Iron-Catalysed C(sp2)-H Borylation Enabled by Carboxylate Activation


1
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh EH9 3FJ, UK
2
GSK Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK
*
Authors to whom correspondence should be addressed.
Molecules 2020, 25(4), 905; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040905
Submission received: 30 January 2020
/
Revised: 13 February 2020
/
Accepted: 14 February 2020
/
Published: 18 February 2020
(This article belongs to the Special Issue Recent Advances in Iron Catalysis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Arene C(sp2)-H bond borylation reactions provide rapid and efficient routes to synthetically versatile boronic esters. While iridium catalysts are well established for this reaction, the discovery and development of methods using Earth-abundant alternatives is limited to just a few examples. Applying an in situ catalyst activation method using air-stable and easily handed reagents, the iron-catalysed C(sp2)-H borylation reactions of furans and thiophenes under blue light irradiation have been developed. Key reaction intermediates have been prepared and characterised, and suggest two mechanistic pathways are in action involving both C-H metallation and the formation of an iron boryl species.
1. Introduction
The development of sustainable methods for the selective C(sp2)-H functionalisation of arenes is an area of intense research but is still dominated by the use of 2nd- and 3rd-row transition metals [1,2,3,4,5,6,7]. Earth-abundant metals offer low toxicity and inexpensive alternatives, with iron being a leading example [8,9,10,11,12]. Direct C(sp2)-H borylation offers a simple and efficient route to aryl-boronic esters, which are key platforms for organic synthesis [13,14,15]. Iridium-based complexes have become a “go-to” for C(sp2)-H borylation reactions [16,17,18,19,20,21,22,23,24], while the discovery and development of Earth-abundant alternatives remains comparatively rare [25,26,27,28,29,30,31,32,33,34,35,36,37].
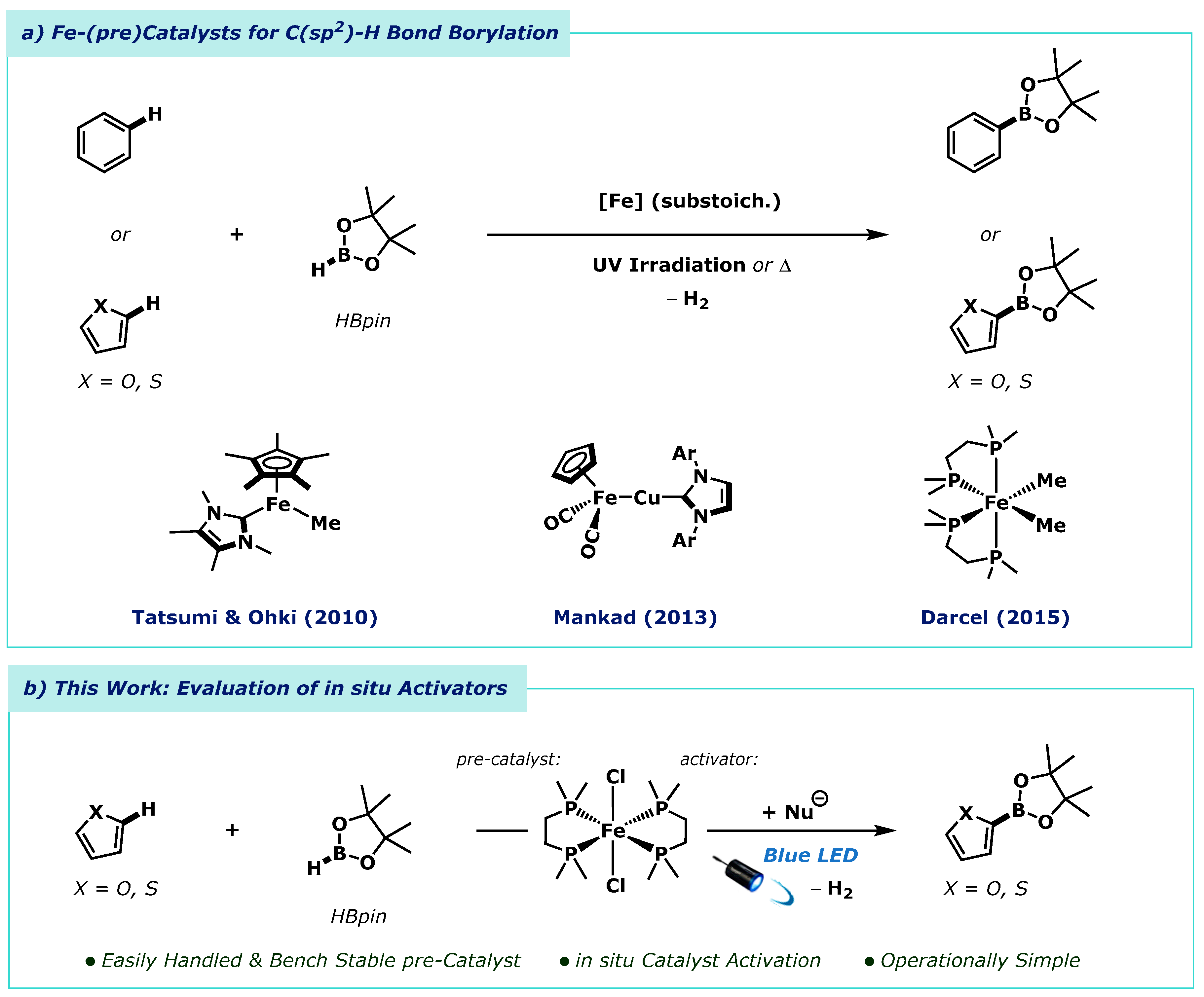
Tatsumi and Ohki showed that arenes would undergo thermally promoted C(sp2)-H borylation using an N-heterocyclic carbene cyclopentadienyl iron(II) alkyl complex [NHC(Cp*)FeMe] as a catalyst in the presence of tert-butylethylene (Scheme 1a) [35]. Mankad applied heterobimetallic Fe-Cu and Fe-Zn complexes under continuous ultraviolet light irradiation to arene C(sp2)-H borylation [36]. Similarly, Darcel and co-workers reported the use of a bis(diphosphino) iron(II) dialkyl and dihydride complexes for arene C(sp2)-H borylation, again under continuous ultraviolet light irradiation [37]. While these landmark reports are highly significant developments, all require the prior synthesis of sensitive inorganic complexes which are synthetically challenging and difficult to handle for the non-specialist practitioner, thus limiting use by the broader synthetic community.
