
The PPARγ Agonist Rosiglitazone Suppresses Syngeneic Mouse SCC (Squamous Cell Carcinoma) Tumor Growth through an Immune-Mediated Mechanism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
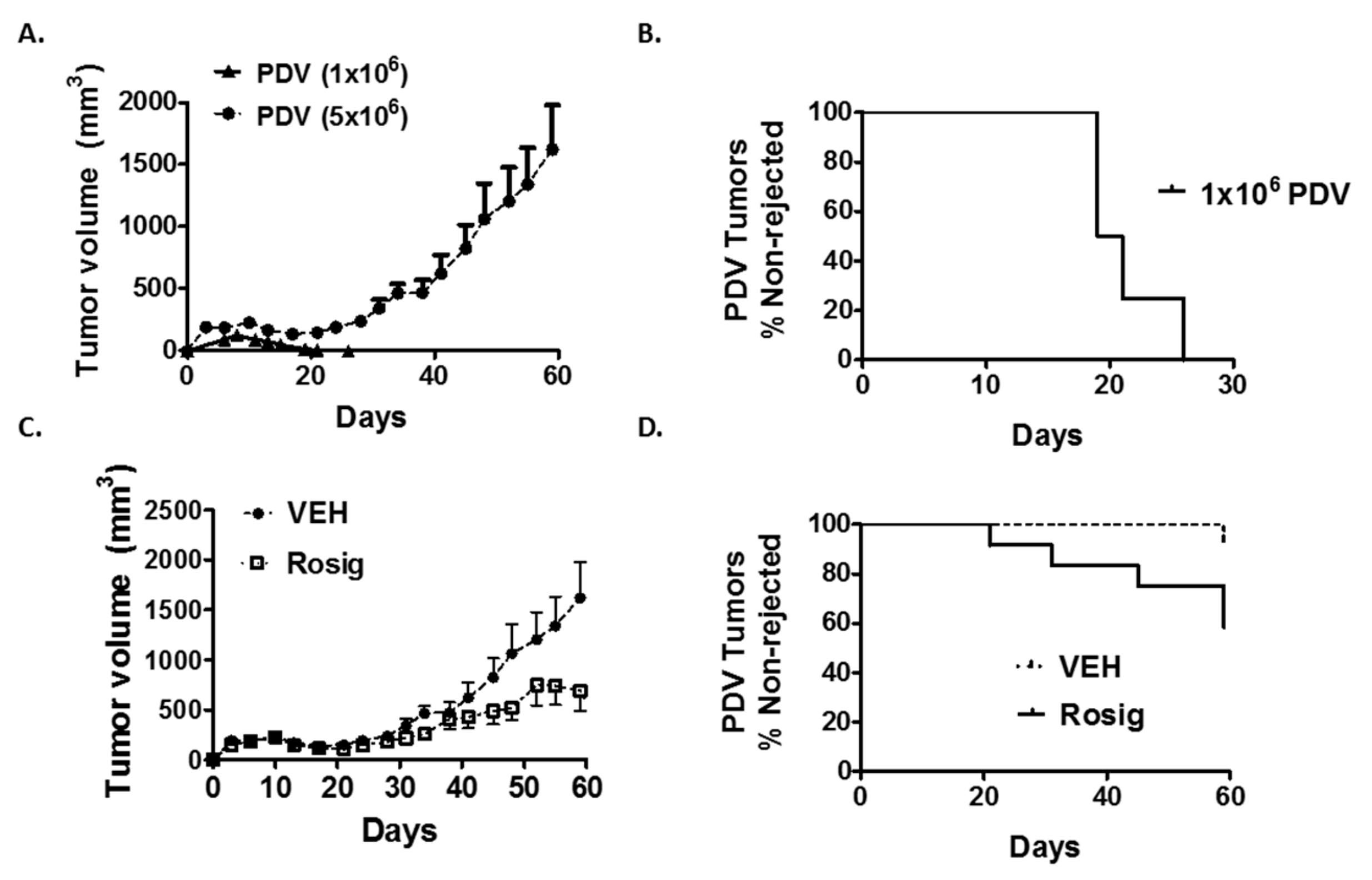
2.1. Rosiglitazone Treatment Suppresses Immunogenic PDV Tumor Growth and Promotes Tumor Regression
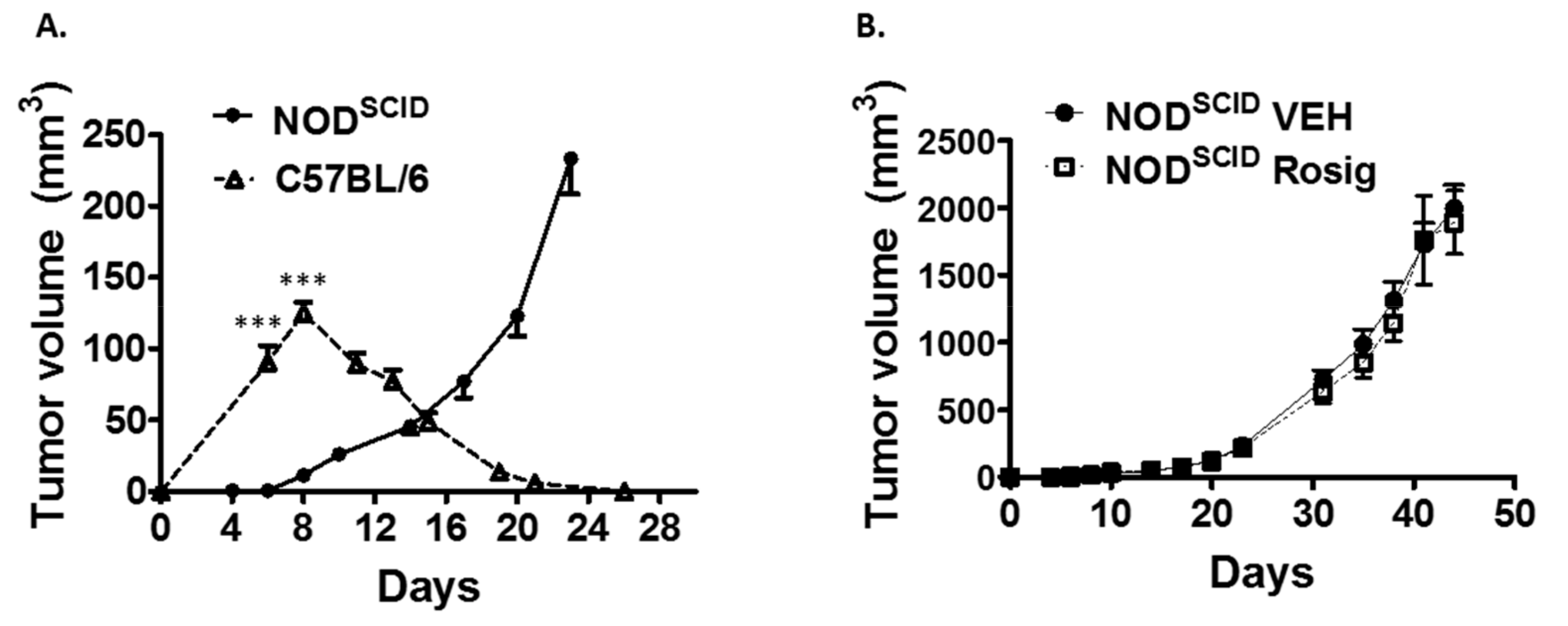
2.2. Both the Failure of PDV Tumor Cells to Establish Durable Tumors at Low Cell Numbers, as Well as the Ability of Rosiglitazone to Suppress Durable Tumor Formation, Is Dependent on an Intact Immune System
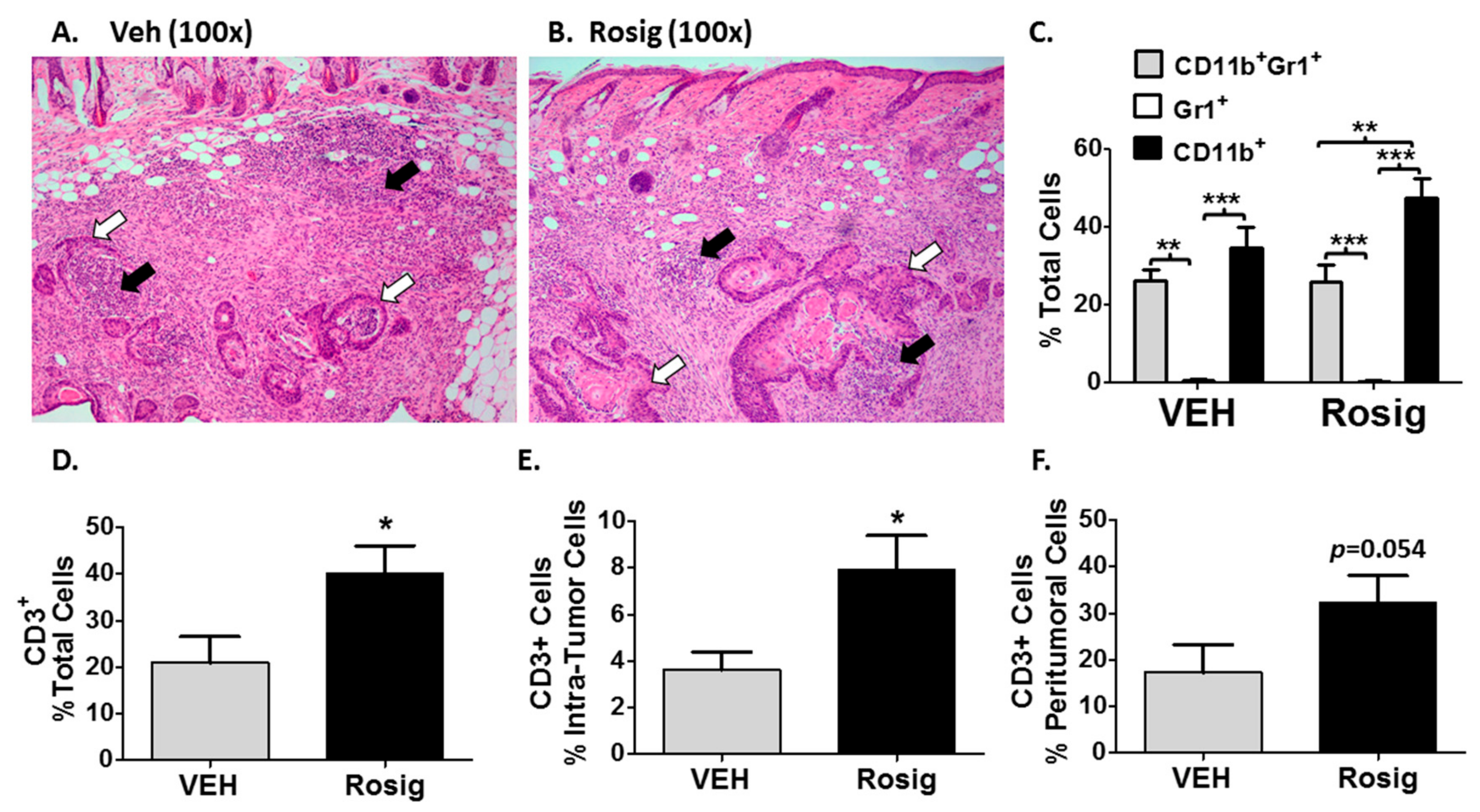
2.3. Immunogenic PDV Tumors Elicit an Early Inflammatory Phase That Rosiglitazone Treatment Alters by Promoting an Increase in Tumor-Infiltrating T-Cells
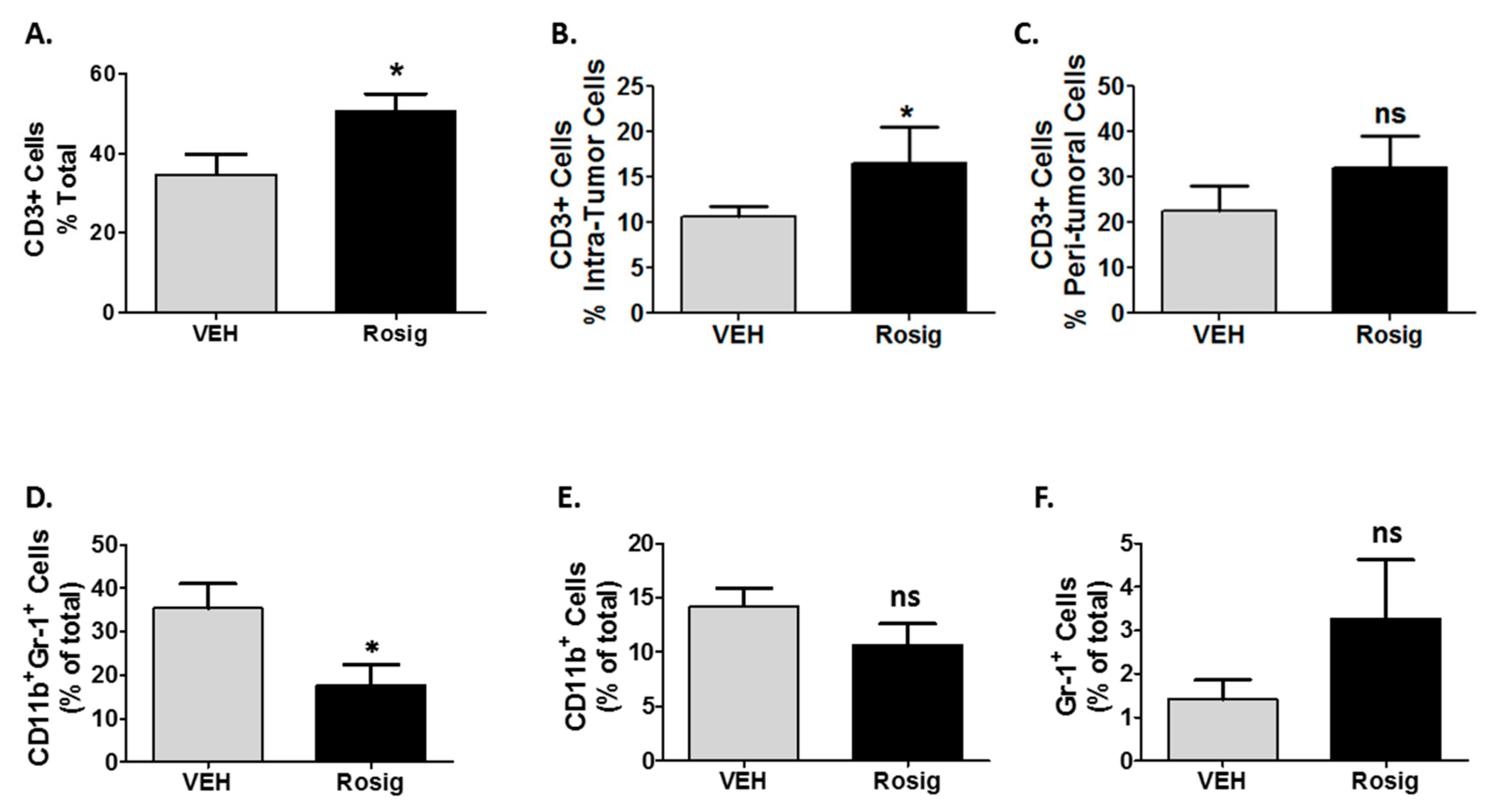
2.4. Rosiglitazone Alters Both the CD3 and Myeloid Cell Infiltration in PDV Tumors at Later Stages in Tumor Growth
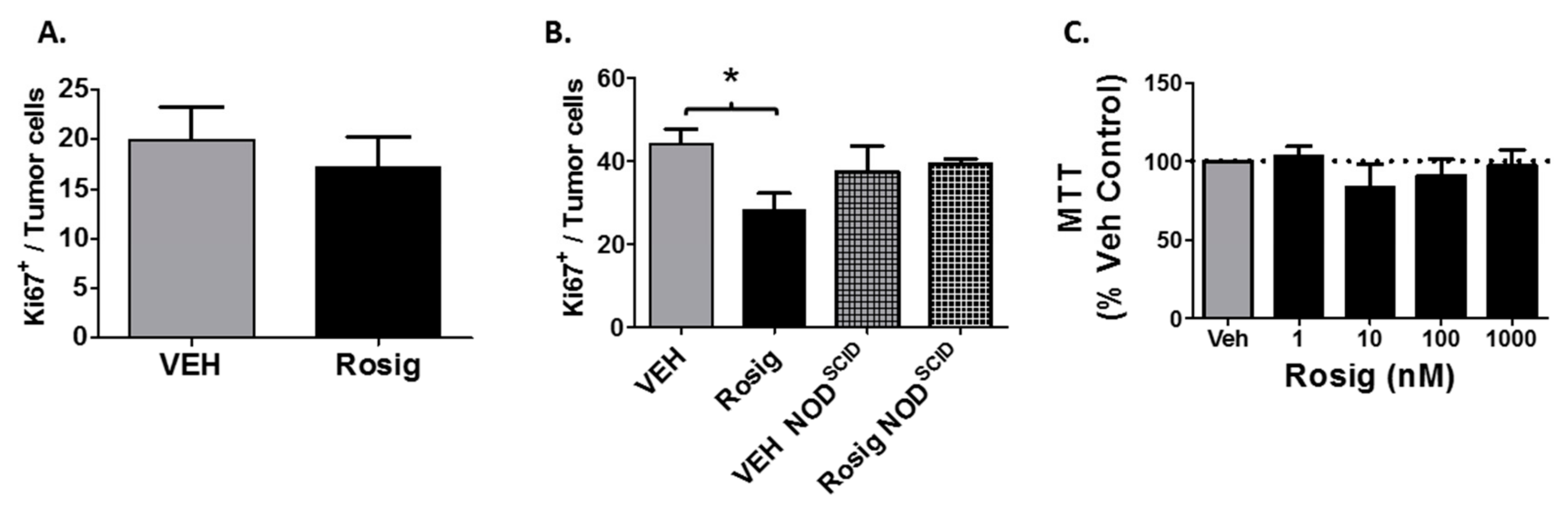
2.5. Rosiglitazone Treatment Suppresses Cell Proliferation in PDV Tumors Grown in Immune Competent Mice, but Not in Immune Deficient Mice or in PDV Cells Grown in Cell Culture
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Cell Line and In Vitro Proliferation Studies
4.3. Animal Studies
4.4. PDV Tumor Cell Injection and Rosiglitazone Treatment
4.5. PDV Tumor Size Measurement
4.6. Immunofluorescence
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Konger, R.L.; Derr-Yellin, E.; Travers, J.B.; Ocana, J.A.; Sahu, R.P. Epidermal PPARγ influences subcutaneous tumor growth and acts through TNF-α to regulate contact hypersensitivity and the acute photoresponse. Oncotarget 2017, 8, 98184–98199. [Google Scholar] [CrossRef] [PubMed]
