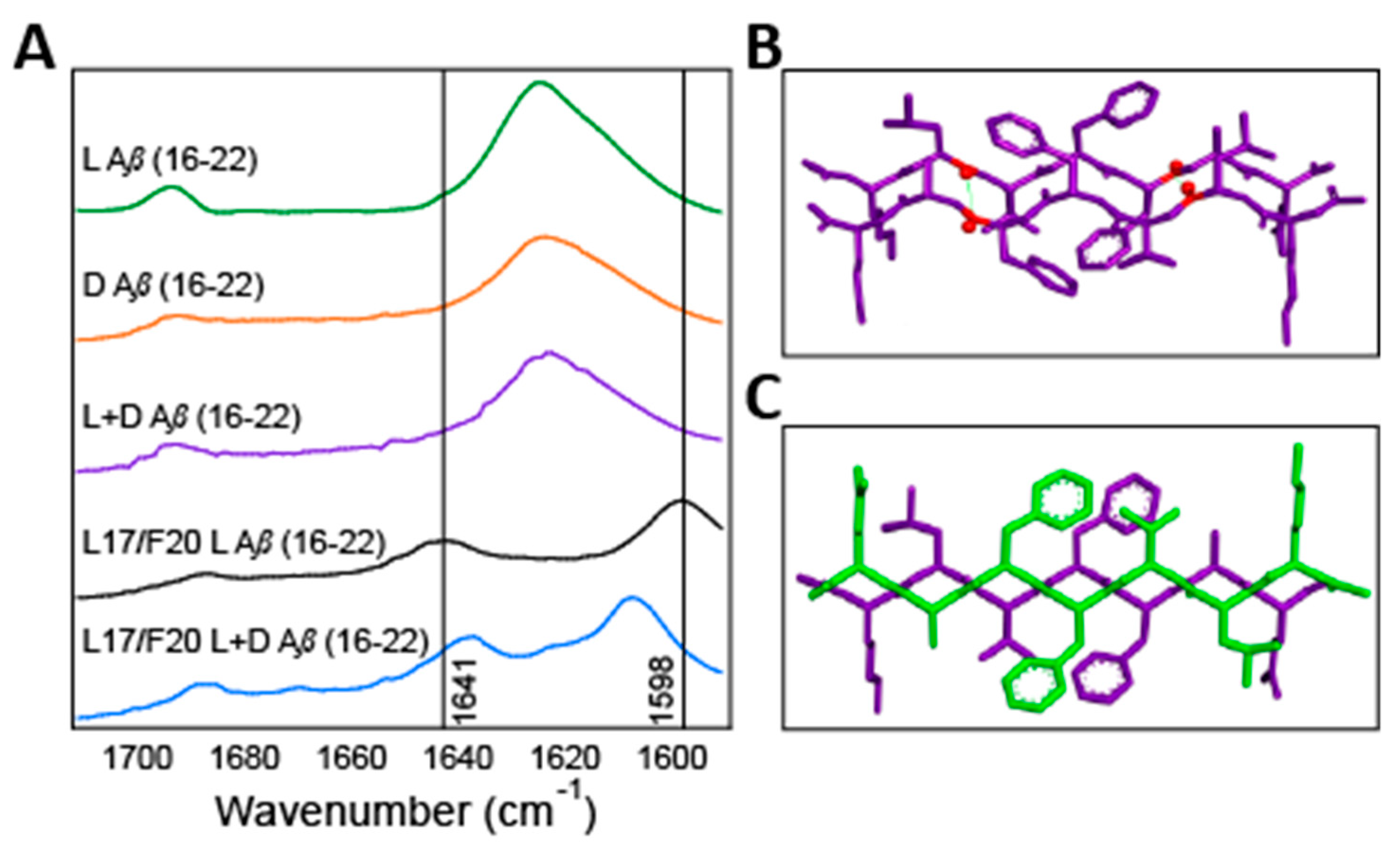{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Sedimentation Analysis
2.2. Transmission and Scanning Electron Microscopy
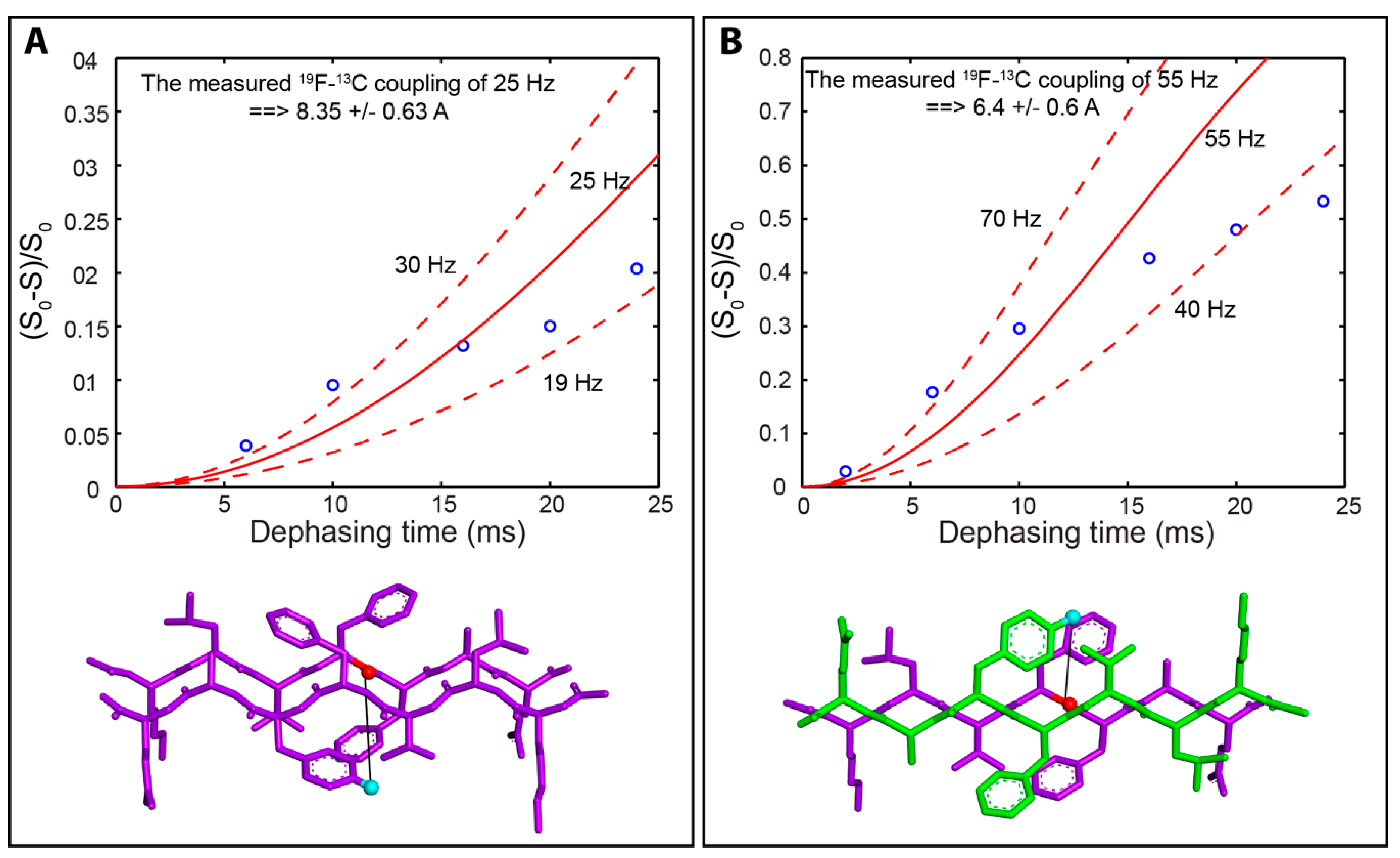
2.3. Isotope Edited Infrared Spectroscopy
2.4. Solid-State Nuclear Magnetic Resonance Spectroscopy
3. Conclusions
4. Materials and Methods
4.1. Peptide Synthesis
4.2. Peptide Purification and Characterization
4.3. Peptide Disaggregation Protocol
4.4. Sedimentation Assays
4.5. Solid-State Fourier Transform Infrared Spectroscopy (FTIR)
4.6. Transmission Electron Microscopy (TEM)
4.7. Scanning Electron Microscopy (SEM)
4.8. Solid State Nuclear Magnetic Resonance (ssNMR) Spectroscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ow, S.Y.; Dunstan, D.E. A brief overview of amyloids and Alzheimer’s disease. Protein Sci. 2014, 23, 1315–1331. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W. Peptide fibrillization. Angew. Chem. Int. Ed. Engl. 2007, 46, 8128–8147. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.S.; Sharpe, P.C.; Singh, Y.; Fairlie, D.P. Amyloid peptides and proteins in review. Rev. Physiol. Biochem. Pharmacol. 2007, 159, 1–77. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Wang, M.; Audas, T.E.; Lee, S. Disentangling a Bad Reputation: Changing Perceptions of Amyloids. Trends Cell Biol. 2017, 27, 465–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonets, K.S.; Nizhnikov, A.A. Amyloids and prions in plants: Facts and perspectives. Prion 2017, 11, 300–312. [Google Scholar] [CrossRef] [Green Version]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef]
- Som Chaudhury, S.; Das Mukhopadhyay, C. Functional amyloids: Interrelationship with other amyloids and therapeutic assessment to treat neurodegenerative diseases. Int. J. Neurosci. 2017, 128, 1–15. [Google Scholar] [CrossRef]
- Eskandari, S.; Guerin, T.; Toth, I.; Stephenson, R.J. Recent advances in self-assembled peptides: Implications for targeted drug delivery and vaccine engineering. Adv. Drug Deliv. Rev. 2017, 110–111, 169–187. [Google Scholar] [CrossRef]