To reduce the synthetic challenges, and need for organometallic reagents, we questioned whether the C(sp2)-H borylation chemistry reported previously could be simplified by in situ catalyst activation using only bench stable reagents. In the example reported by Darcel and co-workers the bis[1,2-bis(dimethylphosphino)ethane-P,P′]dimethyliron(II) pre-catalyst (dmpe2FeMe2) was generated by the addition of methyllithium to the corresponding iron(II) dichloride complex (dmpe2FeCl2) [37]. Similarly, the catalytically active bis[1,2-bis(dimethylphosphino)ethane-P,P′]iron(II) dihydride (dmpe2FeH2) could be accessed using either LiHBEt3 or LiAlH4 [37,38]. Given our previous work on the in situ generation of hydride donors formed by the combination of alkoxide salts and pinacolborane (HBpin) [39], we postulated that the active C(sp2)-H borylation pre-catalyst, dmpe2FeH2, may be accessible by the same method. Reaction of substoichiometric alkoxide salt with HBpin, the boron source used for this borylation, would generate a hydride reductant in situ to activate the dmpe2FeCl2 pre-catalyst to dmpe2FeH2, the active borylation catalyst, and thus initiate catalysis. Importantly, the dmpe2FeCl2 complex displays much greater air- and moisture stability compared to the dihydride and dialkyl analogues. Herein, we report the in situ activation of dmpe2FeCl2 and application to the C(sp2)-H borylation reaction of heteroarenes (Scheme 1b).
2. Results
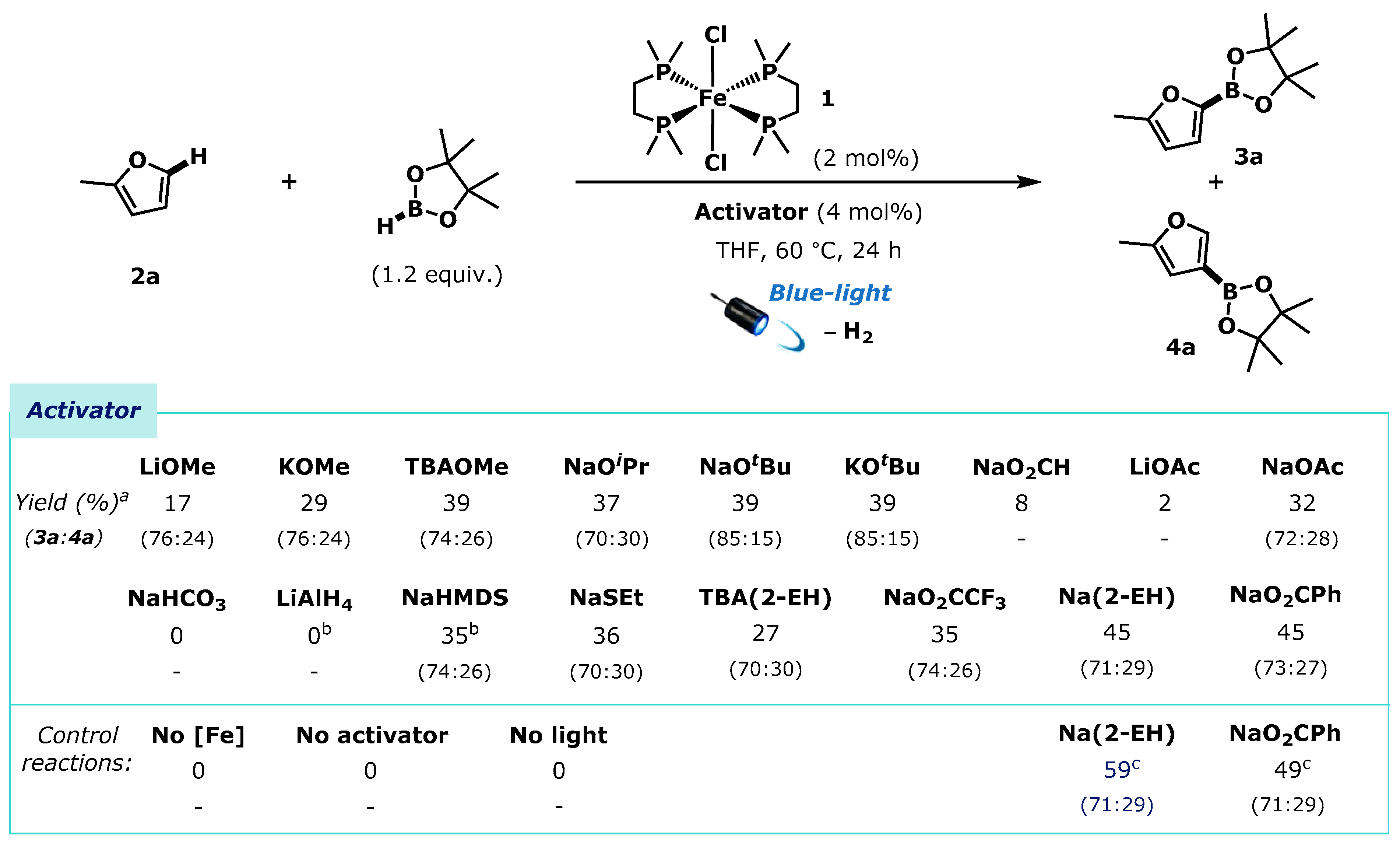
Guided by the work of Darcel and co-workers, we selected 2-methylfuran 2a as an ideal test substrate for our investigations. Darcel and co-workers showed that dmpe2FeMe2 could be used as a pre-catalyst for the borylation of furan 2a (3 equiv.) using HBpin (1 equiv.) under continuous ultraviolet light irradiation to give a regioisomeric mixture of 5- and 4-borylated furans, 3a and 4a respectively (67%, 3a:4a = 82:18) [37]. Using our alkoxide activation strategy we found the use of ultraviolet light for this reaction was not necessary, instead operating with lower energy blue light (Kessil A160 WE, 40 W Blue LED). Additionally, we used an inverted stoichiometry of arene (1 equiv.) and HBpin (1.2 equiv) and a reduced catalyst loading. Using these reaction parameters, we assessed the ability of a selection of potential activators to initiate catalysis alongside the dmpe2FeCl2 1 pre-catalyst. (Scheme 2).
Any of LiOMe, KOMe, TBAOMe (TBA = tetra-n-butylammonium), NaOiPr, NaOtBu or KOtBu triggered pre-catalyst activation and the formation of both furyl boronic ester regioisomers, 3a and 4a, albeit in modest yields (17% to 39%) and with varying regioselectivity, after 24 h. The use of carboxylate salts also initiated catalysis; NaO2CH, LiOAc, NaOAc, Na(2-EH) (2-EH = 2-ethylhexanoate), TBA(2-EH), NaO2CPh, NaO2CCF3 all successfully initiated catalysis with varying efficiency (2% to 45%). Na(2-EH) and NaO2CPh outperformed all alkoxide salts, and the yields obtained using these activators could be increased with prolonged reaction times to give a mixture of furyl boronic esters 3a and 4a in good yield and regioselectivity (Na(2-EH), 59%, 3a:4a = 71:29). Control reactions with no catalyst, no added activator, and with no light irradiation showed no reactivity, highlighting the necessity of each reaction component.