- Ondrey, F. Peroxisome proliferator-activated receptor γ pathway targeting in carcinogenesis: Implications for chemoprevention. Clinical Cancer Res. 2009, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.R.; Chen, C.Y.; Pinzone, J.J.; Ringel, M.D.; Chen, C.S. Beyond peroxisome proliferator-activated receptor gamma signaling: the multi-facets of the antitumor effect of thiazolidinediones. Endocr. Relat. Cancer 2006, 13, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Kim, M.; Tanaka, T. PPAR Ligands for cancer chemoprevention. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Wang, L.H.; Farrar, W.L. A Role for PPARγ in the regulation of cytokines in immune cells and cancer. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Cheng, S.W.; Yang, R.; Wang, B.; Liu, J. Combination chemoprevention: future direction of colorectal cancer prevention. Eur. J. Cancer Prev. 2012, 21, 231–240. [Google Scholar] [CrossRef]
- Sawayama, H.; Ishimoto, T.; Watanabe, M.; Yoshida, N.; Sugihara, H.; Kurashige, J.; Hirashima, K.; Iwatsuki, M.; Baba, Y.; Oki, E.; et al. Small molecule agonists of PPAR-γ exert therapeutic effects in esophageal cancer. Cancer Res. 2014, 74, 575–585. [Google Scholar] [CrossRef]
- Vella, V.; Nicolosi, M.L.; Giuliano, S.; Bellomo, M.; Belfiore, A.; Malaguarnera, R. PPAR-γ agonists as antineoplastic agents in cancers with dysregulated IGF axis. Front. Endocrinol. 2017, 8, 31. [Google Scholar] [CrossRef]
- Burgermeister, E.; Seger, R. PPARγ and MEK interactions in cancer. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef]