- Waku, T.; Tanaka, N. Recent advances in nanofibrous assemblies based on β-sheet-forming peptides for biomedical applications. Polym. Int. 2017, 66, 277–288. [Google Scholar] [CrossRef]
- King, P.J.; Giovanna Lizio, M.; Booth, A.; Collins, R.F.; Gough, J.E.; Miller, A.F.; Webb, S.J. A modular self-assembly approach to functionalised beta-sheet peptide hydrogel biomaterials. Soft Matter 2016, 12, 1915–1923. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B. Two Rippled-Sheet Configurations of Polypeptide Chains, and a Note about the Pleated Sheets. Proc. Natl. Acad. Sci. USA 1953, 39. [Google Scholar] [CrossRef] [PubMed]
- Fuhrhop, J.-H.; Krull, M.; Büldt, G. Precipitates with β-Pleated Sheet Structure by Mixing Aqueous Solutions of Helical Poly(D-lysine) and Poly(L-lysine). Angew. Chem. Int. Ed. 1987, 26, 699–700. [Google Scholar] [CrossRef]
- Dzwolak, W.; Ravindra, R.; Nicolini, C.; Jansen, R.; Winter, R. The diastereomeric assembly of polylysine is the low-volume pathway for preferential formation of beta-sheet aggregates. J. Am. Chem. Soc. 2004, 126, 3762–3768. [Google Scholar] [CrossRef]
- Yamaoki, Y.; Imamura, H.; Fulara, A.; Wojcik, S.; Bozycki, L.; Kato, M.; Keiderling, T.A.; Dzwolak, W. An FT-IR study on packing defects in mixed beta-aggregates of poly(L-glutamic acid) and poly(D-glutamic acid): A high-pressure rescue from a kinetic trap. J. Phys. Chem. B 2012, 116, 5172–5178. [Google Scholar] [CrossRef]
- Swanekamp, R.J.; Welch, J.J.; Nilsson, B.L. Proteolytic stability of amphipathic peptide hydrogels composed of self-assembled pleated beta-sheet or coassembled rippled beta-sheet fibrils. Chem. Commun. 2014, 50, 10133–10136. [Google Scholar] [CrossRef]
- Swanekamp, R.J.; DiMaio, J.T.; Bowerman, C.J.; Nilsson, B.L. Coassembly of enantiomeric amphipathic peptides into amyloid-inspired rippled beta-sheet fibrils. J. Am. Chem. Soc. 2012, 134, 5556–5559. [Google Scholar] [CrossRef]