2.1. Substrate Scope
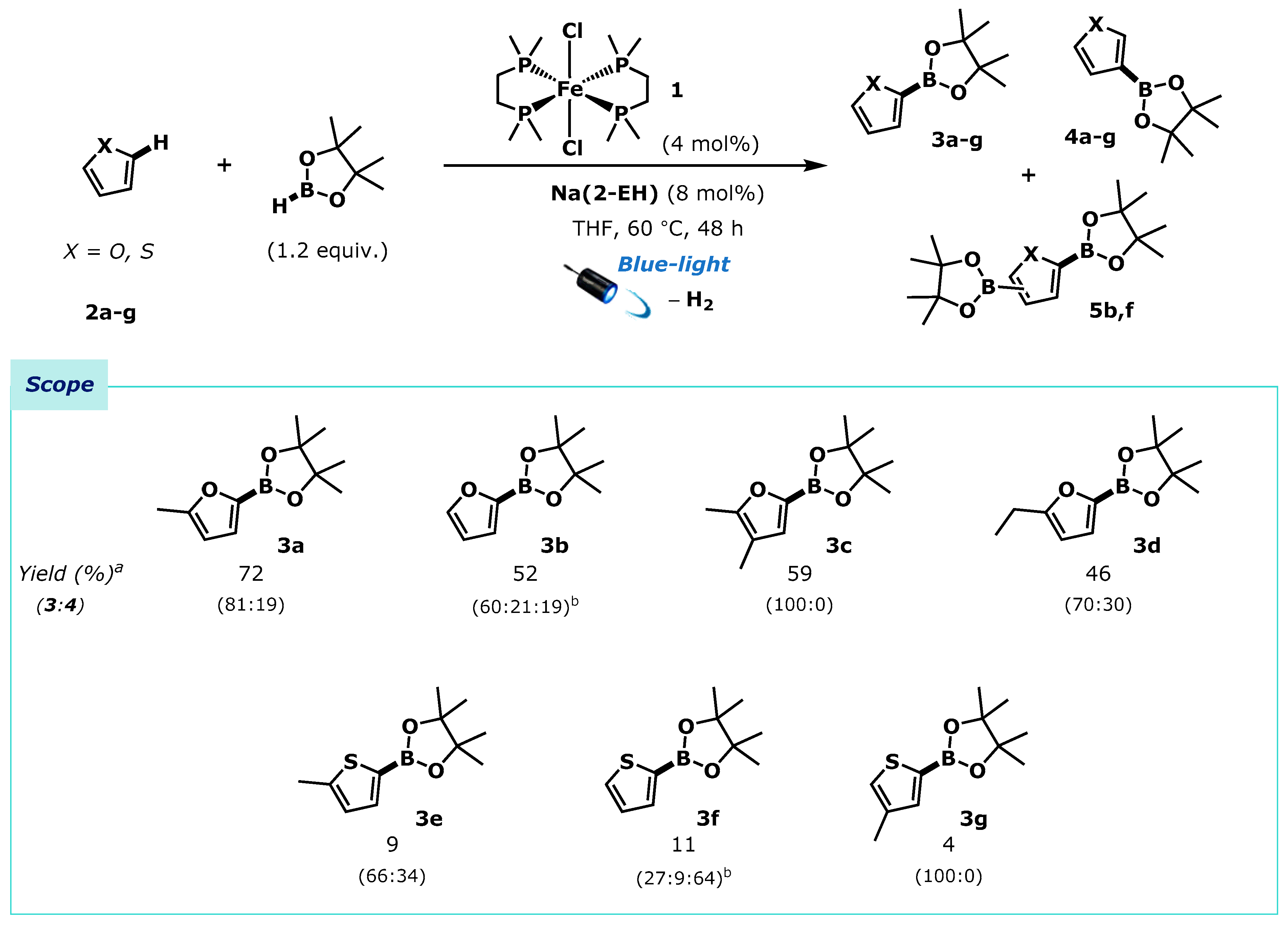
With optimised reaction conditions established using dmpe2FeCl2 (4 mol%), Na(2-EH) (8 mol%), arene (1.0 equiv.) and HBpin (1.2 equiv.) in THF under blue light irradiation, we assessed the reactivity of the system by application to a subset of furan and thiophene derivatives (Scheme 3). 2-Methylfuran 2a underwent efficient borylation to generate a mixture of 5- and 4-borylated regioisomers 3a and 4a in good yield and regioselectivity (72%, 81:19). The parent, unsubstituted furan 2b, also underwent successful borylation but gave a regioisomeric mixture of the 2- and 3-substituted boronic ester regioisomers 3b and 4b, and additionally the bis-boryl furans 5b. Borylation of 2,3-dimethylfuran 2c gave the corresponding 5-boryl regioisomer 3c exclusively in good yield. 2-Ethylfuran 2d reacted similarly to the 2-methylfuran analogue 2a giving a mixture of 4- and 5-substituted boronic esters 3d and 4d. Unfortunately, application to thiophenes demonstrated limited reactivity under the established reaction conditions, giving only low yields of boryl-arenes 3e-g and 4e-g, again as a mixture of regioisomers, and bis-borylated product when the parent thiophene was used [40].
2.2. Mechanistic Investigations
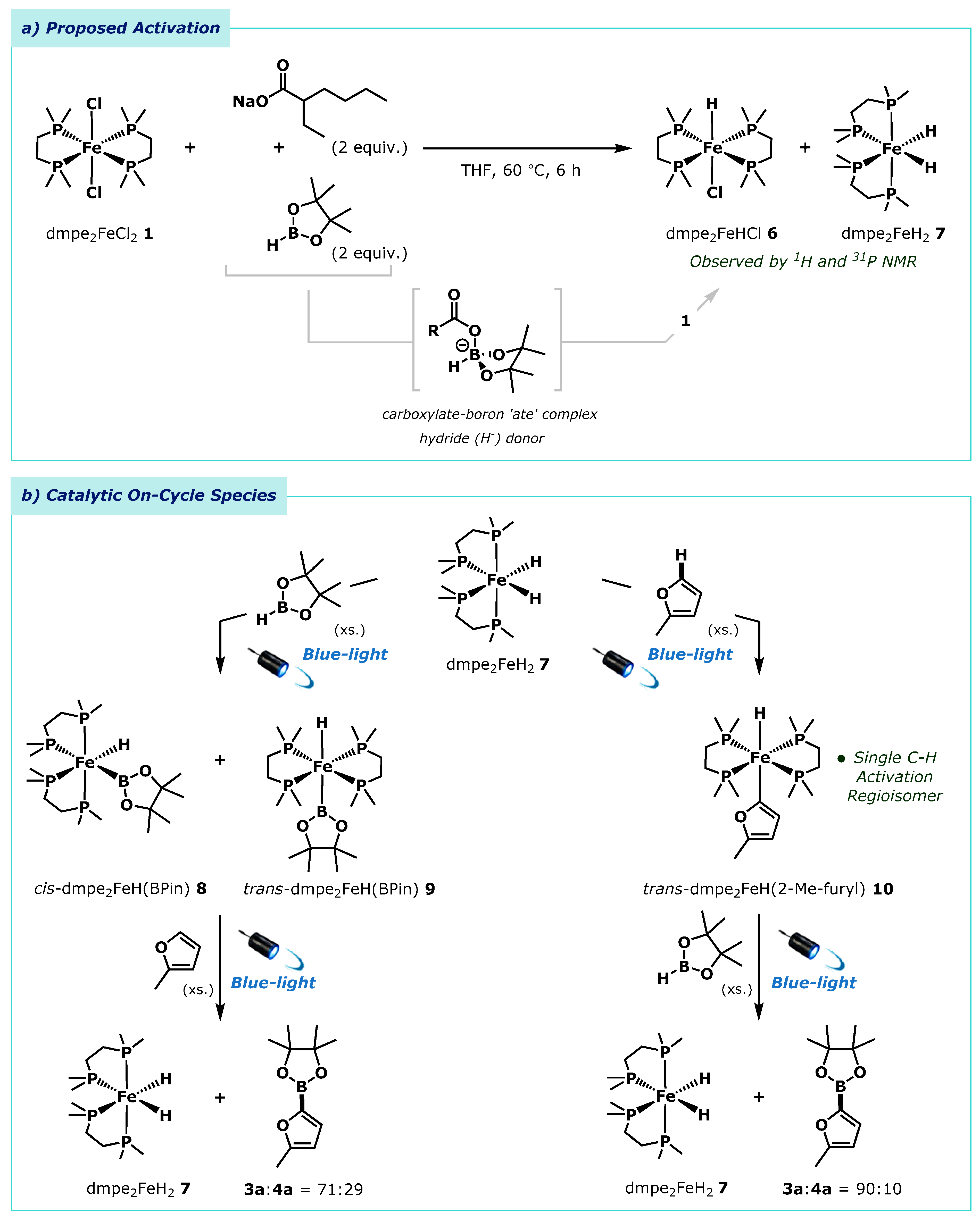
On the basis of successful catalysis we presumed that our in situ activation system provided access to the active iron(II) dihydride complex dmpe2FeH2 7, which had been shown to be catalytically active by Darcel and co-workers [37]. To support this, we combined each component in the absence of light or arene, i.e., the reaction of dmpe2FeCl2 1, Na(2-EH) and HBpin (Scheme 4a). This showed the formation of both the monohydride product dmpe2FeHCl 6 and the expected dihydride, dmpe2FeH2 7, as observed by 31P-NMR spectroscopy (see Supplementary Materials, S16). Reaction of the activator, Na(2-EH), and HBpin in the absence of pre-catalyst showed ligand redistribution to a mixture of boron-containing species, including boron “ate” complexes, BH3 and [BH4]-, as observed by 11B-NMR spectroscopy (see Supplementary Materials, S3). This reactivity is in accordance with that when using other nucleophiles such as alkoxide salts [39,41]. Taken together, these observations are indicative of an in situ activation process, whereby the added carboxylate reagent Na(2-EH) triggers hydride transfer from boron to iron to form the dihydride dmpe2FeH2 7. Once formed, the iron dihydride dmpe2FeH2 7 can efficiently catalyse the C(sp2)-H borylation reaction.
As the dihydride complex dmpe2FeH2 7 was readily formed using our in situ hydride transfer method, and was observable by 1H and 31P-NMR spectroscopy, we next investigated the fundamental steps of this borylation reaction with the aim of identifying key reaction intermediates. Reaction of the in situ generated dmpe2FeH2 7 with excess HBpin under blue light irradiation led to the formation of both cis-dmpe2FeH(Bpin) 8 and trans-dmpe2FeH(Bpin) 9 boryl iron complexes, as observed by 1H, 11B, and 31P NMR spectroscopy (see Supplementary Materials, S17-19). These complexes were previously reported by Darcel and co-workers, where they were formed from the reaction of the related dialkyl complex, dmpe2FeMe2, with HBpin [37]. Addition of 2-methylfuran 2a to the mixture of cis-dmpe2FeH(Bpin) 8 and trans-dmpe2FeH(Bpin) 9 under blue light irradiation gave the formation of the regioisomeric furyl boronic esters 3a and 4a (3a:4a = 71:29), notably in a different ratio to that observed during catalysis (vide supra, 3a:4a = 81:19).
