
Solid-State NMR Techniques for the Structural Characterization of Cyclic Aggregates Based on Borane–Phosphane Frustrated Lewis Pairs

 ,
,
Abstract
:1. Introduction
2. Solid-State NMR—Basics and Methodology
2.1. Fundamental Principles
2.2. Internal Nuclear Interactions
2.2.1. Magnetic Shielding Effects
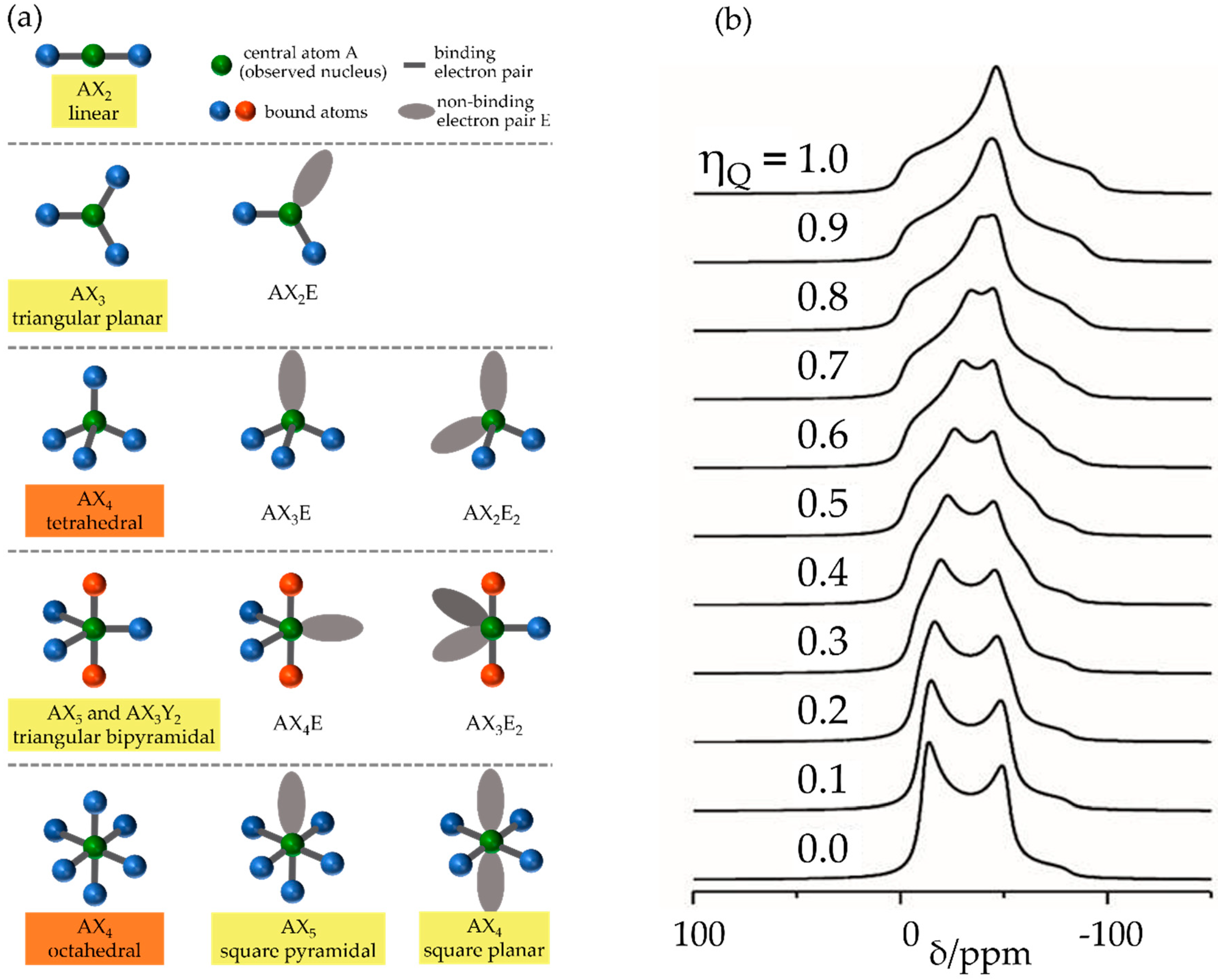
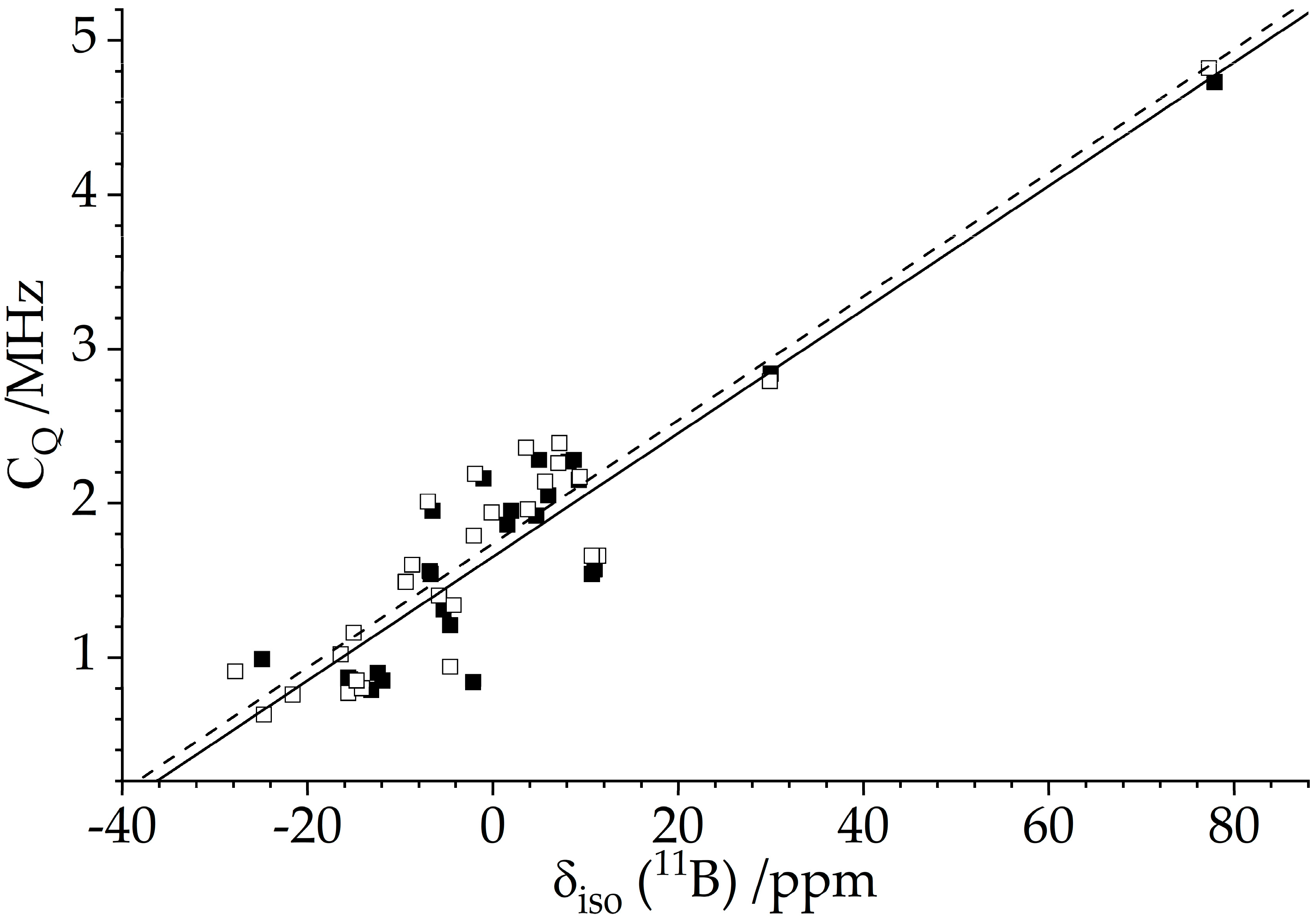
2.2.2. Nuclear Electric Quadrupole Interactions
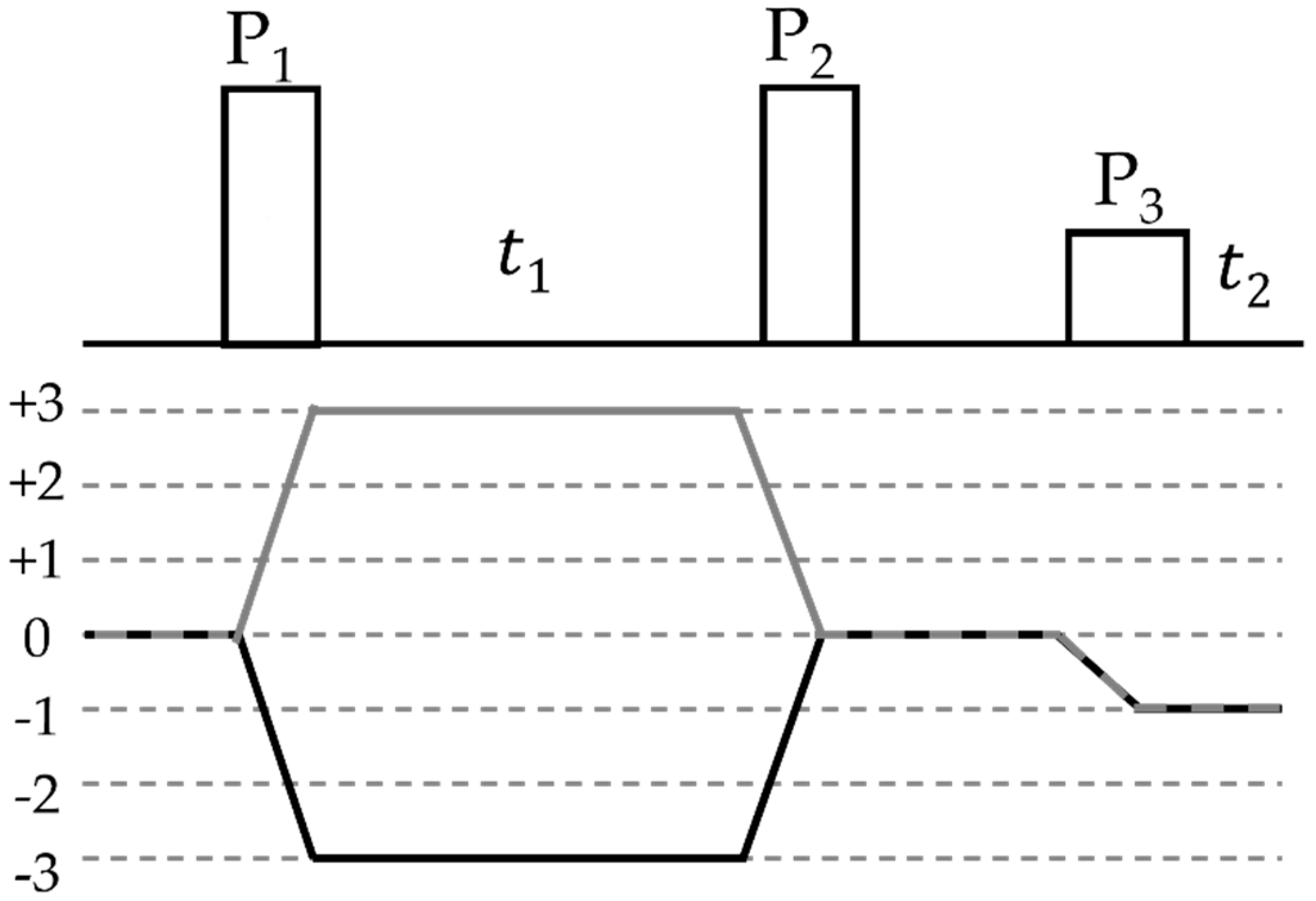
2.2.3. Indirect Spin–Spin Coupling (“J-coupling”)
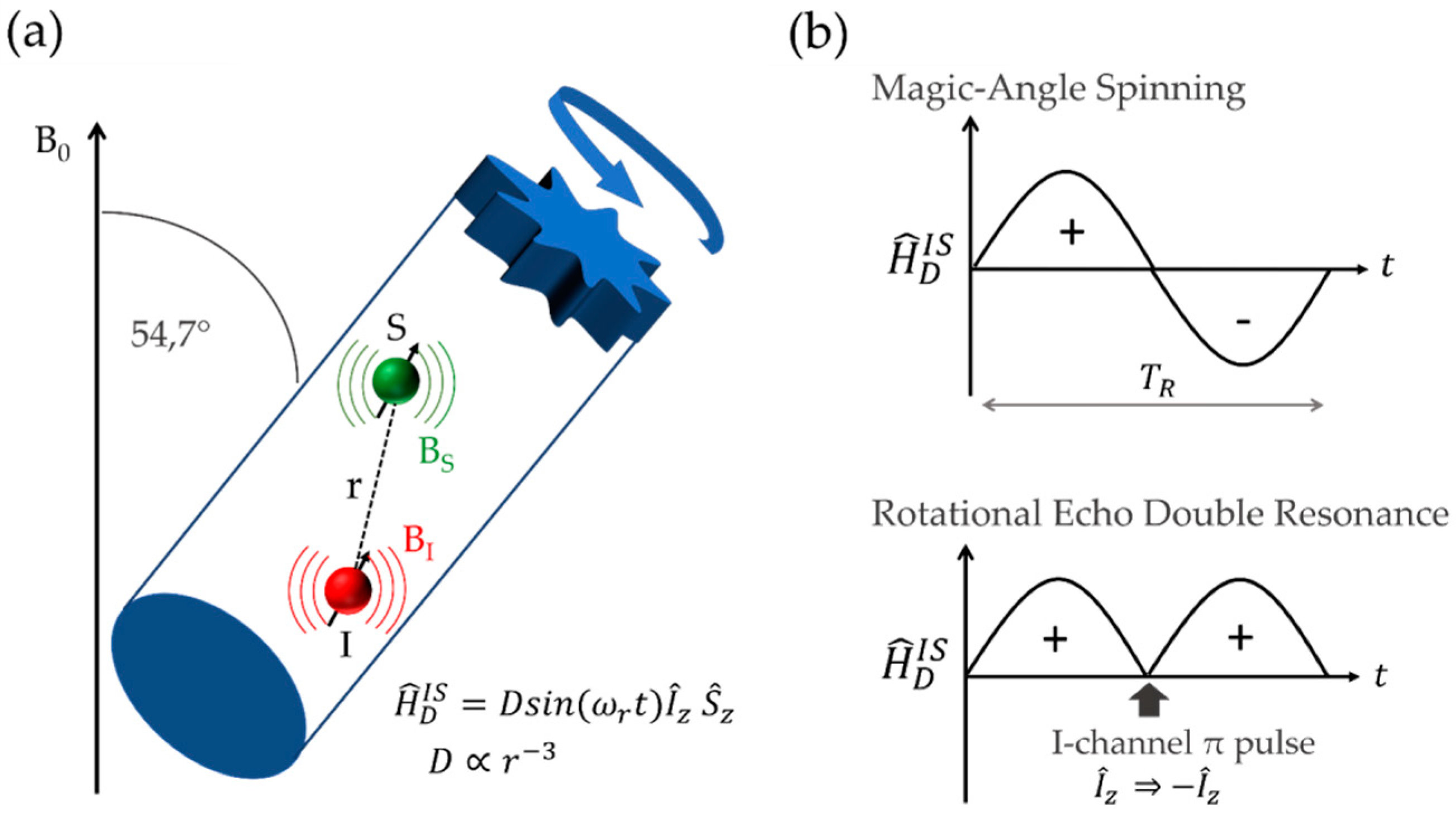
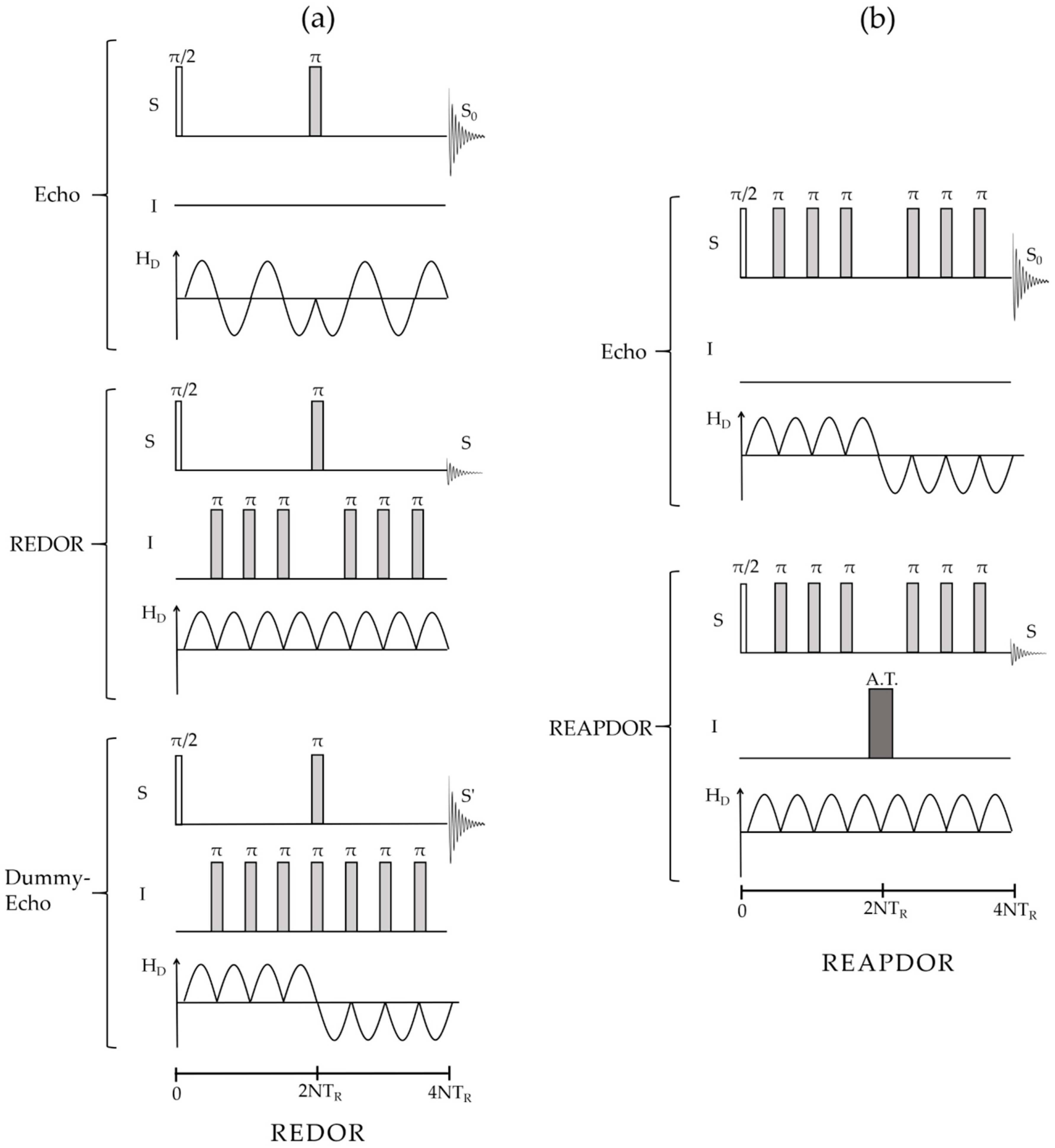
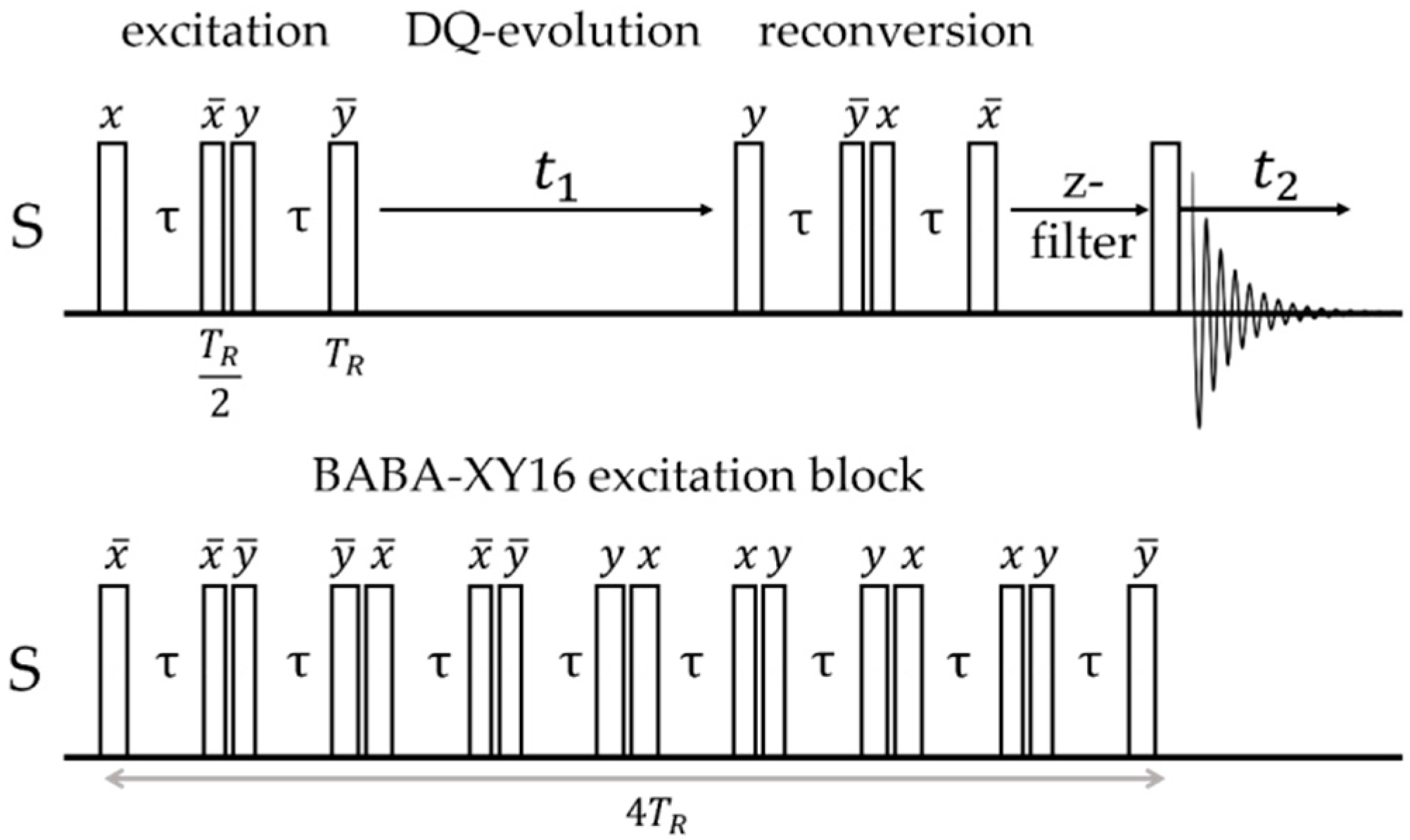
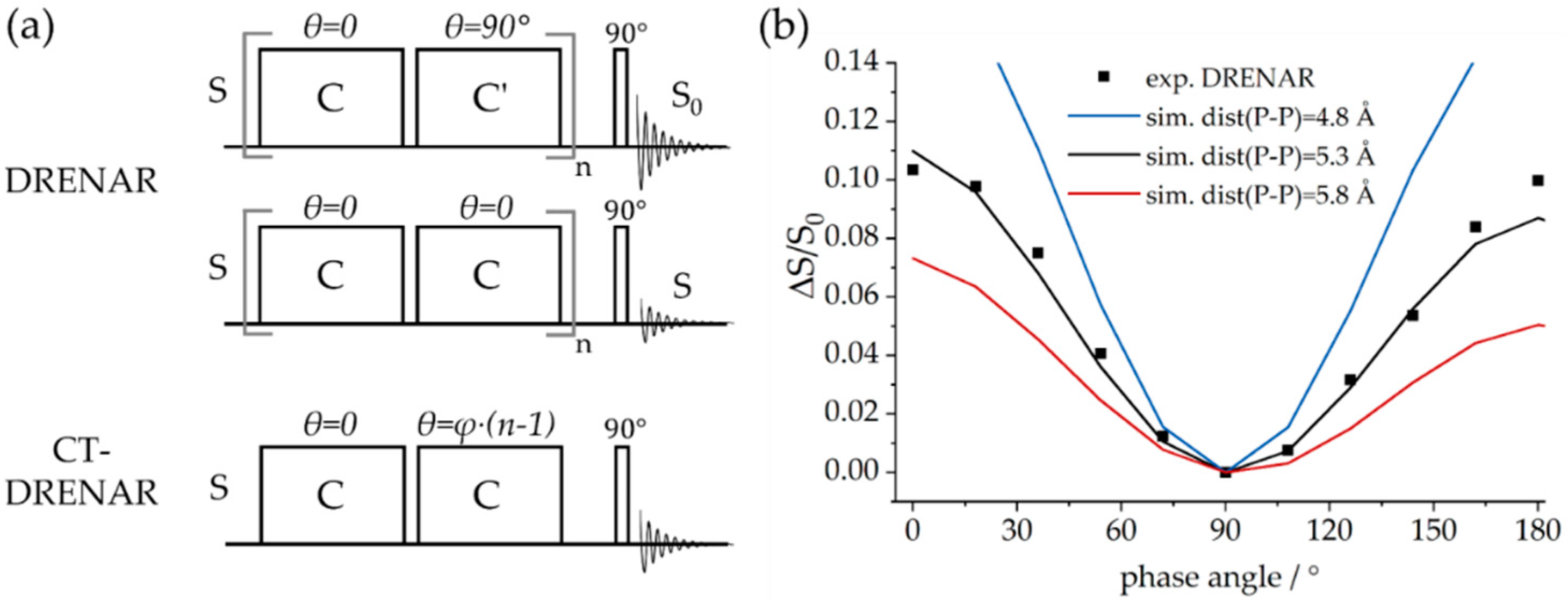
2.2.4. Direct Spin–Spin (Dipolar) Coupling
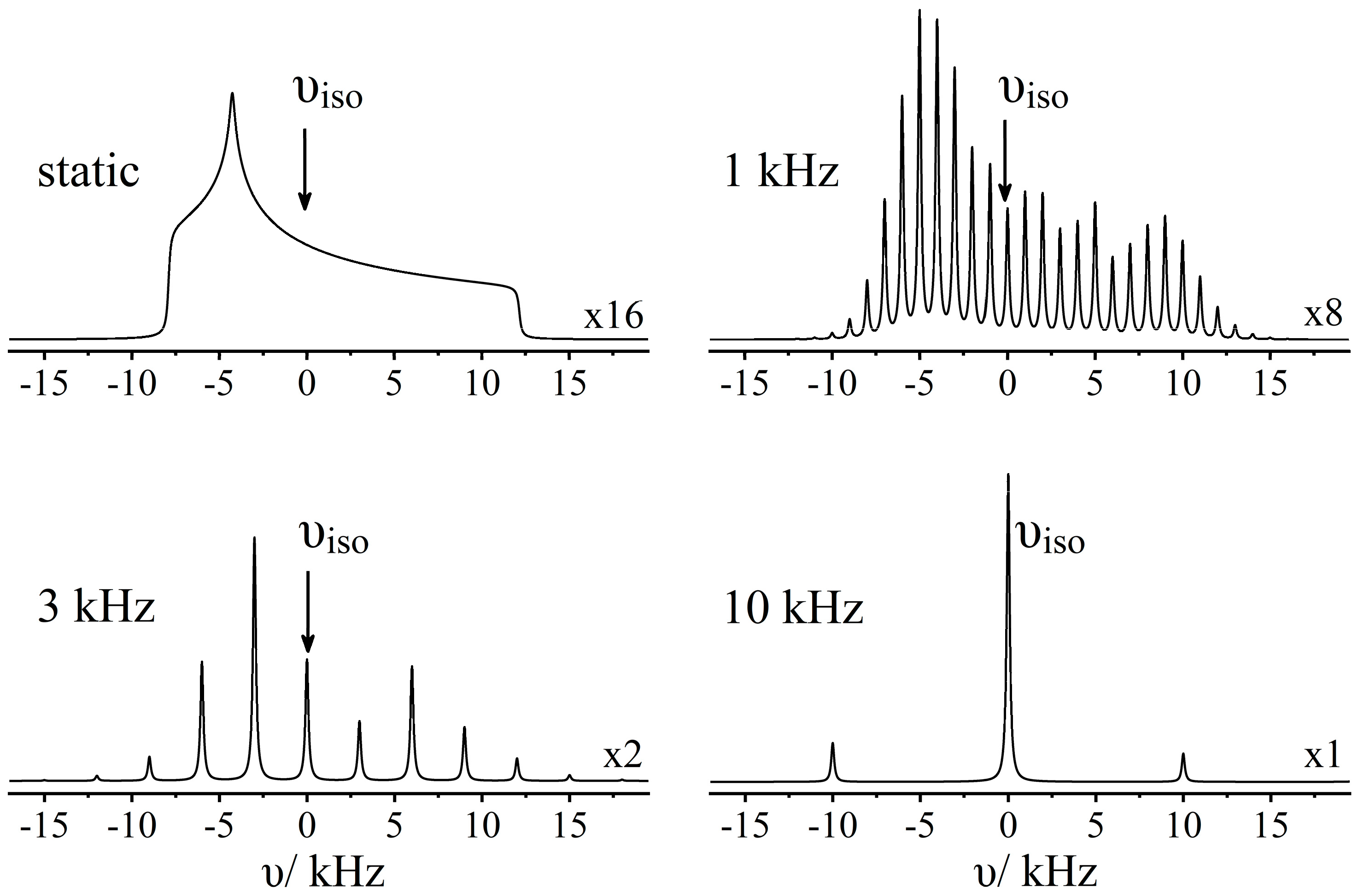
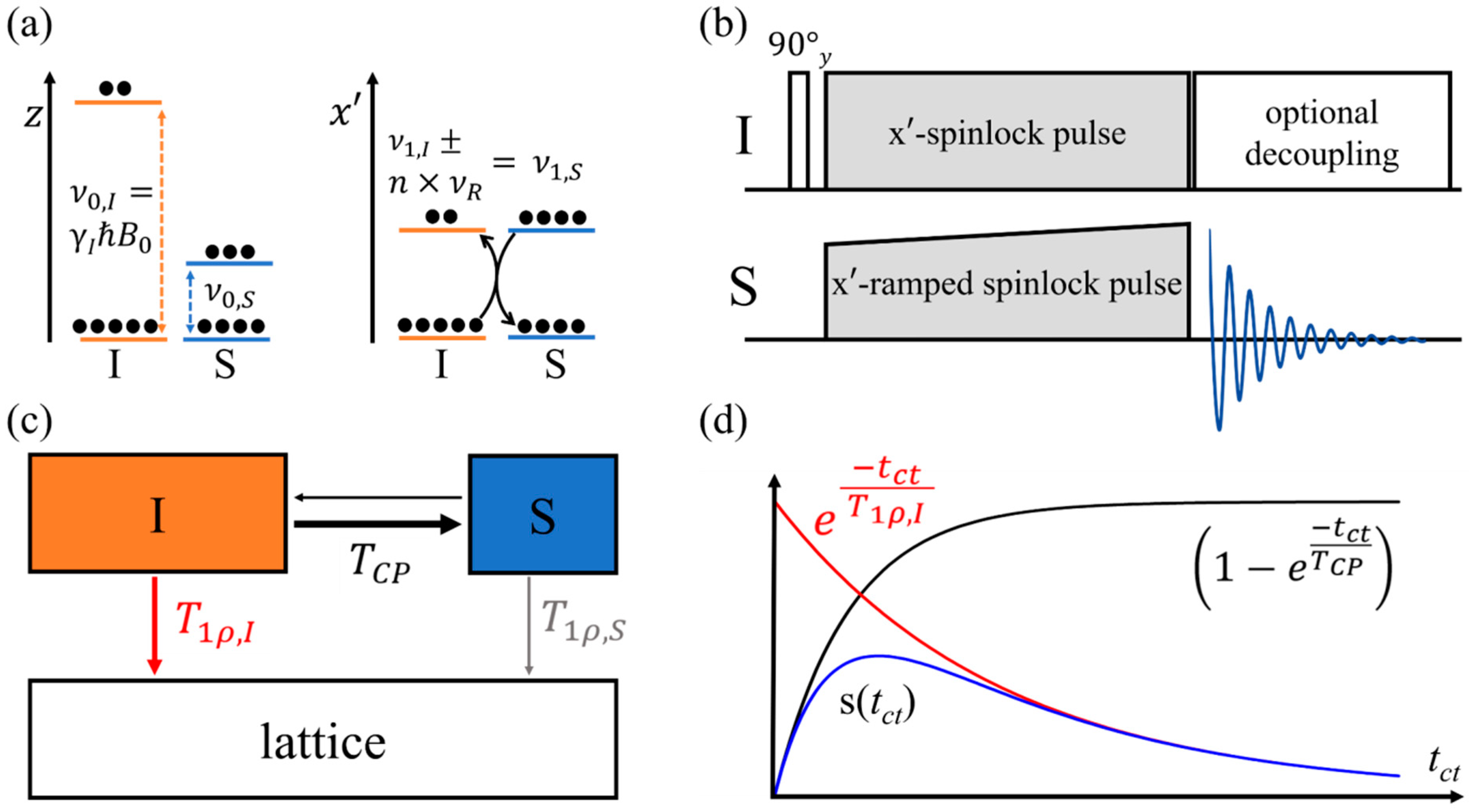
2.3. Cross-Polarization
3. Results and Discussion
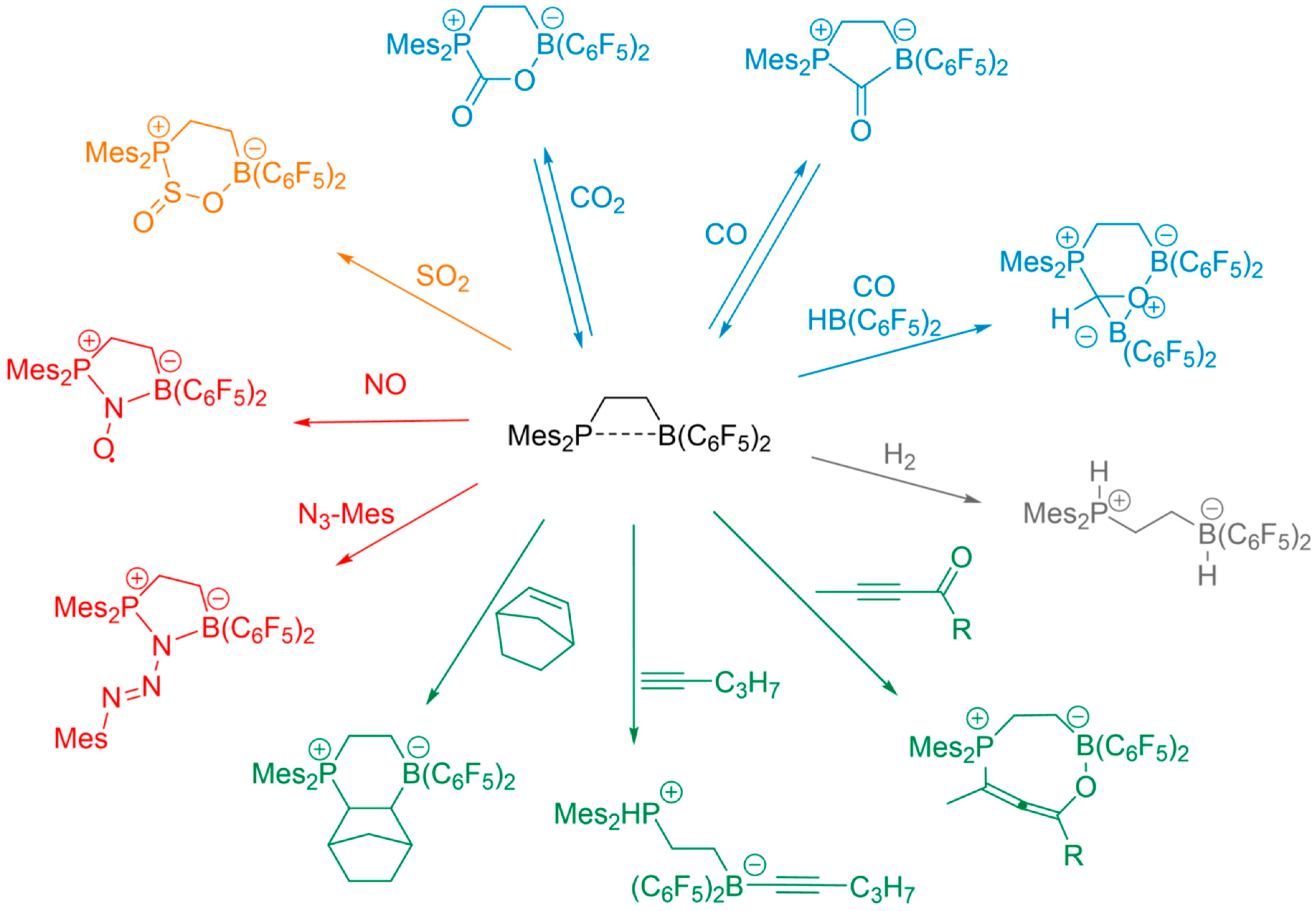
3.1. NMR Criteria of FLP Behavior
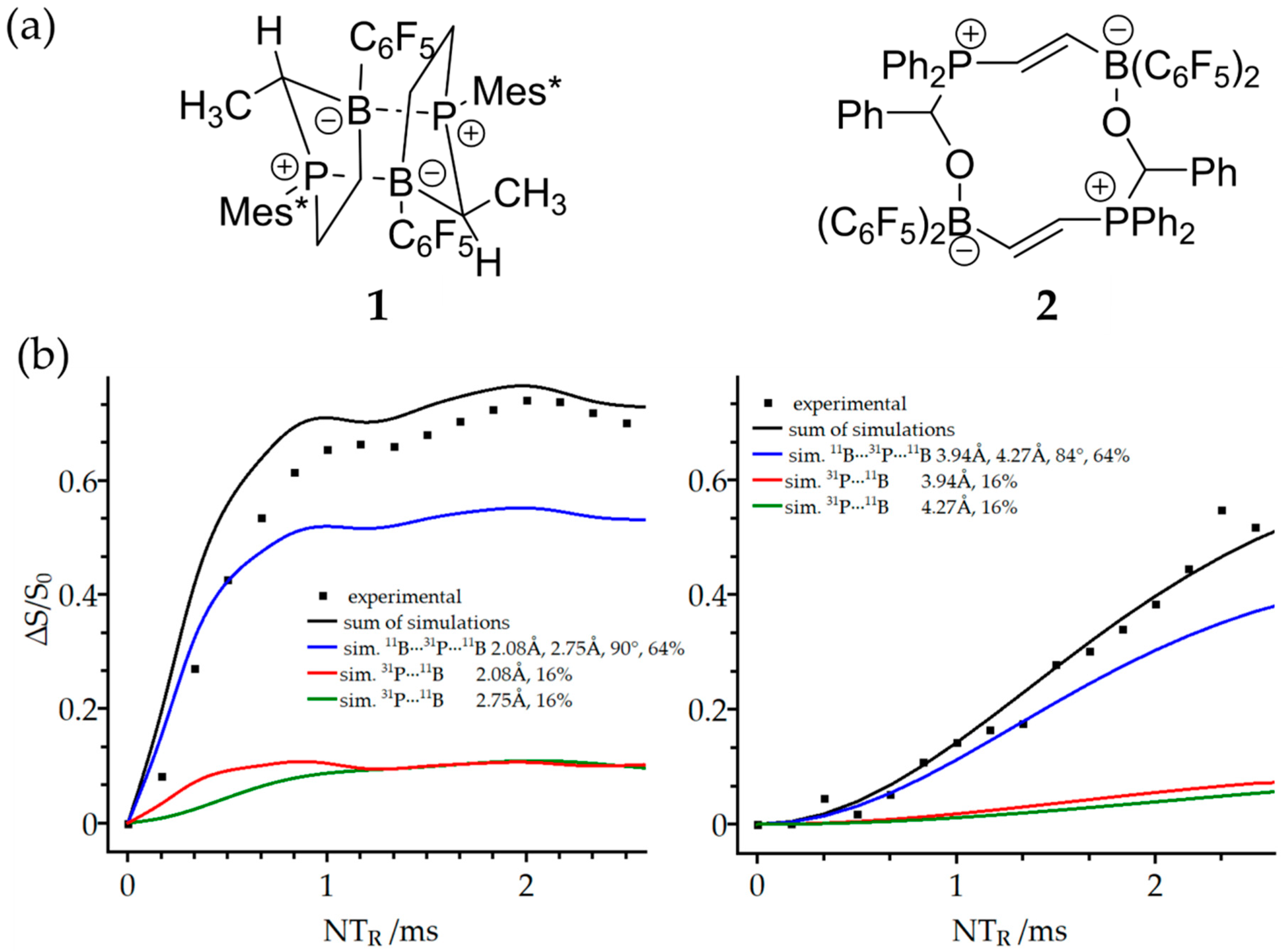
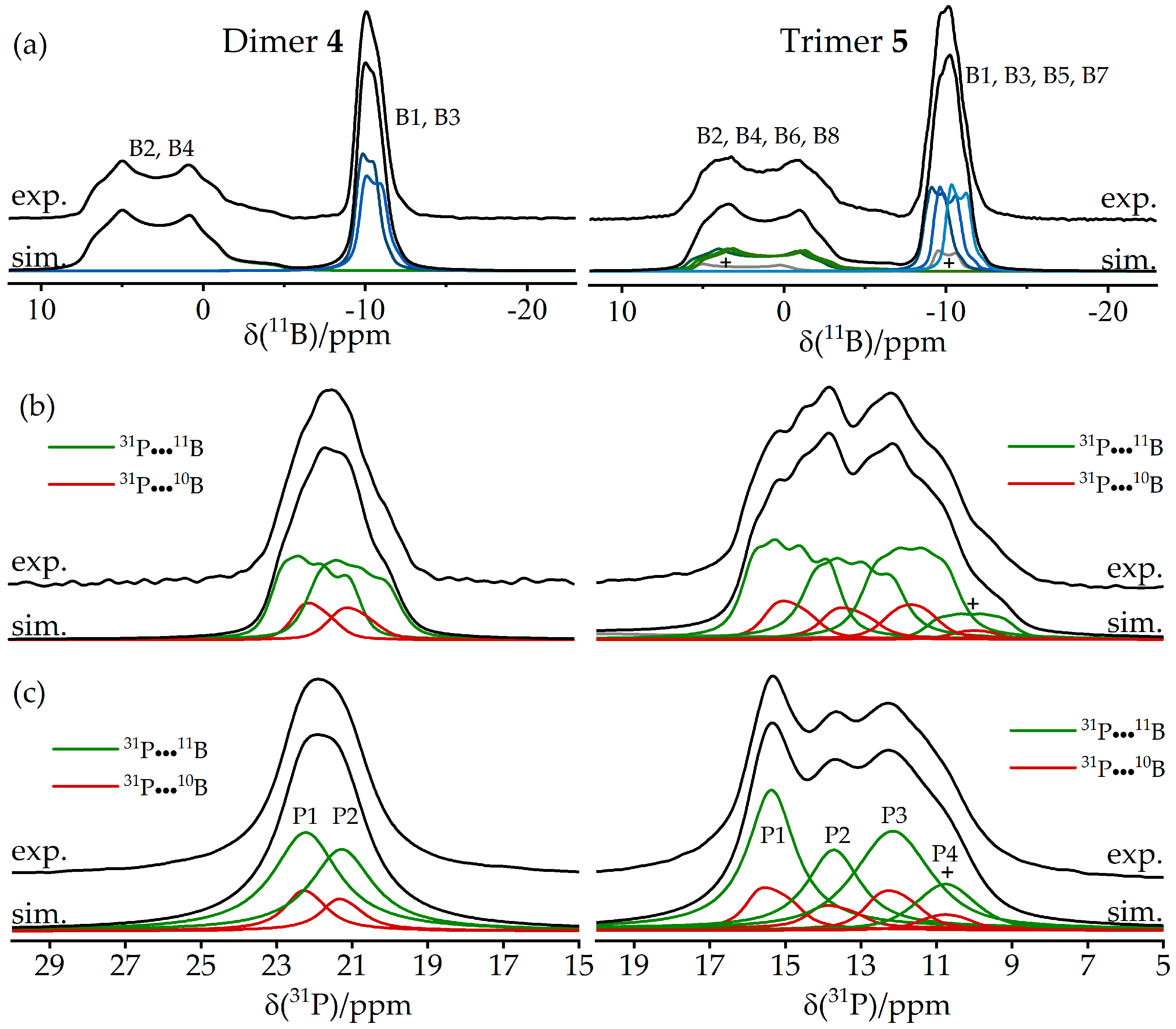
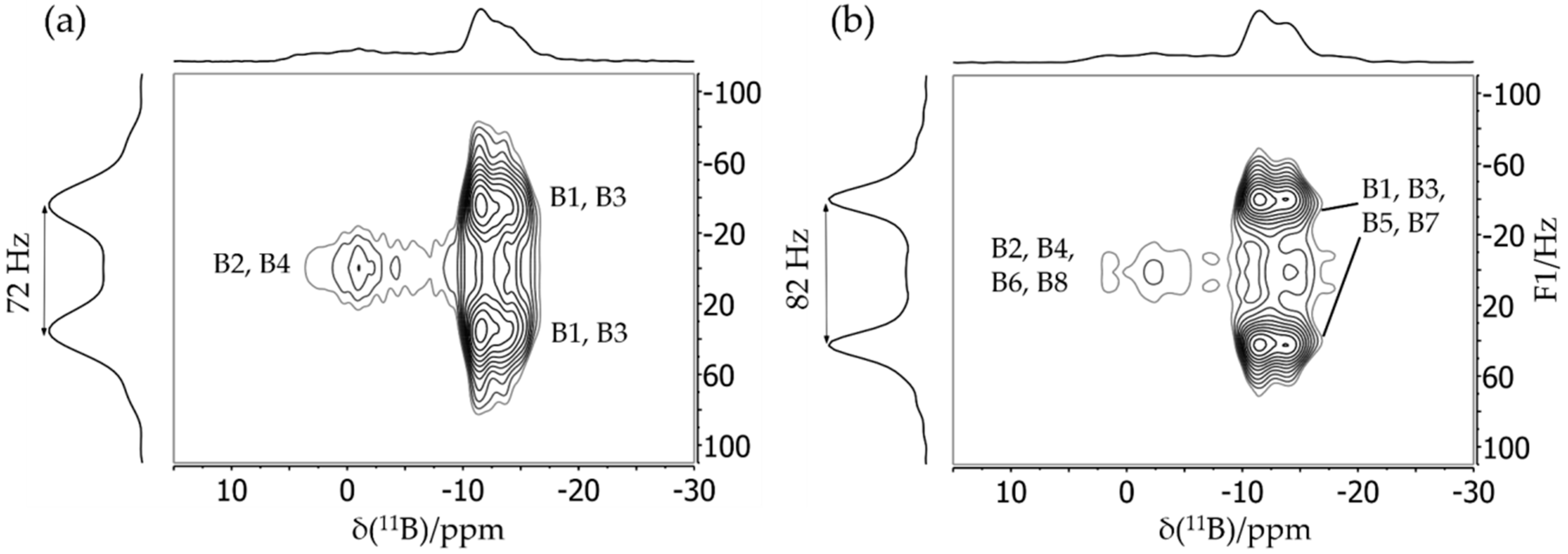
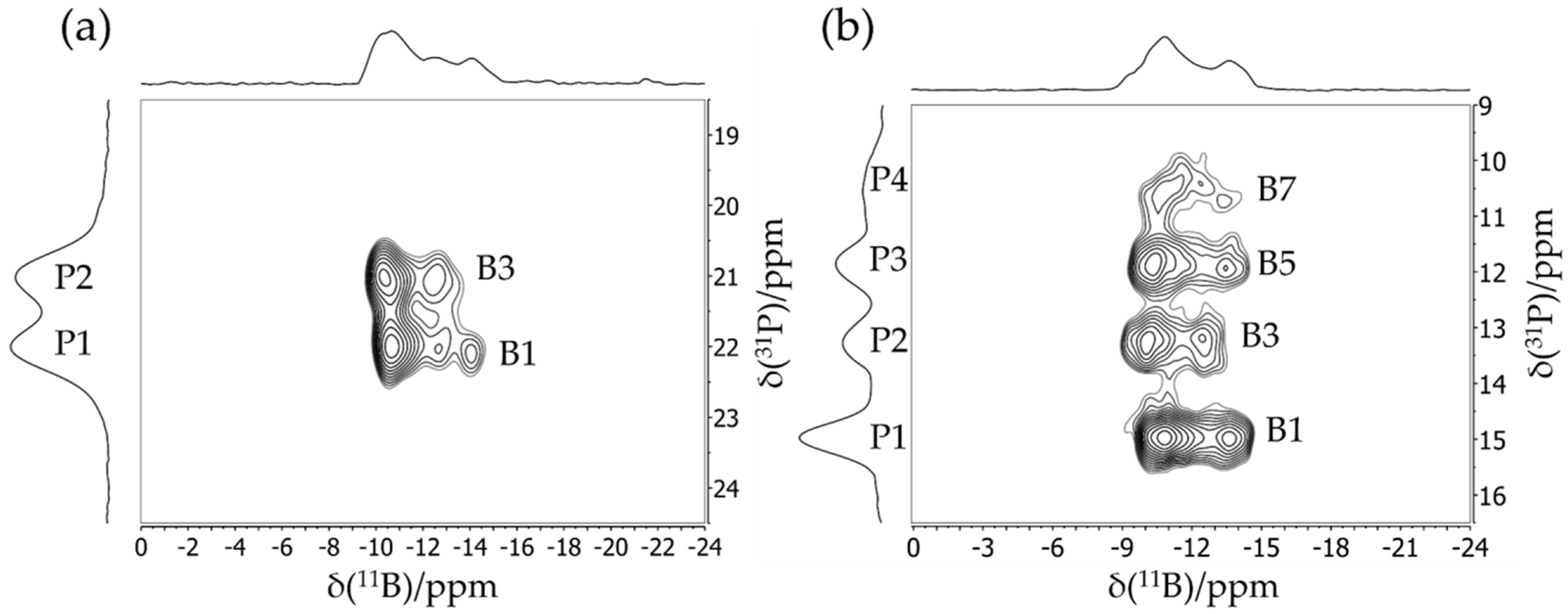
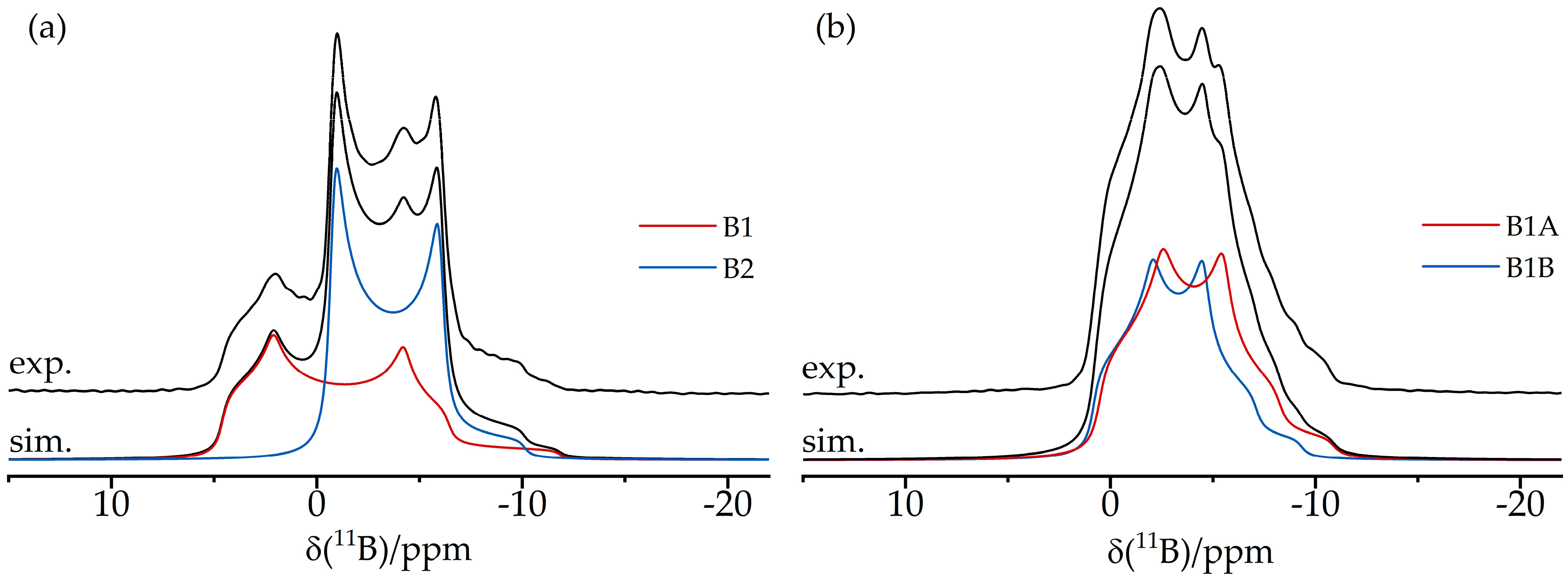
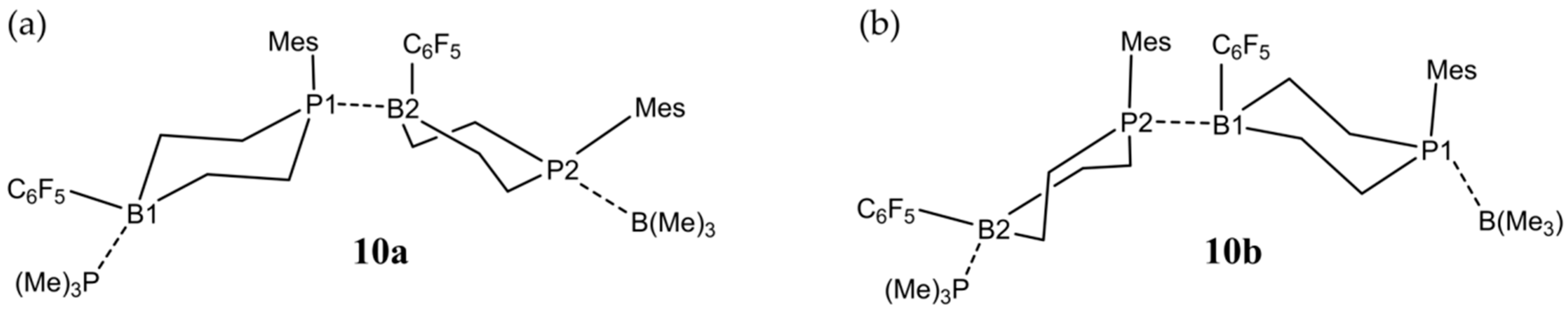
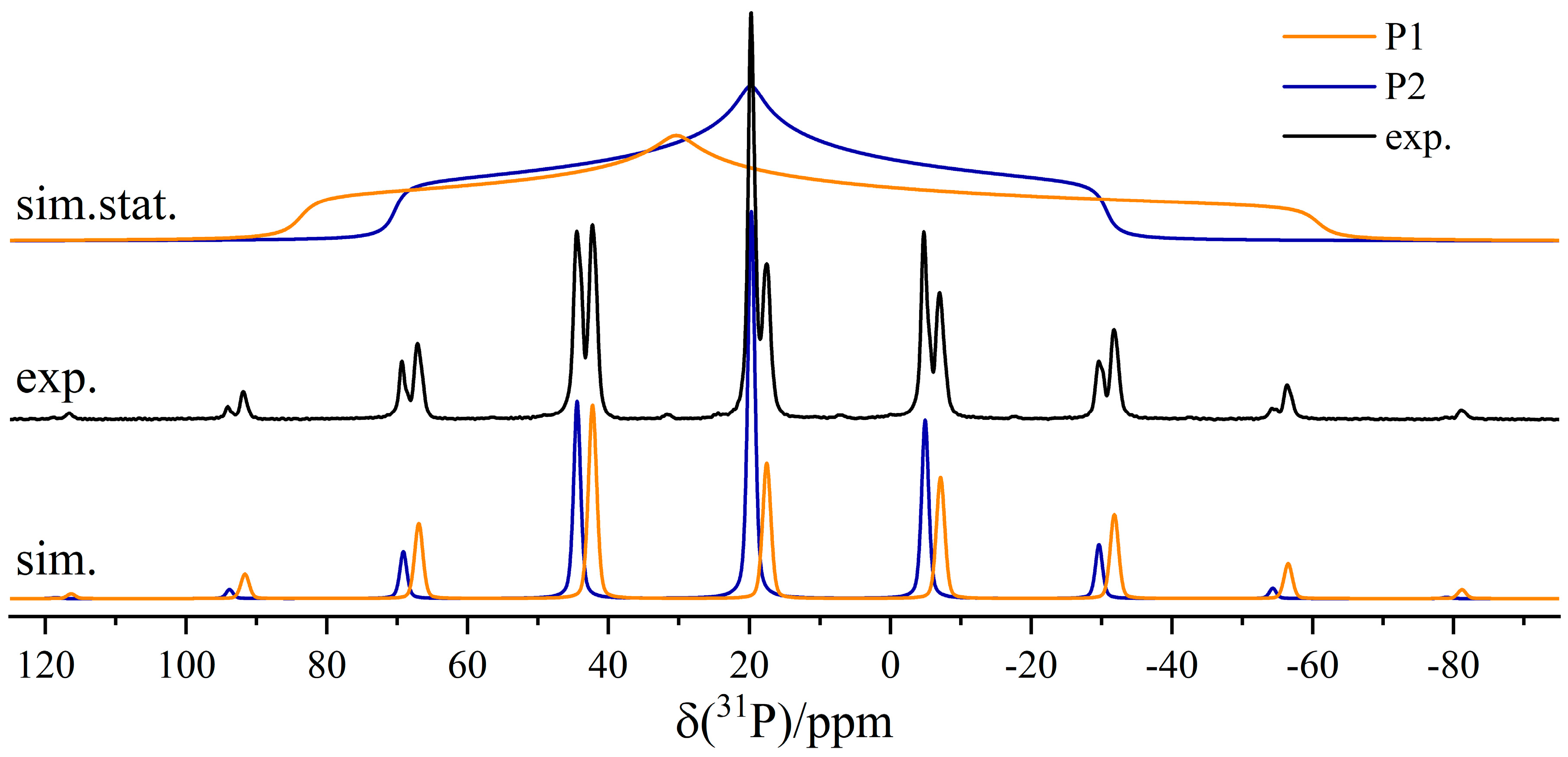
3.2. Dimeric Structures
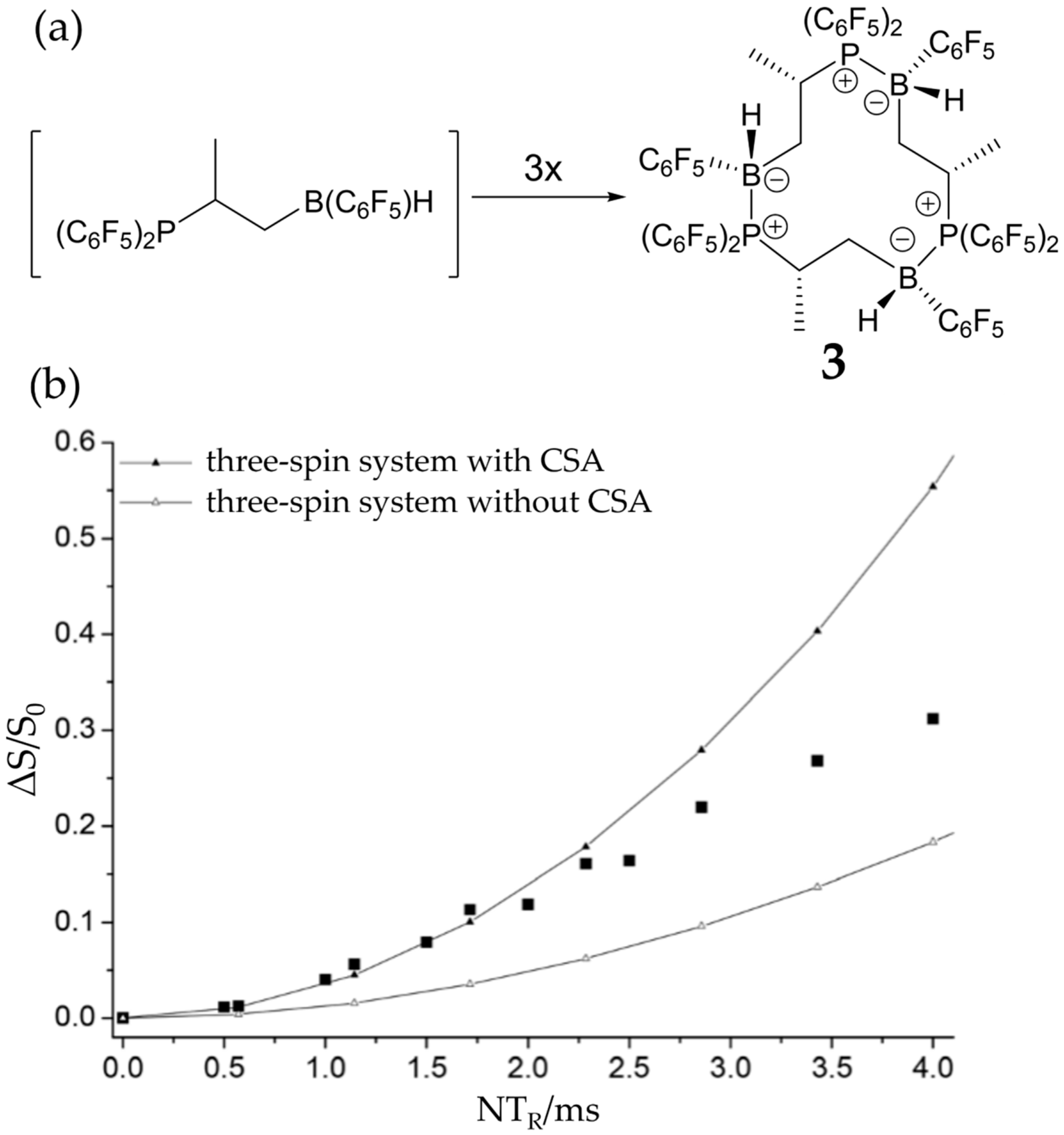
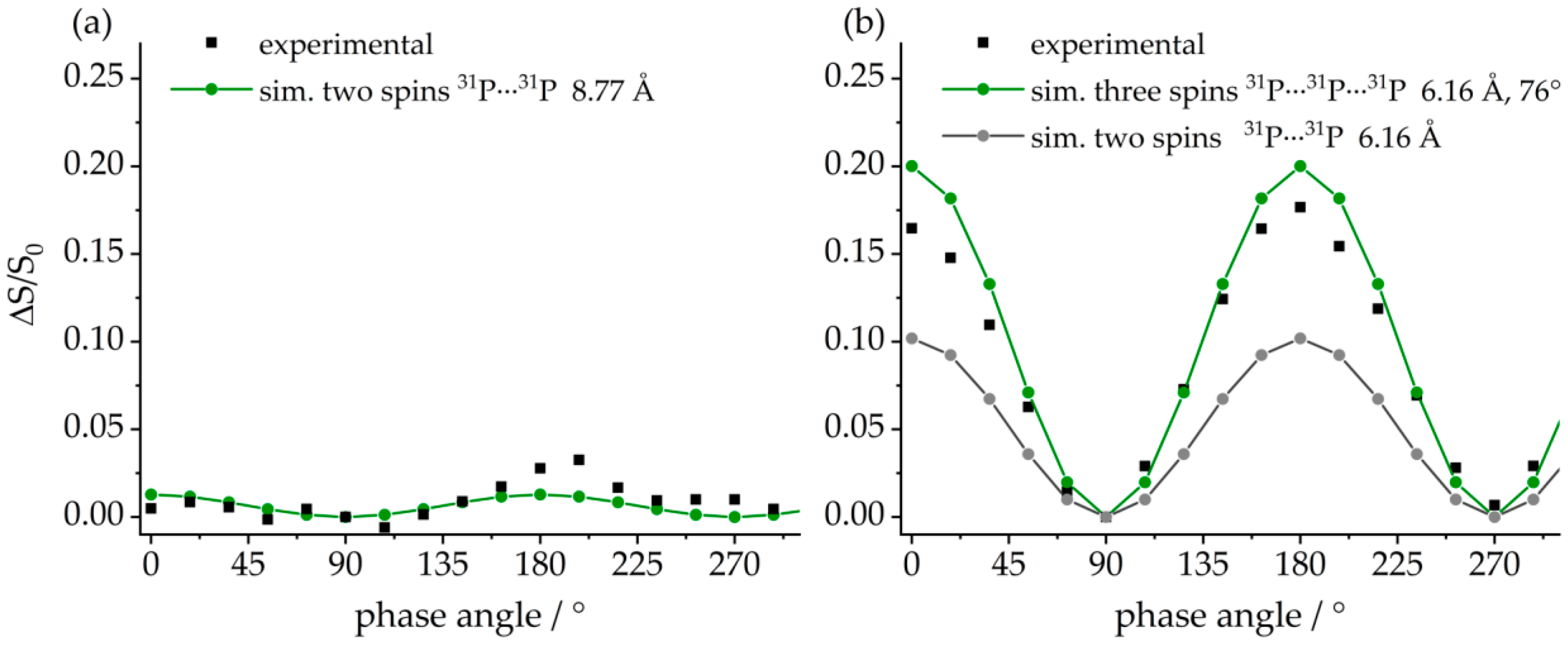
3.3. FLP-Cyclotrimer
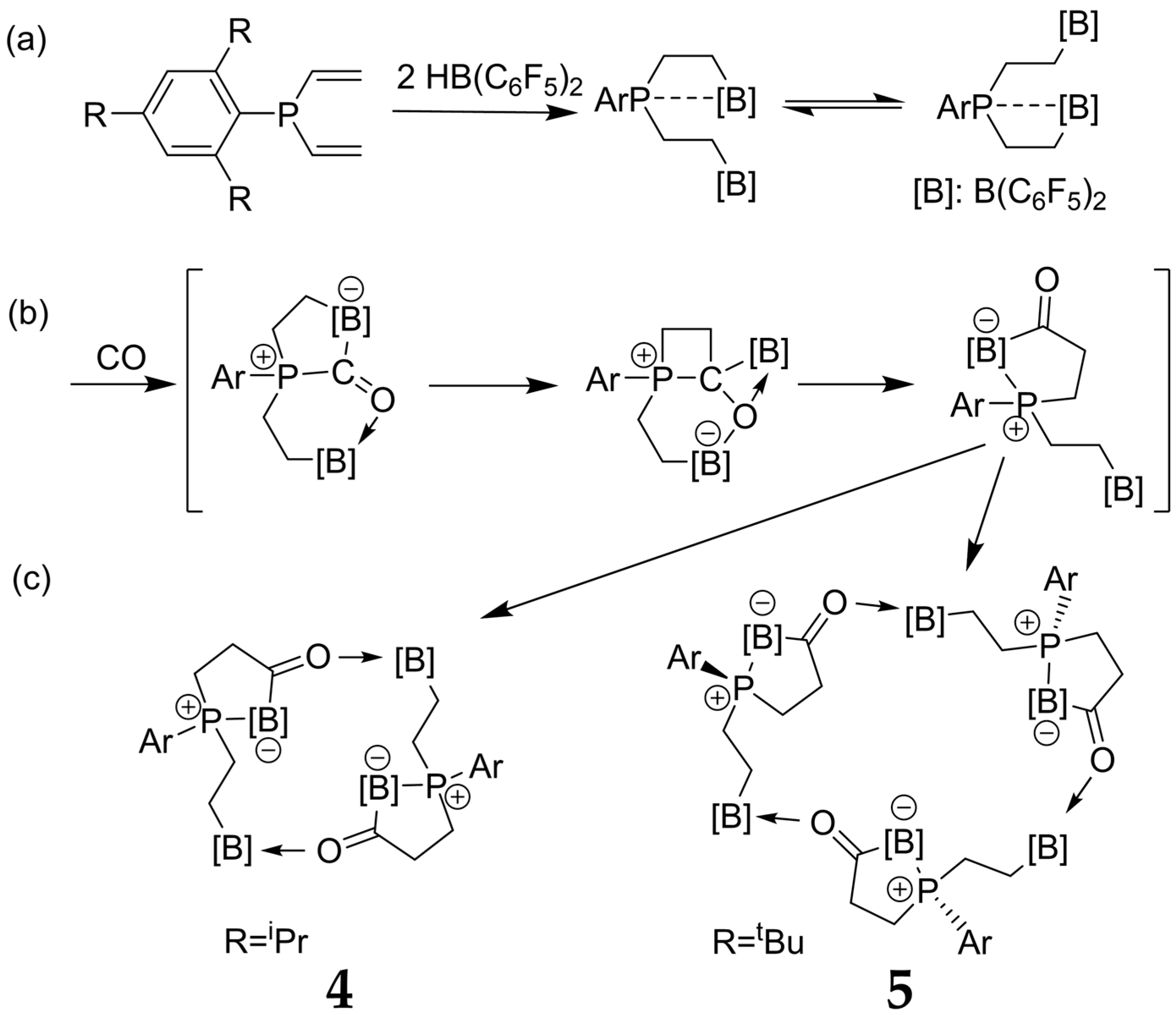
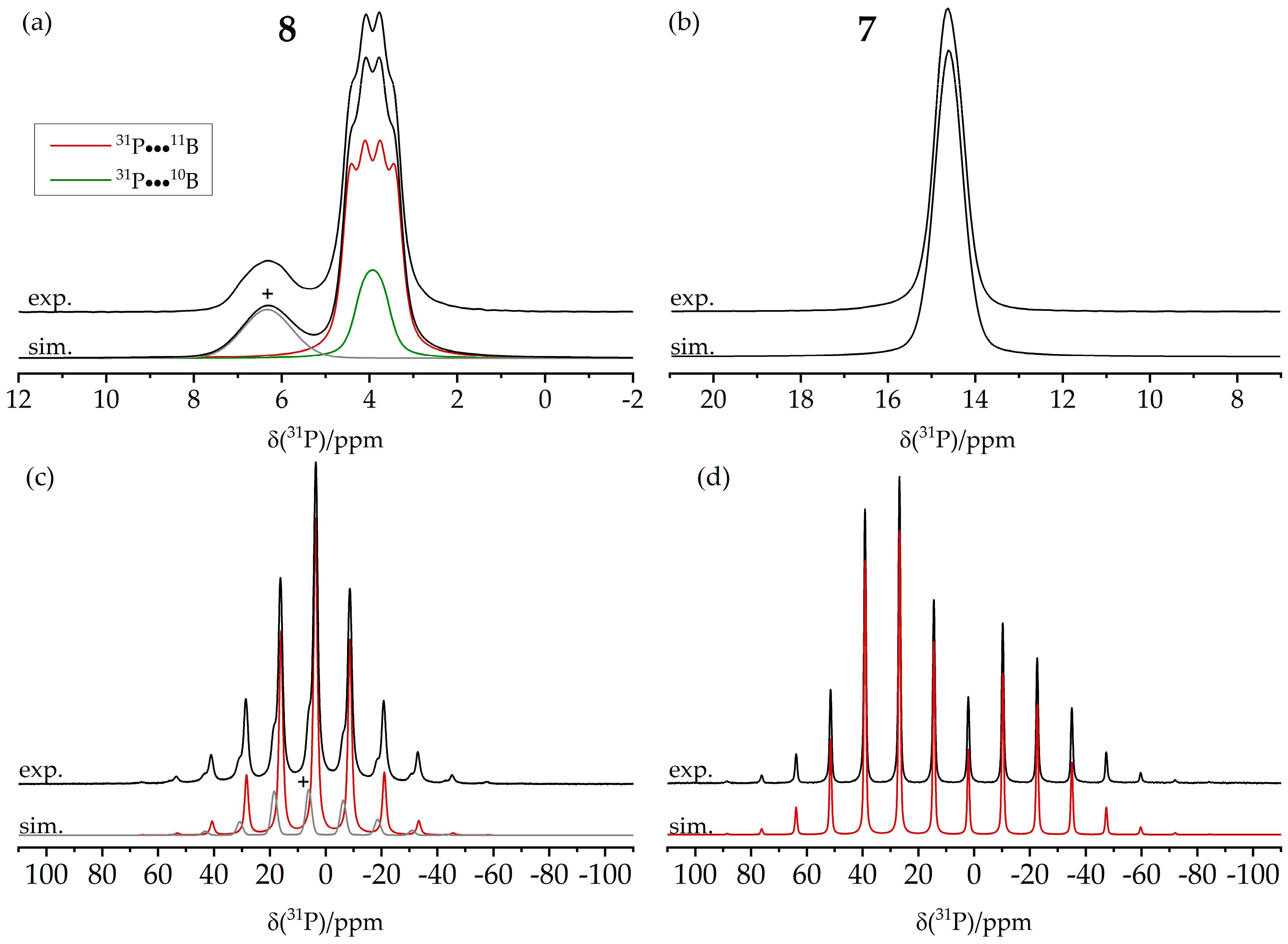
3.4. Dimeric and Trimeric CO Adducts
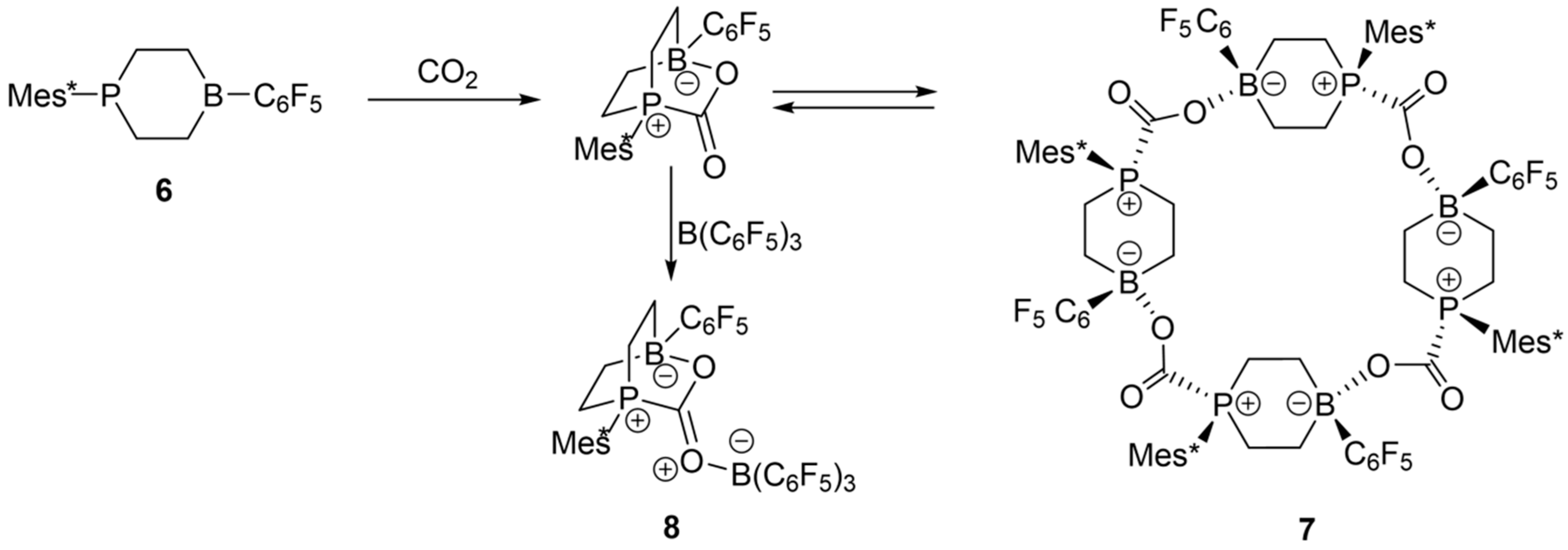
3.5. Tetrameric B/P FLP–CO2 Macrocycles.
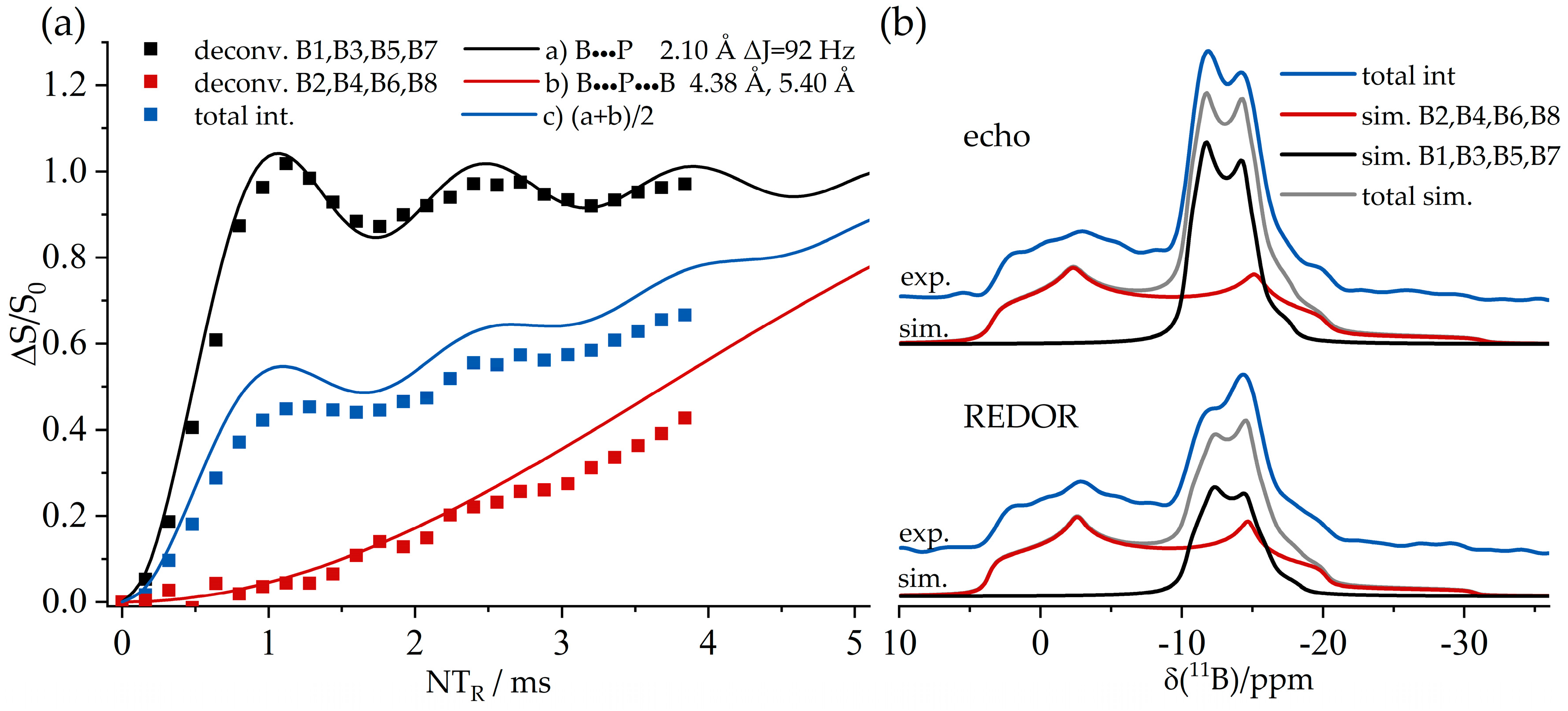
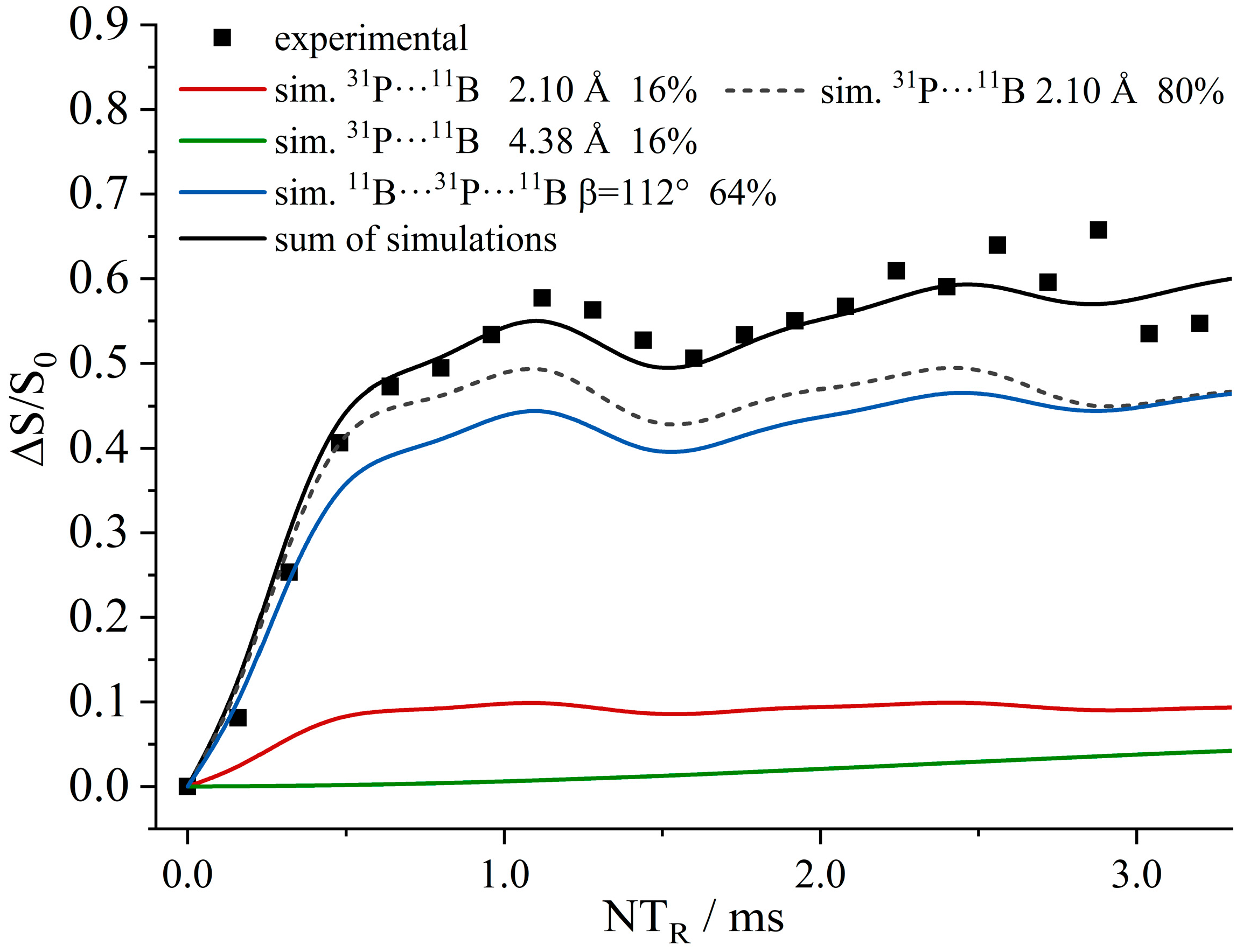
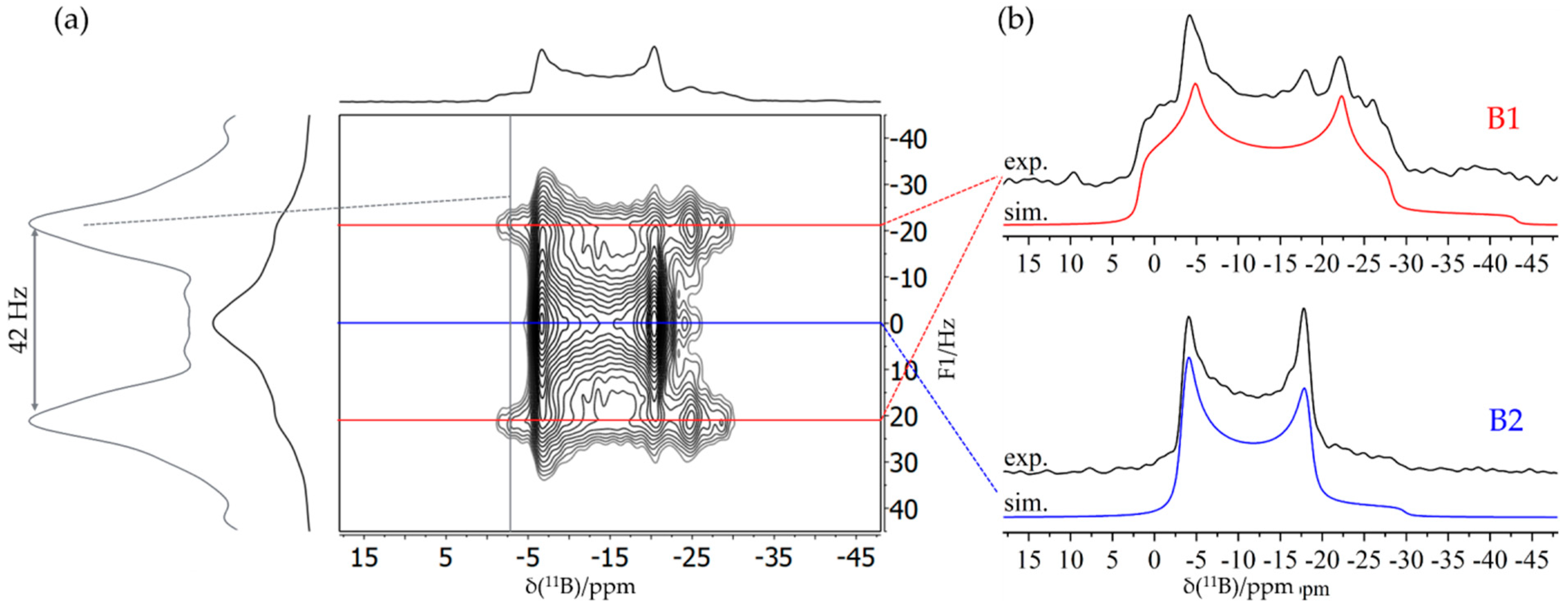
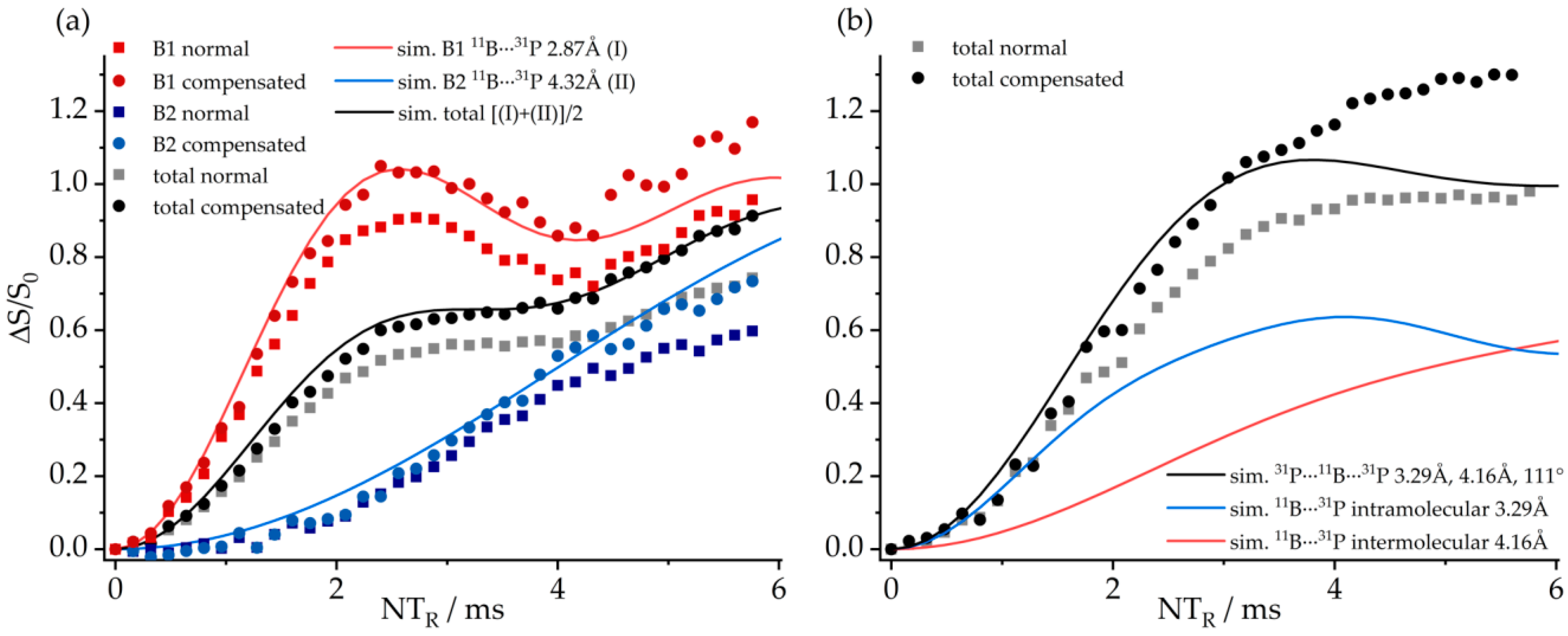
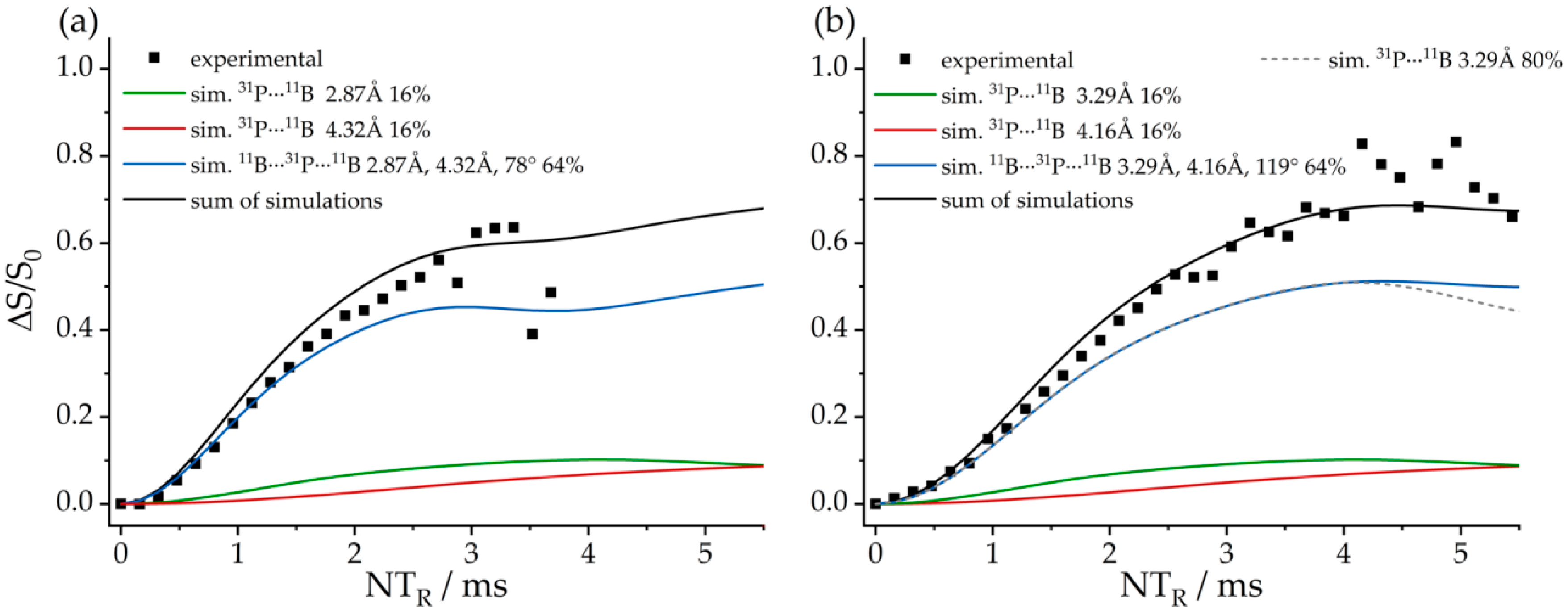
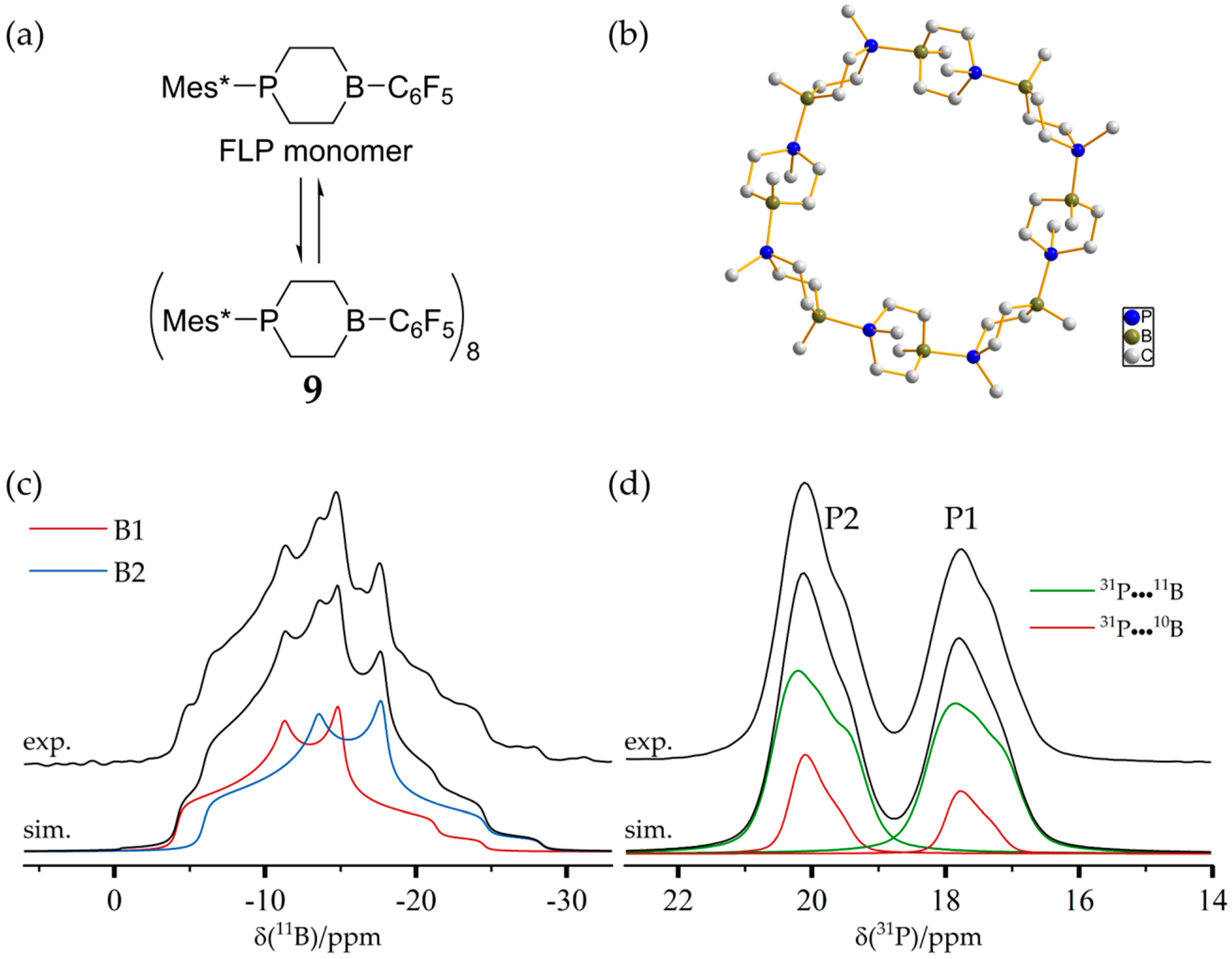
3.6. An FLP-Cyclooctamer
4. Conclusions
5. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wiegand, T.; Eckert, H.; Grimme, S. Frustrated Lewis Pairs I: Uncovering and Understanding; Erker, G., Stephan, D.W., Eds.; Topics in Current Chemistry; Springer-Verlag: Berlin, Germany, 2013; ISBN 978-3-642-36696-3. [Google Scholar]
- Welch, G.C.; Juan, R.R.S.; Masuda, J.D.; Stephan, D.W. Reversible, Metal-Free Hydrogen Activation. Science 2006, 314, 1124–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spies, P.; Schwendemann, S.; Lange, S.; Kehr, G.; Fröhlich, R.; Erker, G. Metal-Free Catalytic Hydrogenation of Enamines, Imines, and Conjugated Phosphinoalkenylboranes. Angew. Chem. Int. Ed. 2008, 47, 7543–7546. [Google Scholar] [CrossRef] [PubMed]
- Spies, P.; Kehr, G.; Bergander, K.; Wibbeling, B.; Fröhlich, R.; Erker, G. Metal-free Dihydrogen Activation Chemistry: Structural and Dynamic Features of Intramolecular P/B pairs. Dalton Trans. 2009, 9, 1534–1541. [Google Scholar] [CrossRef]
- Stephan, D.W. “Frustrated Lewis pair” Hydrogenations. Org. Biomol. Chem. 2012, 10, 5740–5746. [Google Scholar] [CrossRef]
- Erős, G.; Nagy, K.; Mehdi, H.; Pápai, I.; Nagy, P.; Király, P.; Tárkányi, G.; Soós, T. Catalytic Hydrogenation with Frustrated Lewis Pairs: Selectivity Achieved by Size-Exclusion Design of Lewis Acids. Chem. Eur. J. 2012, 18, 574–585. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More. Angew. Chem. Int. Ed. 2010, 49, 46–76. [Google Scholar] [CrossRef]