- Nagy-Smith, K.; Beltramo, P.J.; Moore, E.; Tycko, R.; Furst, E.M.; Schneider, J.P. Molecular, Local, and Network-Level Basis for the Enhanced Stiffness of Hydrogel Networks Formed from Coassembled Racemic Peptides: Predictions from Pauling and Corey. ACS Cent. Sci. 2017, 3, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Nagy, K.J.; Giano, M.C.; Jin, A.; Pochan, D.J.; Schneider, J.P. Enhanced mechanical rigidity of hydrogels formed from enantiomeric peptide assemblies. J. Am. Chem. Soc. 2011, 133, 14975–14977. [Google Scholar] [CrossRef]
- Balbach, J.J.; Ishii, Y.; Antzutkin, O.N.; Leapman, R.D.; Rizzo, N.W.; Dyda, F.; Reed, J.; Tycko, R. Amyloid Fibril Formation by Aβ16–22, a Seven-Residue Fragment of the Alzheimer’s β-Amyloid Peptide, and Structural Characterization by Solid State NMR. Biochemistry 2000, 39, 13748–13759. [Google Scholar] [CrossRef] [PubMed]
- Senguen, F.T.; Doran, T.M.; Anderson, E.A.; Nilsson, B.L. Clarifying the influence of core amino acid hydrophobicity, secondary structure propensity, and molecular volume on amyloid-beta 16–22 self-assembly. Mol. Biosyst. 2011, 7, 497–510. [Google Scholar] [CrossRef]
- Senguen, F.T.; Lee, N.R.; Gu, X.; Ryan, D.M.; Doran, T.M.; Anderson, E.A.; Nilsson, B.L. Probing aromatic, hydrophobic, and steric effects on the self-assembly of an amyloid-beta fragment peptide. Mol. Biosyst. 2011, 7, 486–496. [Google Scholar] [CrossRef]
- Wood, S.J.; Wetzel, R.; Martin, J.D.; Hurle, M.R. Prolines and amyloidogenicity in fragments of the Alzheimer’s peptide beta/A4. Biochemistry 1995, 34, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Hilbich, C.; Kisters-Woike, B.; Reed, J.; Masters, C.L.; Beyreuther, K. Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer’s disease βA4 peptides. J. Mol. Biol. 1992, 228, 460–473. [Google Scholar] [CrossRef]
- Inouye, H.; Gleason, K.A.; Zhang, D.; Decatur, S.M.; Kirschner, D.A. Differential effects of Phe19 and Phe20 on fibril formation by amyloidogenic peptide A beta 16–22 (Ac-KLVFFAE-NH2). Proteins 2010, 78, 2306–2321. [Google Scholar] [CrossRef]
- Klimov, D.K.; Thirumalai, D. Dissecting the Assembly of Aβ16–22 Amyloid Peptides into Antiparallel β Sheets. Structure 2003, 11, 295–307. [Google Scholar] [CrossRef]