Blue light irradiation of the dihydride complex dmpe2FeH2 7 in the presence of excess 2-methylfuran 2a led to exclusive C(sp2)-H bond metallation at the 5-position to give trans-dmpe2FeH(2-Me-furyl) 10, as observed by 1H and 31P NMR spectroscopy (see Supplementary Materials, S22-24). Addition of HBpin to trans-dmpe2FeH(2-Me-furyl) 10 and irradiation with blue light induced formation of the furyl boronic esters 3a and 4a (3a:4a = 90:10). Again, in a different ratio to that observed under catalysis. As the ratio of regioisomers observed under catalytic conditions (3a:4a = 81:19) appears to be a combination of the ratios observed in the stoichiometric studies (3a:4a = 71:29, and 90:10 respectively), it is suggestive that both the C-H metallation and iron boryl pathways are operative. Specifically, the reaction can precede by C(sp2)-H bond metallation to give dmpe2FeH(2-Me-Furyl) 10, followed by C(sp2)-B bond formation, or by direct reaction of arene with the iron boryl species cis-dmpe2FeH(Bpin) 8 and trans-dmpe2FeH(Bpin) 9. The relative ratios of the furyl boronic ester regioisomers indicate both pathways are equally accessible for the activated catalyst. (53% by C-H metallation, 47% by the iron boryl species).
3. Conclusions
In summary, we have investigated the applicability of several alkoxide, carboxylate and other, common bench stable reagents towards the in situ activation of an iron(II) pre-catalyst for C(sp2)-H bond borylation. We found a sodium carboxylate salt Na(2-EH) in combination with HBpin to be a potent pre-catalysts activator generating the iron dihydride dmpe2FeH2 7 in situ. The validity of this method was demonstrated by the generation of catalytically relevant species that were used as mechanistic probes. These suggest two C-H borylation pathways are operating to give the aryl boronic ester products; C-H metallation followed by borylation, and formation of an iron boryl species followed by arylation.
4. Materials and Methods
4.1. General Information
All compounds reported in the manuscript are commercially available or have been previously described in the literature unless indicated otherwise. All experiments involving iron were performed using standard Schlenk techniques under argon or nitrogen atmosphere. All yields refer to yields determined by 1H-NMR spectroscopy of crude reaction mixtures using an internal standard. All product ratios refer to product ratios determined by 1H-NMR spectroscopy of the crude reaction mixtures. 1H-NMR and 13C-NMR data are given for all compounds when possible in the experimental section for characterisation purposes. Spectroscopic data matched those reported previously.
4.2. Activator Synthesis
Tetra-n-butylammonium 2-ethylhexanoate TBA(2-EH)
A suspension of KH (80 mg, 2 mmol) in anhydrous THF (20 mL) was prepared under an N2 atmosphere, 2-ethylhexanoic acid (0.32 mL, 2 mmol) was added dropwise whilst stirring. n.b. gas evolution (H2). The solution was stirred for 3 h at room temperature, and the THF removed in vacuo to give an amorphous colourless solid. The solid was re-dissolved in MeOH (20 mL) and tetra-n-butylammonium chloride (556 mg, 2 mmol) was added, the solution was stirred for 16 h, filtered through a glass frit and dried in vacuo without further purification to give tetra-n-butylammonium 2-ethylhexanoate (0.72 g, 1.86 mmol, 93%) as an amorphous white solid.