- Mömming, C.M.; Otten, E.; Kehr, G.; Fröhlich, R.; Grimme, S.; Stephan, D.W.; Erker, G. Reversible Metal-Free Carbon Dioxide Binding by Frustrated Lewis Pairs. Angew. Chem. Int. Ed. 2009, 48, 6643–6646. [Google Scholar] [CrossRef] [Green Version]
- Sajid, M.; Klose, A.; Birkmann, B.; Liang, L.; Schirmer, B.; Wiegand, T.; Eckert, H.J.; Lough, A.; Fröhlich, R.; G. Daniliuc, C.; et al. Reactions of Phosphorus/Boron Frustrated Lewis Pairs with SO2. Chem. Sci. 2013, 4, 213–219. [Google Scholar] [CrossRef]
- Sajid, M.; Stute, A.; Cardenas, A.J.P.; Culotta, B.J.; Hepperle, J.A.M.; Warren, T.H.; Schirmer, B.; Grimme, S.; Studer, A.; Daniliuc, C.G.; et al. N,N-Addition of Frustrated Lewis Pairs to Nitric Oxide: An Easy Entry to a Unique Family of Aminoxyl Radicals. J. Am. Chem. Soc. 2012, 134, 10156–10168. [Google Scholar] [CrossRef]
- Sajid, M.; Elmer, L.-M.; Rosorius, C.; Daniliuc, C.G.; Grimme, S.; Kehr, G.; Erker, G. Facile Carbon Monoxide Reduction at Intramolecular Frustrated Phosphane/Borane Lewis Pair Templates. Angew. Chem. Int. Ed. 2013, 52, 2243–2246. [Google Scholar] [CrossRef]
- Ekkert, O.; Miera, G.G.; Wiegand, T.; Eckert, H.; Schirmer, B.; Petersen, J.L.; Daniliuc, C.G.; Fröhlich, R.; Grimme, S.; Kehr, G.; et al. Remarkable Coordination Behavior of Alkyl Isocyanides Toward Unsaturated Vicinal Frustrated P/B Lewis Pairs. Chem. Sci. 2013, 4, 2657–2664. [Google Scholar] [CrossRef]
- Ménard, G.; Stephan, D.W. H2 Activation and Hydride Transfer to Olefins by Al(C6F5)3-Based Frustrated Lewis Pairs. Angew. Chem. Int. Ed. 2012, 51, 8272–8275. [Google Scholar] [CrossRef] [PubMed]
- Dureen, M.A.; Brown, C.C.; Stephan, D.W. Deprotonation and Addition Reactions of Frustrated Lewis Pairs with Alkynes. Organometallics 2010, 29, 6594–6607. [Google Scholar] [CrossRef]
- Jiang, C.; Blacque, O.; Berke, H. Activation of Terminal Alkynes by Frustrated Lewis Pairs. Organometallics 2010, 29, 125–133. [Google Scholar] [CrossRef]
- Ullrich, M.; Seto, K.S.-H.; Lough, A.J.; Stephan, D.W. 1,4-Addition Reactions of Frustrated Lewis Pairs to 1,3-Dienes. Chem. Commun. 2009, 2335–2337. [Google Scholar] [CrossRef]
- Xu, B.-H.; Yanez, R.A.A.; Nakatsuka, H.; Kitamura, M.; Fröhlich, R.; Kehr, G.; Erker, G. Reaction of Frustrated Lewis Pairs with Ketones and Esters. Chem. Asian J. 2012, 7, 1347–1356. [Google Scholar] [CrossRef]
- Wiegand, T.; Eckert, H.; Grimme, S. Solid-State NMR as a Spectroscopic Tool for Characterizing Phosphane–Borane Frustrated Lewis Pairs. In Frustrated Lewis Pairs I: Uncovering and Understanding; Topics in Current Chemistry; Erker, G., Stephan, D.W., Eds.; Springer: Berlin, Germany, 2013; pp. 291–345. ISBN 978-3-642-36697-0. [Google Scholar]
- Wiegand, T.; Siedow, M.; Eckert, H.; Kehr, G.; Erker, G. Structural Characterization of Frustrated Lewis Pairs and Their Reaction Products Using Modern Solid-State NMR Spectroscopy Techniques. Isr. J. Chem. 2015, 55, 150–178. [Google Scholar] [CrossRef]