- Burton, J.D.; Goldenberg, D.M.; Blumenthal, R.D. Potential of peroxisome proliferator-activated receptor gamma antagonist compounds as therapeutic agents for a wide range of cancer types. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef]
- Hatton, J.L.; Yee, L.D. Clinical use of PPARγ ligands in cancer. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Reichle, A.; Vogt, T. New indications for established drugs: combined tumor-stroma-targeted cancer therapy with PPARgamma agonists, COX-2 inhibitors, mTOR antagonists and metronomic chemotherapy. Curr. Cancer Drug Targets 2005, 5, 393–419. [Google Scholar] [CrossRef] [PubMed]
- Nicol, C.J.; Yoon, M.; Ward, J.M.; Yamashita, M.; Fukamachi, K.; Peters, J.M.; Gonzalez, F.J. PPARgamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis 2004, 25, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Indra, A.K.; Castaneda, E.; Antal, M.C.; Jiang, M.; Messaddeq, N.; Meng, X.; Loehr, C.V.; Gariglio, P.; Kato, S.; Wahli, W.; et al. Malignant transformation of DMBA/TPA-induced papillomas and nevi in the skin of mice selectively lacking retinoid-X-receptor alpha in epidermal keratinocytes. J. Invest. Dermatol. 2007, 127, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.P.; DaSilva, S.C.; Rashid, B.; Martel, K.C.; Jernigan, D.; Mehta, S.R.; Mohamed, D.R.; Rezania, S.; Bradish, J.R.; Armstrong, A.B.; et al. Mice lacking epidermal PPARγ exhibit a marked augmentation in photocarcinogenesis associated with increased UVB-induced apoptosis, inflammation and barrier dysfunction. Int. J. Cancer 2012, 131, E1055–E1066. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nature Rev. Immunol. 2010, 10, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. BBA Mol. Cell Biol. Lipids 2007, 1771, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kripke, M.L. Reflections on the field of photoimmunology. J. Invest. Dermatol. 2013, 133, 27–30. [Google Scholar] [CrossRef]
- Sahu, R.P.; Turner, M.J.; DaSilva, S.C.; Rashid, B.M.; Ocana, J.A.; Perkins, S.M.; Konger, R.L.; Touloukian, C.E.; Kaplan, M.H.; Travers, J.B. The environmental stressor ultraviolet B radiation inhibits murine antitumor immunity through its ability to generate platelet-activating factor agonists. Carcinogenesis 2012, 33, 1360–1367. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Wang, G.; Qu, P.; Yan, C.; Du, H. Overexpression of dominant negative peroxisome proliferator-activated receptor-γ (PPARγ) in alveolar type II epithelial cells causes inflammation and T-Cell suppression in the lung. Am. J. Pathol. 2011, 178, 2191–2204. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Yan, C.; Czader, M.; Foreman, O.; Blum, J.S.; Kapur, R.; Du, H. Inhibition of peroxisome proliferator-activated receptor-γ in myeloid lineage cells induces systemic inflammation, immunosuppression and tumorigenesis. Blood 2012, 119, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Ngiow, S.F.; von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.L.; Smyth, M.J. Anti-TIM3 antibody promotes T Cell IFN-γ–mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef] [PubMed]