1H-NMR (500 MHz, CDCl3) δ 3.51–3.35 (m, 8H), 2.09 (tt, J = 8.4, 5.4 Hz, 1H), 1.66 (m, 8H), 1.62–1.54 (m, 2H), 1.48–1.39 (m, 8H), 1.39–1.23 (m, 6H), 0.98 (t, J = 7.3 Hz, H), 0.91 (t, J = 7.4 Hz, 3H), 0.88–0.82 (m, 3H). 13C-NMR (126 MHz, CDCl3) δ 181.2, 59.0, 51.1, 33.1, 30.5, 26.4, 24.3, 23.2, 19.8, 14.2, 13.7, 12.7.
4.3. Pre-catalyst Synthesis
dmpe2FeCl2 1 [42]
Anhydrous iron dichloride (0.21 g, 1.67 mmol) was charged to a Schlenk flask and dissolved in anhydrous THF (10 mL), dmpe [(bis(dimethylphosphino)ethane]; 0.50 g, 3.33 mmol) were added to the flask under an Ar atmosphere and the solution left to stir for 48 h at room temperature. The solvent was removed in vacuo, and in an argon-filled glove box, the residue was re-dissolved in dichloromethane (5 mL) and filtered through glass wool. The filtrate was reduced in vacuo to produce a green amorphous solid (0.549 g, 1.29 mmol, 77%).
1H-NMR (400 Hz, d8-THF) δ 2.18 (s, 8H), 1.42 (s, 24H). 31P-NMR (202 MHz, CDCl3) δ 59.0.
4.4. General Borylation Procedure
In an argon-filled glovebox, dmpe2FeCl2 1 (8.6 mg, 0.02 mmol), sodium 2-ethylhexanoate (6.6 mg, 0.04 mmol), HBpin (87 µL, 0.6 mmol), substrate (0.5 mmol), and THF (1 mL) were added to a 1.7 mL sample vial and shaken to ensure full dissolution. The vial was placed under blue light radiation for 48 h and then allowed to cool to room temperature. Yields determined by 1H-NMR spectroscopy of the crude reaction mixtures using 1,3,5-trimethoxybenzene as an internal standard [0.5 mL; standard solution = 1,3,5-trimethoxybenzene (0.336 g, 2.0 mmol) in diethyl ether (10 mL)]. Product ratios were determined by 1H-NMR spectroscopy of the crude reaction mixtures.
4.5. Characterisation of Borylated Products
4.5.1. 2-Methylfuran Derivatives
4,4,5,5-Tetramethyl-2-(5-methylfuran-2-yl)-1,3,2-dioxaborolane 3a [37], 4,4,5,5-tetramethyl-2-(5-methylfuran-3-yl)-1,3,2-dioxaborolane 4a [37]
Following the general procedure; 2-methylfuran 2a (41 mg, 44 µL, 0.5 mmol). Yield = 72%. 3a:4a = 81:19. 1H-NMR (500 MHz, CDCl3) 3a: δ 6.99 (d, J = 3.2 Hz, 1H), 6.06–6.01 (m, 1H), 2.36 (s, 3H), 1.34 (s, 12H). 4a: δ 7.62 (d, J = 0.9 Hz, 1H), 6.15 (t, J = 1.0 Hz, 1H), 2.29 (d, J = 1.1 Hz, 3H), 1.31 (s, 12H). 13C-NMR (126 MHz, CDCl3) 3a: δ 157.8, 124.8, 106.9, 84.0, 24.7, 13.9. 4a: δ 152.7, 149.7, 108.8, 83.3, 24.9, 13.1. 11B-NMR (160 MHz, CDCl3) 3a: δ 27.1. 4a: δ 29.8.
4.5.2. Furan Derivatives
4,4,5,5-Tetramethyl-2-(furanyl-2-yl)-1,3,2-dioxaborolane 3b [43], 4,4,5,5-tetramethyl-2-(furanyl-3-yl)-1,3,2-dioxaborolane 4b [44], 2,5-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)furan 5ba [45], 2,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)furan 5bb [45]
Following the general procedure; furan 2b (34 mg, 36 µL, 0.5 mmol). Yield = 52%. 3b:4b:5ba:5bb 60:21:4:5. 1H-NMR (600 MHz, CDCl3) 3b: δ 7.65 (d, J = 1.7 Hz, 1H), 7.07 (d, J = 3.4 Hz, 1H), 6.44 (dd, J = 3.4, 1.6 Hz, 1H), 1.35 (s, 12H). 4b: δ 7.78 (s, 1H), 7.46 (m, J = 1.6 Hz, 1H), 6.59 (d, J = 1.7 Hz, 1H), 1.32 (s, 1H). 5ba: δ 7.06 (s, 2H), 1.33 (s, 24H). 5bb: δ 7.78 (s, 1H), 7.28 (s, 1H), 1.30 (s, 24H). 13C-NMR (126 MHz, CDCl3) 3b: δ 147.3, 123.2, 110.3, 84.2, 24.8. 4b: δ 151.2, 142.9, 113.1, 83.5, 24.9. 5ba: δ 123.2, 83.5, 24.8. 5bb: δ 151.2, 83.2, 75.1, 24.6. 11B-NMR (160 MHz, CDCl3) 3b: δ 27.2. 4b: δ 29.8.
4.5.3. 2.3-Dimethylfuran Derivatives
4,4,5,5-Tetramethyl-2-(4,5-dimethylfuran-2-yl)-1,3,2-dioxaborolane 3c [46]
Following the general procedure; 2,3-dimethylfuran 2c (48 mg, 53 µL, 0.5 mmol). Yield = 46%. 3c:4c = 100:0. 1H-NMR (500 MHz, CDCl3) δ 6.87 (s, 1H), 2.26 (s, 3H), 1.94 (s, 3H), 1.33 (s, 12H). 13C-NMR (126 MHz, CDCl3) δ 153.4, 127.1, 115.2, 83.9, 24.7, 11.8, 9.7. 11B-NMR (160 MHz, CDCl3) δ 27.2.
4.5.4. 2-Ethylfuran Derivatives
4,4,5,5-Tetramethyl-2-(5-ethylfuran-2-yl)-1,3,2-dioxaborolane 3d [47], 4,4,5,5-tetramethyl-2-(5-methylfuran-3-yl)-1,3,2-dioxaborolane 4d [47]
Following the general procedure; 2-ethylfuran 2d (48 mg, 53 µL, 0.5 mmol). Yield = 59%. 3d:3d = 70:30. 1H-NMR (500 MHz, CDCl3) 3d: δ 7.01 (d, J = 3.3 Hz, 1H), 6.05 (d, J = 3.1, 1H), 2.72 (q, J = 7.6 Hz, 2H), 1.34 (s, 12H), 1.25 (m, 3H). 4d: δ 7.64 (d, J = 0.8 Hz, 1H), 6.18 (d, J = 1.1 Hz, 1H), 2.6 (q, J = 7.5, 2H), 1.31 (s, 12H), 1.25 (m, 3H). 13C-NMR (126 MHz, CDCl3) 3d: δ 163.6, 124.7, 105.2, 84.0, 24.7, 21.6, 12.2. 4d: δ 163.6, 149.6, 107.2, 83.3, 24.9, 21.1, 12.1. 11B-NMR (160 MHz, CDCl3) 3d: δ 27.2. 4d: δ 29.9.