- Wiegand, T.; Eckert, H.; Ekkert, O.; Fröhlich, R.; Kehr, G.; Erker, G.; Grimme, S. New Insights into Frustrated Lewis Pairs: Structural Investigations of Intramolecular Phosphane–Borane Adducts by Using Modern Solid-State NMR Techniques and DFT Calculations. J. Am. Chem. Soc. 2012, 134, 4236–4249. [Google Scholar] [CrossRef]
- Elmer, L.-M.; Kehr, G.; Daniliuc, C.G.; Siedow, M.; Eckert, H.; Tesch, M.; Studer, A.; Williams, K.; Warren, T.H.; Erker, G. The Chemistry of a Non-Interacting Vicinal Frustrated Phosphane/Borane Lewis Pair. Chem. Eur. J. 2017, 23, 6056–6068. [Google Scholar] [CrossRef]
- Sajid, M.; Kehr, G.; Wiegand, T.; Eckert, H.; Schwickert, C.; Pöttgen, R.; Cardenas, A.J.P.; Warren, T.H.; Fröhlich, R.; Daniliuc, C.G.; et al. Noninteracting, Vicinal Frustrated P/B-Lewis Pair at the Norbornane Framework: Synthesis, Characterization, and Reactions. J. Am. Chem. Soc. 2013, 135, 8882–8895. [Google Scholar] [CrossRef]
- Liedtke, R.; Scheidt, F.; Ren, J.; Schirmer, B.; Cardenas, A.J.P.; Daniliuc, C.G.; Eckert, H.; Warren, T.H.; Grimme, S.; Kehr, G.; et al. Frustrated Lewis Pair Modification by 1,1-Carboboration: Disclosure of a Phosphine Oxide Triggered Nitrogen Monoxide Addition to an Intramolecular P/B Frustrated Lewis Pair. J. Am. Chem. Soc. 2014, 136, 9014–9027. [Google Scholar] [CrossRef] [PubMed]
- Ge, F.; Türkyilmaz, F.; Daniliuc, C.G.; Siedow, M.; Eckert, H.; Kehr, G.; Erker, G. A 1,1-Carboboration Route to Bora-Nazarov Systems. Chem. Asian J. 2015, 10, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Wang, L.; Wang, T.; Daniliuc, C.G.; Brinkkötter, M.; Eckert, H.; Kehr, G.; Erker, G. Formation and Reactions of Active Five-membered Phosphane/Borane Frustrated Lewis Pair Ring Systems. Dalton Trans. 2018, 47, 4449–4454. [Google Scholar] [CrossRef] [PubMed]
- Apperley, D.C.; Harris, R.K.; Hodgkinson, P. Solid-State NMR: Basic Principles and Practice; Momentum Press: New York, NY, USA, 2012; ISBN 978-1-60650-352-2. [Google Scholar]
- Abragam, P.A. Principles of Nuclear Magnetism; Oxford University Press: New York, NY, USA, 1961; ISBN 978-0-19-852014-6. [Google Scholar]
- Keeler, J. Understanding NMR Spectroscopy; John Wiley & Sons: Chichester, UK, 2011; ISBN 978-1-119-96493-3. [Google Scholar]
- Duer, M.J. Solid State NMR Spectroscopy: Principles and Applications; Wiley-Blackwell: Malden, MA, USA, 2002; ISBN 978-0-632-05351-3. [Google Scholar]
- Slichter, C.P. Principles of Magnetic Resonance, 3rd ed.; Springer Series in Solid-State Sciences; Springer-Verlag: Berlin, Germany, 1990; ISBN 978-3-540-50157-2. [Google Scholar]
- Pell, A.J.; Pintacuda, G.; Grey, C.P. Paramagnetic NMR in Solution and the Solid State. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 1, 111. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral De Menezes, S.M.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further Conventions for NMR Shielding and Chemical Shifts IUPAC Recommendations 2008. Solid State Nucl. Magn. Reson. 2008, 33, 41–56. [Google Scholar] [CrossRef]
- Andrew, E.R.; Bradbury, A.; Eades, R.G. Nuclear Magnetic Resonance Spectra from a Crystal Rotated at High Speed. Nature 1958, 182, 1659. [Google Scholar] [CrossRef]