- O’Nuallain, B.; Thakur, A.K.; Williams, A.D.; Bhattacharyya, A.M.; Chen, S.; Thiagarajan, G.; Wetzel, R. Kinetics and Thermodynamics of Amyloid Assembly Using a High-Performance Liquid Chromatography–Based Sedimentation Assay. Methods Enzymol. 2006, 413, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Foley, A.R.; Warner, C.J.A.; Zhang, X.; Rolandi, M.; Abrams, B.; Raskatov, J.A. Suppression of Oligomer Formation and Formation of Non-Toxic Fibrils upon Addition of Mirror-Image Abeta42 to the Natural l-Enantiomer. Angew. Chem. Int. Ed. Engl. 2017, 56, 11506–11510. [Google Scholar] [CrossRef] [PubMed]
- Childers, W.S.; Mehta, A.K.; Lu, K.; Lynn, D.G. Templating molecular arrays in amyloid’s cross-beta grooves. J. Am. Chem. Soc. 2009, 131, 10165–10172. [Google Scholar] [CrossRef] [PubMed]
- Krysmann, M.J.; Castelletto, V.; Kelarakis, A.; Hamley, I.W.; Hule, R.A.; Pochan, D.J. Self-assembly and hydrogelation of an amyloid peptide fragment. Biochemistry 2008, 47, 4597–4605. [Google Scholar] [CrossRef]
- Tjernberg, L.O.; Callaway, D.J.E.; Tjernberg, A.; Hahne, S.; Lilliehöök, C.; Terenius, L.; Thyberg, J.; Nordstedt, C. A Molecular Model of Alzheimer Amyloid β-Peptide Fibril Formation. J. Biol. Chem. 1999, 274, 12619–12625. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Schaefer, J.; Waugh, J.S. Magic-angle spinning and polarization transfer in proton-enhanced NMR. J. Magn. Reson. 1977, 28, 105–112. [Google Scholar] [CrossRef]
- Kotecha, M.; Wickramasinghe, N.P.; Ishii, Y. Efficient low-power heteronuclear decoupling in 13C high-resolution solid-state NMR under fast magic angle spinning. Magn. Reson. Chem. 2007, 45 (Suppl. 1), S221–S230. [Google Scholar] [CrossRef]
- Wickramasinghe, N.P.; Ishii, Y. Sensitivity enhancement, assignment, and distance measurement in 13C solid-state NMR spectroscopy for paramagnetic systems under fast magic angle spinning. J. Magn. Reson. 2006, 181, 233–243. [Google Scholar] [CrossRef]
- Yannoni, C.S. High-resolution NMR in solids: The CPMAS experiment. Acc. Chem. Res. 2002, 15, 201–208. [Google Scholar] [CrossRef]