4.5.5. 2-Methylthiophene Derivatives
4,4,5,5-Tetramethyl-2-(5-methylthiophen-2-yl)-1,3,2-dioxaborolane 3e [47], 4,4,5,5-tetramethyl-2-(5-methylthiophen-3-yl)-1,3,2-dioxaborolane 4e [47]
Following the general procedure; 2-methylthiophene 2e (49 mg, 48 µL, 0.5 mmol). Yield = 9%. 3e:4e = 66:34. 1H-NMR (500 MHz, CDCl3) 3e: δ 7.45 (d, J = 3.4 Hz, 1H), 6.84 (d, J = 3.4, 1H), 2.53 (s, 3H), 1.33 (s, 12H). 4e: δ 7.67 (d, J = 1.2 Hz, 1H), 7.04 (s, 1H), 2.49 (d, J = 1.1 Hz, 3H), 1.32 (s, 12H). 13C-NMR (126 MHz, CDCl3) 3e: δ 147.5, 137.6, 127.0, 83.9, 24.9, 15.4. 4e: not observed. 11B-NMR (160 MHz, CDCl3) δ 28.7.
4.5.6. Thiophene Derivatives
4,4,5,5-Tetramethyl-2-(thiophen-2-yl)-1,3,2-dioxaborolane 3f [48], 4,4,5,5-tetramethyl-2-(thiophen-3-yl)-1,3,2-dioxaborolane 4f [49], 2,5-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophene 5f [48]
Following the general procedure; thiophene 2f (42 mg, 40 µL, 0.5 mmol). Yield = 11%. 3f:4f:5f = 27:9:64. 1H-NMR (500 MHz, CDCl3) 3f: δ 7.64 (m, 1H), 7.63 (d, J = 4.8 Hz, 1H), 7.19 (d, J = 4.8 Hz, 1H), 1.35 (s, 12H). 4f: δ 7.92 (d, J = 2.6 Hz, 1H), 7.41 (d, J = 4.9 Hz, 1H), 7.34 (dd, J = 4.8, 2.7 Hz, 1H), 1.35 (s, 12H). 5f: δ 7.66 (s, 2H), 1.34 (s, 24H). 13C-NMR (126 MHz, CDCl3) 3f: δ 137.2, 132.4, 128.2, 83.2, 24.9. 4f: δ 136.5, 129.0, 125.3, 83.2, 24.8. 5f: δ 137.7, 84.1, 24.8. 11B-NMR (160 MHz, CDCl3) δ 29.2.
4.5.7. 3-Methylthiophene Derivatives
4,4,5,5-Tetramethyl-2-(4-methylthiophen-2-yl)-1,3,2-dioxaborolane 3g [17]
Following the general procedure; 3-methylthiophene 2g (49 mg, 48 µL, 0.5 mmol). Yield = 4%. 3g:4g = 100:0. 1H-NMR (500 MHz, CDCl3) δ 7.44 (d, J = 1.2 Hz, 1H), 7.19 (t, J = 1.1 Hz, 1H), 2.29 (d, J = 0.9 Hz, 3H), 1.34 (s, 12H). 13C-NMR (126 MHz, CDCl3) δ 139.5, 139.0, 128.2, 84.0, 24.9, 15.1. 11B-NMR (160 MHz, CDCl3) δ 29.1.
4.6. Mechanistic Investigations
dmpe2FeH2 7 [37]
dmpe2FeCl2 1 (10 mg, 0.023 mmol), sodium 2-ethylhexanoate (7.6 mg, 0.046 mmol), and HBpin (7 µL, 0.046 mmol) were added to a Young’s NMR tube under an Ar atmosphere and heated at 60 °C for 3 days. 1H-NMR (600 MHz, THF) δ −14.38 (m). 31P-NMR (500 MHz, THF) δ 76.9 (t, J = 28 Hz), 67.7 (t, J = 28 Hz).
dmpe2FeCl2 1 (4.3 mg, 0.001 mmol), sodium 2-ethylhexanoate (3.3 mg, 0.002 mmol), and HBpin (87 µL, 0.6 mmol) were added to a Young’s NMR tube under an Ar atmosphere and irradiated with blue light for 16 h. 1H-NMR (500 MHz, THF) δ −13.1 (p, J = 43.2 Hz), −14.0 (m). 31P-NMR (500 MHz, THF) δ 77.6(m), 77.2 (m), 59.7 (m), 58.9 (m).
dmpe2FeH(2-Me-furyl) 10
dmpe2FeCl2 1 (10 mg, 0.023 mmol), sodium 2-ethylhexanoate (30.4 mg, 0.184 mmol), and HBpin (7 µL, 0.046 mmol) were added to a Young’s NMR tube under an Ar atmosphere and warmed at 60 °C for 24 h. 2-methylfuran (8 µL, 0.092 mmol) was added under an Ar atmosphere and the sample irradiated with blue light for 3 h. This complex was observed in situ. 1H-NMR (500 MHz, THF) δ -18.93 (q, J = 45.8 Hz). 31P-NMR (500 MHz, THF) δ 77.1 (d, J = 38.1 Hz). MS: (HRMS – EI+) Found 438.12041 (C17H38O156Fe1P4), requires 438.12171.
Supplementary Materials
The following are available online.
Author Contributions
L.B. and J.H.D. carried out the experimental work. J.H.D., A.P.D. and S.P.T. conceived and supervised the project. L.B., J.H.D., and S.P.T. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by The Royal Society, UF130393 and RF191015, and GSK/EPSRC, PIII0002.