- Kaupp, M.; Bühl, M.; Malkin, V.G. Calculation of NMR and EPR Parameters: Theory and Applications; John Wiley & Sons: Weinheim, Germany, 2006; ISBN 978-3-527-60496-8. [Google Scholar]
- Herzfeld, J.; Berger, A.E. Sideband Intensities in NMR Spectra of Samples Spinning at the Magic Angle. J. Chem. Phys. 1980, 73, 6021–6030. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Cheng, Y.-S.; Ho, C.-I.; Guo, Z.-H.; Huang, S.-J.; Org, M.-L.; Oss, A.; Samoson, A.; Chan, J.C.C. Preparation of Fibril Nuclei of Beta-Amyloid Peptides in Reverse Micelles. Chem. Commun. 2018, 54, 10459–10462. [Google Scholar] [CrossRef]
- Penzel, S.; Oss, A.; Org, M.-L.; Samoson, A.; Böckmann, A.; Ernst, M.; Meier, B.H. Spinning Faster: Protein NMR at MAS Frequencies up to 126 kHz. J. Biomol. NMR 2019, 73, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Man, P.P. Quadrupolar Interactions. In Encyclopedia of Nuclear Magnetic Resonance; John Wiley & Sons: Chichester, UK, 2007; pp. 3838–3848. ISBN 978-0-471-49082-1. [Google Scholar]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Calvé, S.L.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling One- and Ttwo-Dimensional Solid-State NMR Spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Bak, M.; Rasmussen, J.T.; Nielsen, N.C. SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy. J. Magn. Reson. 2011, 213, 366–400. [Google Scholar] [CrossRef] [PubMed]
- Perras, F.A.; Widdifield, C.M.; Bryce, D.L. QUEST—QUadrupolar Exact SofTware: A fast Graphical Program for the Exact Simulation of NMR and NQR Spectra for Quadrupolar Nuclei. Solid State Nucl. Magn. Reson. 2012, 45–46, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009; Available online: https://gaussian.com (accessed on 18 March 2020).
- TURBOMOLE v6.5 2013, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007. Available online: http://www.turbomole.com (accessed on 18 March 2020).
- Blaha, P.; Schwarz, K.; Madsen, G.K.H.; Kvasnicka, D.; Luitz, J. WIEN2k, An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties, 2nd ed.; Vienna University of Technology: Vienna, Austria, 2001; ISBN 3-9501031-1. [Google Scholar]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-Principles Simulation: Ideas, Illustrations and the CASTEP Code. J. Phys. Cond. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Medek, A.; Harwood, J.S.; Frydman, L. Multiple-Quantum Magic-Angle Spinning NMR: A New Method for the Study of Quadrupolar Nuclei in Solids. J. Am. Chem. Soc. 1995, 117, 12779–12787. [Google Scholar] [CrossRef]
- Amoureux, J.-P.; Fernandez, C.; Frydman, L. Optimized Multiple-Quantum Magic-Angle Spinning NMR Experiments on Half-Integer Quadrupoles. Chem. Phys. Lett. 1996, 259, 347–355. [Google Scholar] [CrossRef]
- Amoureux, J.-P.; Fernandez, C.; Steuernagel, S. Z-Filtering in MQMAS NMR. J. Magn. Reson. A 1996, 123, 116–118. [Google Scholar] [CrossRef]
- Vaara, J.; Jokisaari, J.; Wasylishen, R.E.; Bryce, D.L. Spin-Spin Coupling Tensors as Determined by Experiment and Computational Chemistry. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 41, 233–304. [Google Scholar] [CrossRef]