- Goldbourt, A. Biomolecular magic-angle spinning solid-state NMR: Recent methods and applications. Curr. Opin. Biotechnol. 2013, 24, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.; Gibby, M.G.; Waugh, J.S. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973, 59, 569–590. [Google Scholar] [CrossRef]
- Schaefer, J.; Stejskal, E.O. Carbon-13 nuclear magnetic resonance of polymers spinning at the magic angle. J. Am. Chem. Soc. 1976, 98, 1031–1032. [Google Scholar] [CrossRef]
- Gullion, T.; Schaefer, J. Rotational-echo double-resonance NMR. J. Magn. Reson. 1989, 81, 196–200. [Google Scholar] [CrossRef]
- Kar, K.; Arduini, I.; Drombosky, K.W.; van der Wel, P.C.; Wetzel, R. D-polyglutamine amyloid recruits L-polyglutamine monomers and kills cells. J. Mol. Biol. 2014, 426, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Torbeev, V.; Grogg, M.; Ruiz, J.; Boehringer, R.; Schirer, A.; Hellwig, P.; Jeschke, G.; Hilvert, D. Chiral recognition in amyloid fiber growth. J. Pept. Sci. 2016, 22, 290–304. [Google Scholar] [CrossRef]
- Gullion, T.; Baker, D.B.; Conradi, M.S. New, compensated Carr-Purcell sequences. J. Magn. Reson. 1990, 89, 479–484. [Google Scholar] [CrossRef]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urban, J.M.; Ho, J.; Piester, G.; Fu, R.; Nilsson, B.L. Rippled β-Sheet Formation by an Amyloid-β Fragment Indicates Expanded Scope of Sequence Space for Enantiomeric β-Sheet Peptide Coassembly. Molecules 2019, 24, 1983. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101983
Urban JM, Ho J, Piester G, Fu R, Nilsson BL. Rippled β-Sheet Formation by an Amyloid-β Fragment Indicates Expanded Scope of Sequence Space for Enantiomeric β-Sheet Peptide Coassembly. Molecules. 2019; 24(10):1983. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101983
Chicago/Turabian StyleUrban, Jennifer M., Janson Ho, Gavin Piester, Riqiang Fu, and Bradley L. Nilsson. 2019. "Rippled β-Sheet Formation by an Amyloid-β Fragment Indicates Expanded Scope of Sequence Space for Enantiomeric β-Sheet Peptide Coassembly" Molecules 24, no. 10: 1983. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101983