Acknowledgments
S.P.T. acknowledges the University of Edinburgh and the Royal Society for a University Research Fellowship. J.H.D. and S.P.T. acknowledge GSK, EPSRC, and the University of Edinburgh for post-doctoral funding. L.B. acknowledges the Royal Society and the University of Edinburgh for a PhD studentship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kakiuchi, F.; Chatani, N. Catalytic Methods for C-H Bond Functionalization: Application in Organic Synthesis. Adv. Synth. Catal. 2003, 345, 1077–1101. [Google Scholar] [CrossRef]
- Godula, K.; Sames, D. C-H bond functionalization in complex organic synthesis. Science 2006, 312, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, L.; O’Hara, F.; Gaunt, M.J. Recent developments in natural product synthesis using metal-catalysed C-H bond functionalisation. Chem. Soc. Rev. 2011, 40, 1885–1898. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C-H bond functionalization: Emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Morton, D. Recent Advances in C-H Functionalization. J. Org. Chem. 2016, 81, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roudesly, F.; Oble, J.; Poli, G. Metal-catalyzed C-H activation/functionalization: The fundamentals. J. Mol. Catal. A Chem. 2017, 426, 275–296. [Google Scholar] [CrossRef]
- Su, B.; Cao, Z.C.; Shi, Z.J. Exploration of Earth-Abundant Transition Metals (Fe, Co, and Ni) as Catalysts in Unreactive Chemical Bond Activations. Acc Chem. Res. 2015, 48, 886–896. [Google Scholar] [CrossRef]
- Cera, G.; Ackermann, L. Iron-Catalyzed C–H Functionalization Processes. Top. Curr. Chem. 2016, 374, 57. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed C-H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef] [PubMed]
- Loup, J.; Dhawa, U.; Pesciaioli, F.; Wencel-Delord, J.; Ackermann, L. Enantioselective C–H Activation with Earth-Abundant 3d Transition Metals. Angew. Chem. Int. Ed. 2019, 58, 12803–12818. [Google Scholar] [CrossRef] [PubMed]
- Mkhalid, I.A.I.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C-H activation for the construction of C-B bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.W.B.; Watson, A.J.B. Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks. Chemistry 2017, 3, 31–55. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, G.; Zhang, S.; Wang, H.; Wang, L.; Liu, L.; Jiao, J.; Li, P. Recent advances in catalytic C–H borylation reactions. Tetrahedron 2017, 73, 7123–7157. [Google Scholar] [CrossRef]
- Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N.R.; Hartwig, J.F. Mild iridium-catalyzed borylation of arenes. High turnover numbers, room temperature reactions, and isolation of a potential intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. [Google Scholar] [CrossRef]
- Chotana, G.A.; Kallepalli, V.A.; Maleczka, R.E.; Smith, M.R. Iridium-catalyzed borylation of thiophenes: Versatile, synthetic elaboration founded on selective C-H functionalization. Tetrahedron 2008, 64, 6103–6114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, J.F. Regioselectivity of the borylation of alkanes and arenes. Chem. Soc. Rev. 2011, 40, 1992–2002. [Google Scholar] [CrossRef]
- Hartwig, J.F. Borylation and silylation of C-H bonds: A platform for diverse C-H bond functionalizations. Acc. Chem. Res. 2012, 45, 864–873. [Google Scholar] [CrossRef]
- Preshlock, S.M.; Plattner, D.L.; Maligres, P.E.; Krska, S.W.; Maleczka, R.E.; Smith, M.R. A traceless directing group for C-H borylation. Angew. Chem. Int. Ed. 2013, 52, 12915–12919. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.A.; Hartwig, J.F. Iridium-catalyzed C-H borylation of heteroarenes: Scope, regioselectivity, application to late-stage functionalization, and mechanism. J. Am. Chem. Soc. 2014, 136, 4287–4299. [Google Scholar] [CrossRef] [PubMed]
- Sadler, S.A.; Tajuddin, H.; Mkhalid, I.A.I.; Batsanov, A.S.; Albesa-Jove, D.; Cheung, M.S.; Maxwell, A.C.; Shukla, L.; Roberts, B.; Blakemore, D.C.; et al. Iridium-catalyzed C-H borylation of pyridines. Org. Biomol. Chem. 2014, 12, 7318–7327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Segawa, Y.; Itami, K. Para C-H borylation of benzene derivatives by a bulky iridium catalyst. J. Am. Chem. Soc. 2015, 137, 5193–5198. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Semba, K.; Nakao, Y. Para-Selective C–H Borylation of (Hetero)Arenes by Cooperative Iridium/Aluminum Catalysis. Angew. Chem. Int. Ed. 2017, 56, 4853–4857. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Jiang, Y.; Kuang, C.; Wang, S.; Liu, H.; Zhang, Y.; Wang, J. Nano-Fe2O3-catalyzed direct borylation of arenes. Chem. Commun. 2010, 46, 3170–3172. [Google Scholar] [CrossRef]
- Zhang, H.; Hagihara, S.; Itami, K. Aromatic C-H borylation by nickel catalysis. Chem. Lett. 2015, 44, 779–781. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, T.; Tobisu, M.; Chatani, N. Nickel-catalyzed borylation of arenes and indoles via C-H bond cleavage. Chem. Commun. 2015, 51, 6508–6511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzacano, T.J.; Mankad, N.P. Thermal C-H borylation using a CO-free iron boryl complex. Chem. Commun. 2015, 51, 5379–5382. [Google Scholar] [CrossRef]
- Obligacion, J.V.; Semproni, S.P.; Pappas, I.; Chirik, P.J. Cobalt-Catalyzed C(sp2)-H Borylation: Mechanistic Insights Inspire Catalyst Design. J. Am. Chem. Soc. 2016, 138, 10645–10653. [Google Scholar] [CrossRef]
- Léonard, N.G.; Bezdek, M.J.; Chirik, P.J. Cobalt-Catalyzed C(sp2)-H borylation with an air-stable, readily prepared terpyridine cobalt(II) Bis(acetate) precatalyst. Organometallics 2017, 36, 142–150. [Google Scholar] [CrossRef]
- Yoshigoe, Y.; Kuninobu, Y. Iron-Catalyzed ortho-Selective C-H Borylation of 2-Phenylpyridines and Their Analogs. Org. Lett. 2017, 19, 3450–3453. [Google Scholar] [CrossRef] [PubMed]
- Obligacion, J.V.; Bezdek, M.J.; Chirik, P.J. C(sp2)-H Borylation of Fluorinated Arenes Using an Air-Stable Cobalt Precatalyst: Electronically Enhanced Site Selectivity Enables Synthetic Opportunities. J. Am. Chem. Soc. 2017, 139, 2825–2832. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, M.; Kusaka, H.; Yuge, H. Iron-catalyzed versatile and efficient C(sp2)-H borylation. Chem. Lett. 2019, 48, 898–901. [Google Scholar] [CrossRef] [Green Version]
- Agahi, R.; Challinor, A.J.; Dunne, J.; Docherty, J.H.; Carter, N.B.; Thomas, S.P. Regiodivergent hydrosilylation, hydrogenation, [2π + 2π]-cycloaddition and C-H borylation using counterion activated earth-abundant metal catalysis. Chem. Sci. 2019, 10, 5079–5084. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, T.; Ohki, Y.; Tatsumi, K. C-H bond activation/borylation of furans and thiophenes catalyzed by a half-sandwich iron N-heterocyclic carbene complex. Chemistry 2010, 5, 1657–1666. [Google Scholar] [CrossRef]