- Caulin, C.; Bauluz, C.; Gandarillas, A.; Cano, A.; Quintanilla, M. Changes in keratin expression during malignant progression of transformed mouse epidermal keratinocytes. Exp. Cell Res. 1993, 204, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Girardi, M.; Oppenheim, D.; Glusac, E.J.; Filler, R.; Balmain, A.; Tigelaar, R.E.; Hayday, A.C. Characterizing the protective component of the αβ T cell response to transplantable squamous cell carcinoma. J. Invest. Dermatol. 2004, 122, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Whang, M.I.; Guerra, N.; Raulet, D.H. Costimulation of dendritic epidermal γδ T Cells by a new NKG2D ligand expressed specifically in the skin. J. Immunol. 2009, 182, 4557–4564. [Google Scholar] [CrossRef]
- Girardi, M.; Oppenheim, D.E.; Steele, C.R.; Lewis, J.M.; Glusac, E.; Filler, R.; Hobby, P.; Sutton, B.; Tigelaar, R.E.; Hayday, A.C. Regulation of cutaneous malignancy by γδ T cells. Science 2001, 294, 605–609. [Google Scholar] [CrossRef]
- Christensen, J.E.; Andreasen, S.Ø.; Christensen, J.P.; Thomsen, A.R. CD11b expression as a marker to distinguish between recently activated effector CD8+ T cells and memory cells. Int. Immunol. 2001, 13, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, S.; Smith, C.W.; Shardonofsky, F.R.; Burns, A.R. The role of Mac-1 (CD11b/CD18) in antigen-induced airway eosinophilia in mice. Am. J. Respir. Cell Mol. Biol. 2001, 25, 170–177. [Google Scholar] [CrossRef]
- Goyal, G.; Wong, K.; Nirschl, C.J.; Souders, N.; Neuberg, D.; Anandasabapathy, N.; Dranoff, G. PPAR-γ contributes to immunity by cancer vaccines that secrete GM-CSF. Cancer Immunol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- De Gruijl, F.R. UV-induced immunosuppression in the balance†. Photochem. Photobiol. 