- Bryce, D.L.; Wasylishen, R.E. Indirect Nuclear Spin-Spin Coupling Tensors in Diatomic Molecules: A Comparison of Results Obtained by Experiment and First Principles Calculations. J. Am. Chem. Soc. 2000, 122, 3197–3205. [Google Scholar] [CrossRef]
- Olivieri, A.C. Quadrupolar Effects in the CPMAS NMR Spectra of Spin-1/2 Nuclei. J. Magn. Reson. 1989, 81, 201–205. [Google Scholar]
- Harris, R.K.; Olivieri, A.C. Quadrupolar Effects Transferred to Spin-1/2 Magic-Angle Spinning Spectra of Solids. Prog. Nucl. Magn. Reson. Spectrosc. 1992, 24, 435–456. [Google Scholar] [CrossRef]
- Hierso, J.-C. Indirect Nonbonded Nuclear Spin–Spin Coupling: A Guide for the Recognition and Understanding of “Through-Space” NMR J Constants in Small Organic, Organometallic, and Coordination Compounds. Chem. Rev. 2014, 114, 4838–4867. [Google Scholar] [CrossRef]
- Wiegand, T.; Eckert, H.; Ren, J.; Brunklaus, G.; Fröhlich, R.; Daniliuc, C.G.; Lübbe, G.; Bussmann, K.; Kehr, G.; Erker, G.; et al. Indirect “No-Bond” 31P···31P Spin–Spin Couplings in P,P-[3]Ferrocenophanes: Insights from Solid-State NMR Spectroscopy and DFT Calculations. J. Phys. Chem. A 2014, 118, 2316–2331. [Google Scholar] [CrossRef]
- Zilm, K.W.; Grant, D.M. High-Resolution NMR Spectra with J-Couplings in Solids. J. Magn. Reson. 1982, 48, 524–526. [Google Scholar] [CrossRef]
- Fyfe, C.A.; Wong-Moon, K.C.; Huang, Y.; Grondey, H. INEPT Experiments in Solid-State NMR. J. Am. Chem. Soc. 1995, 117, 10397–10398. [Google Scholar] [CrossRef]
- Franke, D.; Hudalla, C.; Eckert, H. Heteronuclear X-Y Double Quantum MAS NMR in Crystalline Inorganic Solids. Applications for Indirect Detection and Spectral Editing of Rare-Spin Resonances. Solid State Nucl. Magn. Reson. 1992, 1, 33–40. [Google Scholar] [CrossRef]
- Cadars, S.; Sein, J.; Duma, L.; Lesage, A.; Pham, T.N.; Baltisberger, J.H.; Brown, S.P.; Emsley, L. The Refocused INADEQUATE MAS NMR Experiment in Multiple Spin-Systems: Interpreting Observed Correlation Peaks and Optimising Lineshapes. J. Magn. Reson. 2007, 188, 24–34. [Google Scholar] [CrossRef]
- Gullion, T.; Schaefer, J. Rotational-Echo Double-Resonance NMR. J. Magn. Reson. 1989, 81, 196–200. [Google Scholar] [CrossRef]
- Gullion, T.; Vega, A.J. Measuring Heteronuclear Dipolar Couplings for I = 1/2, S > 1/2 Spin Pairs by REDOR and REAPDOR NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2005, 47, 123–136. [Google Scholar] [CrossRef]
- Gullion, T. Rotational-Echo, Double-Resonance NMR. In Modern Magnetic Resonance; Webb, G.A., Ed.; Springer Science & Business Media: Dordrecht, The Netherlands, 2007; ISBN 978-1-4020-3910-2. [Google Scholar]
- Chan, J.C.C.; Eckert, H. Dipolar Coupling Information in Multispin Systems: Application of a Compensated REDOR NMR Approach to Inorganic Phosphates. J. Magn. Reson. 2000, 147, 170–178. [Google Scholar] [CrossRef]
- Gullion, T. Measurement of Dipolar Interactions between Spin-1/2 and Quadrupolar Nuclei by Rotational-Echo, Adiabatic-Passage, Double-Resonance NMR. Chem. Phys. Lett. 1995, 246, 325–330. [Google Scholar] [CrossRef]
- Gullion, T. Detecting 13C–17O Dipolar Interactions by Rotational-Echo, Adiabatic-Passage, Double-Resonance NMR. J. Magn. Reson. 1995, 117, 326–329. [Google Scholar] [CrossRef]