- Mazzacano, T.J.; Mankad, N.P. Base metal catalysts for photochemical C-H borylation that utilize metal-metal cooperativity. J. Am. Chem. Soc. 2013, 135, 17258–17261. [Google Scholar] [CrossRef]
- Dombray, T.; Werncke, C.G.; Jiang, S.; Grellier, M.; Vendier, L.; Bontemps, S.; Sortais, J.B.; Sabo-Etienne, S.; Darcel, C. Iron-catalyzed C-H borylation of arenes. J. Am. Chem. Soc. 2015, 137, 4062–4065. [Google Scholar] [CrossRef]
- Allen, O.R.; Dalgarno, S.J.; Field, L.D.; Jensen, P.; Turnbull, A.J.; Willis, A.C. Addition of CO2 to alkyl iron complexes, Fe(PP)2Me2. Organometallics 2008, 27, 2092–2098. [Google Scholar] [CrossRef]
- Docherty, J.H.; Peng, J.; Dominey, A.P.; Thomas, S.P. Activation and discovery of earth-abundant metal catalysts using sodium tert-butoxide. Nat. Chem. 2017, 9, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Buys, I.E.; Field, L.D.; Hambley, T.W.; McQueen, A.E.D.J. Photochemical reactions of [cis-Fe(H)2(Me2PCH2CH2PMe2)2] with thiophenes: Insertion into C–H and C–S bonds. Chem. Soc. Chem. Commun. 1994, 5, 557–558. [Google Scholar] [CrossRef]
- Query, I.P.; Squier, P.A.; Larson, E.M.; Isley, N.A.; Clark, T.B. Alkoxide-catalyzed reduction of ketones with pinacolborane. J. Org. Chem. 2011, 76, 6452–6456. [Google Scholar] [CrossRef]
- Elton, T.E.; Ball, G.E.; Bhadbhade, M.; Field, L.D.; Colbran, S.B. Evaluation of Organic Hydride Donors as Reagents for the Reduction of Carbon Dioxide and Metal-Bound Formates. Organometallics 2018, 37, 3972–3982. [Google Scholar] [CrossRef]
- Wollenburg, M.; Moock, D.; Glorius, F. Hydrogenation of Borylated Arenes. Angew. Chem. Int. Ed. 2019, 58, 6549–6553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiao, L. Pyridine-Catalyzed Radical Borylation of Aryl Halides. J. Am. Chem. Soc. 2017, 139, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Ishiyama, T. Process for the Production of Hetereoaryl-type Boron Compounds with Iridium Catalyst. European Patent EP1481978A1, 1 December 2004. [Google Scholar]
- Xue, C.; Luo, Y.; Teng, H.; Ma, Y.; Nishiura, M.; Hou, Z. Ortho-Selective C–H Borylation of Aromatic Ethers with Pinacol-borane by Organo Rare-Earth Catalysts. ACS Catalysis. 2018, 8, 5017–5022. [Google Scholar] [CrossRef]
- Kato, T.; Kuriyama, S.; Nakajima, K.; Nishibayashi, Y. Catalytic C–H Borylation Using Iron Complexes Bearing 4,5,6,7-Tetrahydroisoindol-2-ide-Based PNP-Type Pincer Ligand. Chem Asian J. 2019, 14, 2097–2101. [Google Scholar] [CrossRef]
- Tobisu, M.; Igarashi, T.; Chatani, N. Iridium/N-heterocyclic carbene-catalyzed C–H borylation of arenes by diisopropylaminoborane. Beilstein J. Org. Chem. 2016, 12, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Ratniyom, J.; Dechnarong, N.; Yotphan, S.; Kiatisevi, S. Convenient Synthesis of Arylboronates through a Synergistic Pd/Cu-Catalyzed Miyaura Borylation Reaction under Atmospheric Conditions. Eur. J. Org. Chem. 2014, 7, 1381–1385. [Google Scholar] [CrossRef]
Sample Availability: Samples of the pre-catalyst 1 are available from the authors. |
Scheme 1.
Iron-catalysed C-H borylation of arenes. (a) Prior approaches to iron-catalysed C(sp2)-H-bond borylation with pinacolborane (HBpin) using organoiron and iron/copper bimetallic catalysts. (b) This work: C(sp2)-H bond borylation using dmpe2FeCl2 as a pre-catalyst, activated by exogenous nucleophiles, under blue light irradiation.
Scheme 1.
Iron-catalysed C-H borylation of arenes. (a) Prior approaches to iron-catalysed C(sp2)-H-bond borylation with pinacolborane (HBpin) using organoiron and iron/copper bimetallic catalysts. (b) This work: C(sp2)-H bond borylation using dmpe2FeCl2 as a pre-catalyst, activated by exogenous nucleophiles, under blue light irradiation.

Scheme 2.
Activator screening for the borylation of 2-methyl furan by dmpe2FeCl2 1. a Yields determined by 1H-NMR spectroscopy of the crude reaction mixtures using 1,3,5-trimethoxybenzene as an internal standard. Product ratios were determined by 1H-NMR spectroscopy of the crude reaction mixtures. b Reaction time = 15 h. c Reaction time = 48 h. 2-EH = 2-ethylhexanoate. TBA = tetra-n-butylammonium.
Scheme 2.
Activator screening for the borylation of 2-methyl furan by dmpe2FeCl2 1. a Yields determined by 1H-NMR spectroscopy of the crude reaction mixtures using 1,3,5-trimethoxybenzene as an internal standard. Product ratios were determined by 1H-NMR spectroscopy of the crude reaction mixtures. b Reaction time = 15 h. c Reaction time = 48 h. 2-EH = 2-ethylhexanoate. TBA = tetra-n-butylammonium.

Scheme 3.
Na(2-EH) activated borylation of furan and thiophene derivatives using dmpe2FeCl2 1. a Yields determined by 1H-NMR spectroscopy of the crude reaction mixtures using 1,3,5-trimethoxybenzene as an internal standard. Product ratios were determined by 1H-NMR spectroscopy of the crude reaction mixtures. b Values represent the ratio of 2-boryl:3-boryl:bis-boryl products.
Scheme 3.
Na(2-EH) activated borylation of furan and thiophene derivatives using dmpe2FeCl2 1. a Yields determined by 1H-NMR spectroscopy of the crude reaction mixtures using 1,3,5-trimethoxybenzene as an internal standard. Product ratios were determined by 1H-NMR spectroscopy of the crude reaction mixtures. b Values represent the ratio of 2-boryl:3-boryl:bis-boryl products.

Scheme 4.
(a) Pre-catalyst activation and hydride formation. (b) Mechanistic investigations of dmpe2FeH2 7 produced by hydride transfer from HBpin and Na(2-EH).
Scheme 4.
(a) Pre-catalyst activation and hydride formation. (b) Mechanistic investigations of dmpe2FeH2 7 produced by hydride transfer from HBpin and Na(2-EH).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Britton, L.; Docherty, J.H.; Dominey, A.P.; Thomas, S.P. Iron-Catalysed C(sp2)-H Borylation Enabled by Carboxylate Activation. Molecules 2020, 25, 905. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040905
AMA Style
Britton L, Docherty JH, Dominey AP, Thomas SP. Iron-Catalysed C(sp2)-H Borylation Enabled by Carboxylate Activation. Molecules. 2020; 25(4):905. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040905
Chicago/Turabian StyleBritton, Luke, Jamie H. Docherty, Andrew P. Dominey, and Stephen P. Thomas. 2020. "Iron-Catalysed C(sp2)-H Borylation Enabled by Carboxylate Activation" Molecules 25, no. 4: 905. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040905