2008, 84, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Du, H.; Blum, J.S.; Yan, C. Critical role of PPARγ in myeloid-derived suppressor cell-stimulated cancer cell proliferation and metastasis. Oncotarget 2016, 7, 1529–1543. [Google Scholar] [PubMed]
- Huang, G.; Yin, L.; Lan, J.; Tong, R.; Li, M.; Na, F.; Mo, X.; Chen, C.; Xue, J.; Lu, Y. Synergy between peroxisome proliferator-activated receptor gamma agonist and radiotherapy in cancer. Cancer Sci. 2018, 109, 2243–2255. [Google Scholar] [CrossRef]
- Murakami, H.; Ono, A.; Takahashi, T.; Onozawa, Y.; Tsushima, T.; Yamazaki, K.; Jikoh, T.; Boku, N.; Yamamoto, N. Phase I study of Efatutazone, an oral PPARgamma agonist, in patients with metastatic solid tumors. Anticancer Res. 2014, 34, 5133–5141. [Google Scholar] [PubMed]
- Komatsu, Y.; Yoshino, T.; Yamazaki, K.; Yuki, S.; Machida, N.; Sasaki, T.; Hyodo, I.; Yachi, Y.; Onuma, H.; Ohtsu, A. Phase 1 study of efatutazone, a novel oral peroxisome proliferator-activated receptor gamma agonist, in combination with FOLFIRI as second-line therapy in patients with metastatic colorectal cancer. Invest. New Drugs 2014, 32, 473–480. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Marshall, J.L.; Wagner, A.J.; Hwang, J.J.; Malik, S.; Cotarla, I.; Deeken, J.F.; He, A.R.; Daniel, H.; Halim, A.B.; et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer 2012, 118, 5403–5413. [Google Scholar] [CrossRef]