- Chopin, L.; Vega, S.; Gullion, T. A MAS NMR Method for Measuring 13C−17O Distances. J. Am. Chem. Soc. 1998, 120, 4406–4409. [Google Scholar] [CrossRef]
- Nimerovsky, E.; Goldbourt, A. Distance Measurements between Boron and Carbon at Natural Abundance Using Magic Angle Spinning REAPDOR NMR and a Universal Curve. Phys. Chem. Chem. Phys. 2012, 14, 13437–13443. [Google Scholar] [CrossRef]
- Feike, M.; Graf, R.; Schnell, I.; Jäger, C.; Spiess, H.W. Structure of Crystalline Phosphates from 31P Double-Quantum NMR Spectroscopy. J. Am. Chem. Soc. 1996, 118, 9631–9634. [Google Scholar] [CrossRef]
- Saalwächter, K.; Lange, F.; Matyjaszewski, K.; Huang, C.-F.; Graf, R. BaBa-xy16: Robust and Broadband Homonuclear DQ Recoupling for Applications in Rigid and Soft Solids up to the Highest MAS Frequencies. J. Magn. Reson. 2011, 212, 204–215. [Google Scholar] [CrossRef]
- Geen, H.; Gottwald, J.; Graf, R.; Schnell, I.; Spiess, H.W.; Titman, J.J. Elucidation of Dipolar Coupling Networks under Magic-Angle Spinning. J. Magn. Reson. 1997, 125, 224–227. [Google Scholar] [CrossRef]
- Hohwy, M.; Jakobsen, H.J.; Edén, M.; Levitt, M.H.; Nielsen, N.C. Broadband Dipolar Recoupling in the Nuclear Magnetic Resonance of Rotating Solids: A compensated C7 Pulse Sequence. J. Chem. Phys. 1998, 108, 2686–2694. [Google Scholar] [CrossRef]
- Lee, Y.K.; Kurur, N.D.; Helmle, M.; Johannessen, O.G.; Nielsen, N.C.; Levitt, M.H. Efficient Dipolar Recoupling in the NMR of Rotating Solids. A Sevenfold Symmetric Radiofrequency Pulse Sequence. Chem. Phys. Lett. 1995, 242, 304–309. [Google Scholar] [CrossRef]
- Ren, J.; Eckert, H. A Homonuclear Rotational Echo Double-Resonance Method for Measuring Site-Resolved Distance Distributions in I=1/2 Spin Pairs, Clusters, and Multispin Systems. Angew. Chem. Int. Ed. 2012, 51, 12888–12891. [Google Scholar] [CrossRef]
- Ren, J.; Eckert, H. DQ-DRENAR: A New NMR Technique to Measure Site-Resolved Magnetic Dipole-Dipole Interactions in Multispin-1/2 Systems: Theory and Validation on Crystalline Phosphates. J. Chem. Phys. 2013, 138, 164201. [Google Scholar] [CrossRef]
- Ren, J.; Eckert, H. Measurement of Homonuclear Magnetic Dipole–Dipole Interactions in Multiple Spin-1/2 Systems Using Constant-Time DQ-DRENAR NMR. J. Magn. Reson. 2015, 260, 46–53. [Google Scholar] [CrossRef]
- Ren, J.; Eckert, H. DQ-DRENAR with Back-to-Back (BABA) Excitation: Measuring Homonuclear Dipole-Dipole Interactions in Multiple Spin 1/2 Systems. Solid State Nucl. Magn. Reson. 2015, 71, 11–18. [Google Scholar] [CrossRef]
- Pines, A.; Gibby, M.G.; Waugh, J.S. Proton-Enhanced Nuclear Induction Spectroscopy. A Method for High Resolution NMR of Dilute Spins in Solids. J. Chem. Phys. 1972, 56, 1776–1777. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, S.R.; Hahn, E.L. Nuclear Double Resonance in the Rotating Frame. Phys. Rev. 1962, 128, 2042–2053. [Google Scholar] [CrossRef]
- Wiegand, T. Structure, Reactivity, and Cooperativity of Functional Organophosphor Compounds: Development and Application of Modern Solid-State NMR and DFT Strategies. PhD Thesis, University of Münster, Münster, Germany, 7 January 2013. [Google Scholar]
- Siedow, M. Charakterisierung Frustrierter Lewis-Paar-Addukte mittels Kombination moderner Festkörper-NMR-Methoden mit quantenchemischen Berechnungen. PhD Thesis, University of Münster, Münster, Germany, 18 April 2018. [Google Scholar]
- Knitsch, R. Structural Characterization of Novel Organoboron Compounds by Application of Magnetic Resonance Spectroscopy and DFT Calculations. PhD Thesis, University of Münster, Münster, Germany, 24 April 2019. [Google Scholar]
- Erdmann, M.; Wiegand, T.; Blumenberg, J.; Eckert, H.; Ren, J.; Daniliuc, C.G.; Kehr, G.; Erker, G. Formation, Structural Characterization, and Reactions of a Unique Cyclotrimeric Vicinal Lewis Pair Containing (C6F5)2P-Lewis Base and (C6F5)BH-Lewis Acid Components. Dalton Trans. 2014, 43, 15159–15169. [Google Scholar] [CrossRef]
- Sajid, M.; Lawzer, A.; Dong, W.; Rosorius, C.; Sander, W.; Schirmer, B.; Grimme, S.; Daniliuc, C.G.; Kehr, G.; Erker, G. Carbonylation Reactions of Intramolecular Vicinal Frustrated Phosphane/Borane Lewis Pairs. J. Am. Chem. Soc. 2013, 135, 18567–18574. [Google Scholar] [CrossRef]
- Wang, L.; Dong, S.; Daniliuc, C.G.; Liu, L.; Grimme, S.; Knitsch, R.; Eckert, H.; Hansen, M.R.; Kehr, G.; Erker, G. Formation of Macrocyclic Ring Systems by Carbonylation of Trifunctional P/B/B Frustrated Lewis Pairs. Chem. Sci. 2018, 9, 1544–1550. [Google Scholar] [CrossRef] [Green Version]
- Chandran, C.V.; Madhu, P.K.; Kurur, N.D.; Bräuniger, T. Swept-Frequency Two-Pulse Phase Modulation (SWf-TPPM) Sequences with Linear Sweep Profile for Heteronuclear Decoupling in Solid-State NMR. Magn. Reson. Chem. 2008, 46, 943–947. [Google Scholar] [CrossRef]
- Jie, X.; Sun, Q.; Daniliuc, C.G.; Knitsch, R.; Hansen, M.R.; Eckert, H.; Kehr, G.; Erker, G. Cycloaddition Reactions of an Active Cyclic Phosphane/Borane Pair with Alkenes, Alkynes, and Carbon Dioxide. Chem. Eur. J. 2020, 26, 1269–1273. [Google Scholar] [CrossRef]
- Jie, X.; Daniliuc, C.G.; Knitsch, R.; Hansen, M.R.; Eckert, H.; Ehlert, S.; Grimme, S.; Kehr, G.; Erker, G. Aggregation Behavior of a Six-Membered Cyclic Frustrated Phosphane/Borane Lewis Pair: Formation of a Supramolecular Cyclooctameric Macrocyclic Ring System. Angew. Chem. Int. Ed. 2019, 58, 882–886. [Google Scholar] [CrossRef]
- Xing, J.Y.; Buffet, J.C.; Rees, N.H.; Norby, P.; O’Hare, D. Hydrogen Cleavage by Solid-Phase Frustrated Lewis Pairs. Chem. Commun. 2016, 52, 10478–10481. [Google Scholar] [CrossRef]
- Peersen, O.B.; Wu, X.L.; Kustanovich, I.; Smith, S.O. Variable-Amplitude Cross-Polarization MAS NMR. J. Magn. Reson. A 1993, 104, 334–339. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta--Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical Hybrid Density Functional with Perturbative Second-Order Correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Feller, D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Baerends, E.J.; Ziegler, T.; Atkins, A.J.; Autschbach, J.; Bashford, D.; Baseggio, O.; Berces, A.; Bickelhaupt, F.M.; Bo, C.; Boerritger, P.M.; et al. ADF 2017, Theoretical Chemistry, Vrije Universiteit, Amsterdam, documentation. Available online: http://www.scm.com (accessed on 18 March 2020).