- Ondrey, F.G. Pioglitazone in oral leukoplakia: a phase II trial. In Proceedings of the American Association for Cancer Research International Conference: Frontiers in Cancer Prevention Research, Philadelphia, PA, USA, 26–30 October 2007; p. 170. [Google Scholar]
- Ni, J.; Zhou, L.L.; Ding, L.; Zhao, X.; Cao, H.; Fan, F.; Li, H.; Lou, R.; Du, Y.; Dong, S.; et al. PPARgamma agonist efatutazone and gefitinib synergistically inhibit the proliferation of EGFR-TKI-resistant lung adenocarcinoma cells via the PPARgamma/PTEN/Akt pathway. Exp. Cell Res. 2017, 361, 246–256. [Google Scholar] [CrossRef]
- Girnun, G.D.; Naseri, E.; Vafai, S.B.; Qu, L.; Szwaya, J.D.; Bronson, R.; Alberta, J.A.; Spiegelman, B.M. Synergy between PPARγ ligands and platinum-based drugs in cancer. Cancer Cell 2007, 11, 395–406. [Google Scholar] [CrossRef]
- Ren, L.; Konger, R.L. Evidence that peroxisome proliferator-activated receptor γ suppresses squamous carcinogenesis through anti-inflammatory signaling and regulation of the immune response. Mol. Carcinogen. 2019. In press. [Google Scholar] [CrossRef]
- Liu, K.; Black, R.M.; Acton Iii, J.J.; Mosley, R.; Debenham, S.; Abola, R.; Yang, M.; Tschirret-Guth, R.; Colwell, L.; Liu, C.; et al. Selective PPARγ modulators with improved pharmacological profiles. Bioorg. Med. Chem. Lett. 2005, 15, 2437–2440. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Muise, E.S.; Dai, H.; Raubertas, R.; Wong, K.K.; Thompson, G.M.; Wood, H.B.; Meinke, P.T.; Lum, P.Y.; Thompson, J.R.; et al. Novel transcriptome profiling analyses demonstrate that selective PPARγ modulators display attenuated and selective gene regulatory activity in comparison with PPARγ full agonists. Mol. Pharm. 2012. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kleinhenz, D.J.; Rupnow, H.L.; Campbell, A.G.; Thulé, P.M.; Sutliff, R.L.; Hart, C.M. The PPARγ ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vascul. Pharmacol. 2007, 46, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Hindlet, P.; Barraud, C.; Boschat, L.; Farinotti, R.; Bado, A.; Buyse, M. Rosiglitazone and metformin have opposite effects on intestinal absorption of oligopeptides via the proton-dependent PepT1 transporter. Mol. Pharm. 2012, 81, 319–327. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: PDV tumor cells are available from the authors. All other reagents and compounds are commercially available. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konger, R.L.; Derr-Yellin, E.; Ermatov, N.; Ren, L.; Sahu, R.P. The PPARγ Agonist Rosiglitazone Suppresses Syngeneic Mouse SCC (Squamous Cell Carcinoma) Tumor Growth through an Immune-Mediated Mechanism. Molecules 2019, 24, 2192. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112192
Konger RL, Derr-Yellin E, Ermatov N, Ren L, Sahu RP. The PPARγ Agonist Rosiglitazone Suppresses Syngeneic Mouse SCC (Squamous Cell Carcinoma) Tumor Growth through an Immune-Mediated Mechanism. Molecules. 2019; 24(11):2192. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112192
Chicago/Turabian StyleKonger, Raymond L., Ethel Derr-Yellin, Nurmukambed Ermatov, Lu Ren, and Ravi P. Sahu. 2019. "The PPARγ Agonist Rosiglitazone Suppresses Syngeneic Mouse SCC (Squamous Cell Carcinoma) Tumor Growth through an Immune-Mediated Mechanism" Molecules 24, no. 11: 2192. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112192