- Autschbach, J.; Ziegler, T. Nuclear Spin-Spin Coupling Constants from Regular Approximate Relativistic Density Functional Calculations. I. Formalism and Scalar Relativistic Results for Heavy Metal Compounds. J. Chem. Phys. 2000, 113, 936–947. [Google Scholar] [CrossRef]
- Autschbach, J.; Ziegler, T. Nuclear Spin-Spin Coupling Constants from Regular Approximate Relativistic Density Functional Calculations. II. Spin-Orbit Coupling Effects and Anisotropies. J. Chem. Phys. 2000, 113, 9410–9418. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Towards Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Bryce, D.L.; Autschbach, J. Relativistic Hybrid Density Functional Calculations of Indirect Nuclear Spin-Spin Coupling Tensors–Comparison with Experiment for Diatomic Alkali Metal Halides. Can. J. Chem. 2009, 87, 927–941. [Google Scholar] [CrossRef] [Green Version]






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
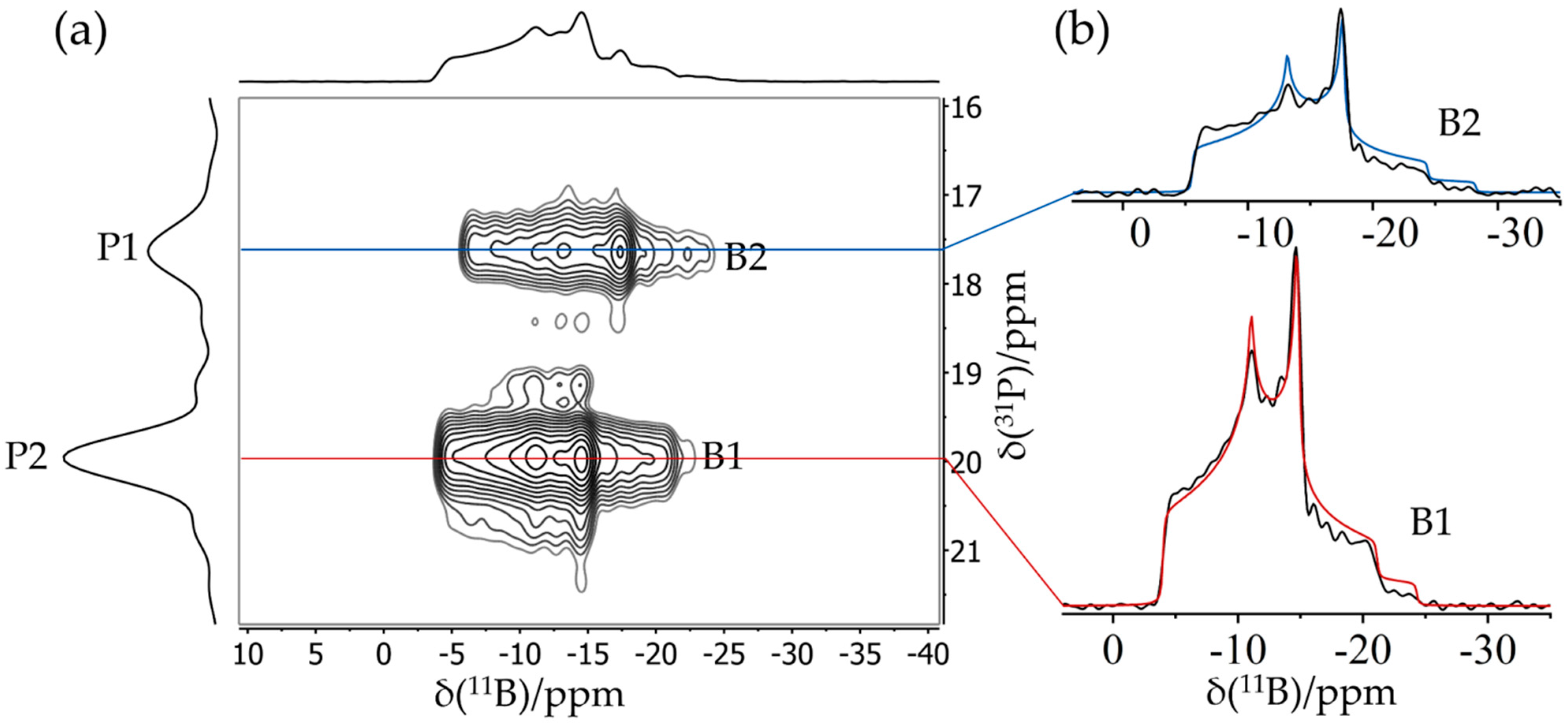{kind=link}
{kind=link}
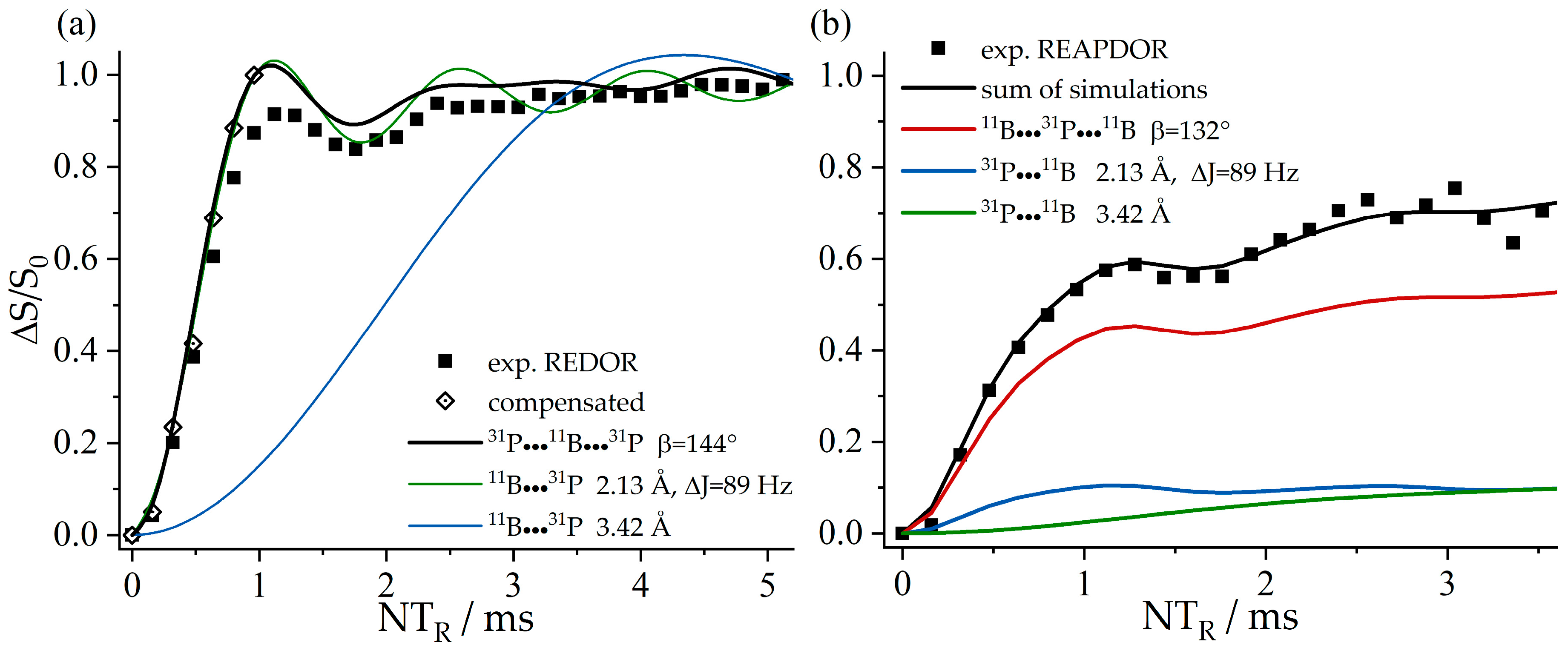{kind=link}
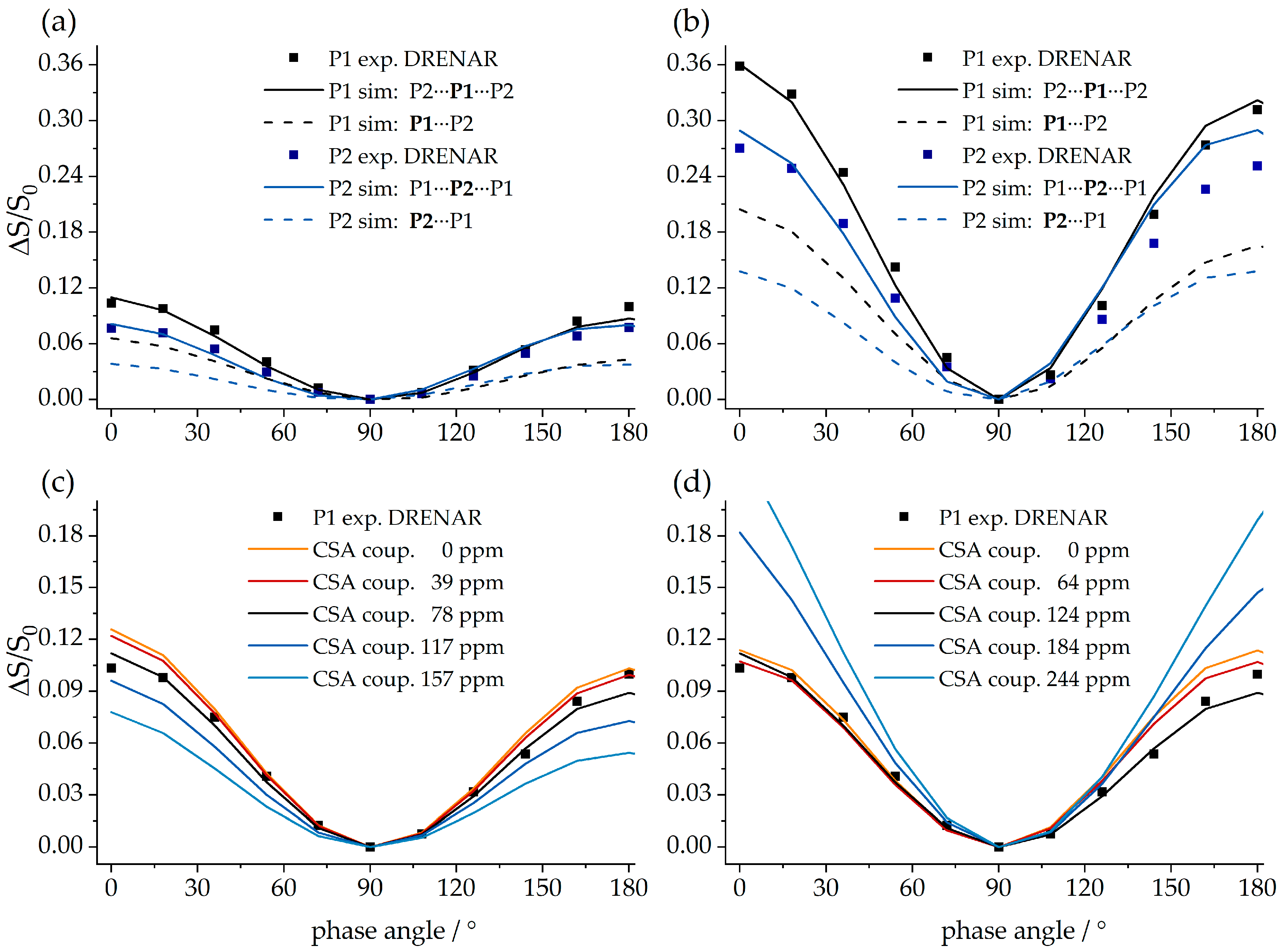{kind=link}
| Comp. | 11B Parameters | 31P Parameters | J-Coupling | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Species | Coord. 31P species | ησ | Calc. ΔJ (Hz) | |||||||
| 1 | B | −4.6 | 1.21 | 0.63 | P | 48.9 | 93.8 | 0.83 | 49 | |
| −4.2 | 1.34 | 0.62 | 57.0 | 120.1 | 0.74 | 41/22 | ||||
| 2 | B | −2.1 | 0.84 | 0.48 | P | 11.7 | 49.5 | 0.43 | ||
| −4.6 | 0.94 | 0.42 | 5.8 | 55.4 | 0.40 | 13/17 | ||||
| 3 | B | −14.6 | 1.69 | 0.35 | P | 17.4 | 99 | 0.57 | ||
| −17.6 | 1.67 | 0.36 | 26.4 | 61.8 | 92 | |||||
| 4 | B1 | −9.5 | 1.10 | 0.20 | P1 | 21.8 | 66 | |||
| −11.3 | 1.11 | 0.25 | 26.6 | 62 | ||||||
| B2, B4 | 8.2 | 2.30 | 0.30 | |||||||
| 6.7/5.8 | 2.32/2.33 | 0.30 | ||||||||
| B3 | −9.3 | 1.00 | 0.20 | P2 | 20.8 | 70 | ||||
| −11.3 | 1.03 | 0.26 | 22.3 | 70 | ||||||
| 5 | B1 | −9.5 | 1.08 | 0.30 | P1 | 14.8 | 80 | |||
| −10.9 | 1.17 | 0.11 | 18.0 | 69 | 92 | |||||
| B2 | 7.3 | 2.38 | 0.25 | |||||||
| 7.5 | 2.49 | 0.26 | ||||||||
| B3 | −8.3 | 1.05 | 0.30 | P2 | 13.2 | 82 | ||||
| −12.0 | 1.04 | 0.17 | 14.1 | 78 | 92 | |||||
| B4 | 6.5 | 2.37 | 0.28 | |||||||
| 5.92 | 2.27 | 0.33 | ||||||||
| B5 | −8.9 | 1.10 | 0.30 | P3 | 11.7 | 78 | ||||
| −13.0 | 1.13 | 0.06 | 12.7 | 75 | 92 | |||||
| B6 | 6.9 | 2.36 | 0.28 | |||||||
| 6.13 | 2.24 | 0.33 | ||||||||
| B7 | −8.9 | 1.09 | 0.20 | P4 | 10.1 | 70 | ||||
| B8 | 7.5 | 2.21 | 0.15 | |||||||
| 7 | B1A | 1.3 | 2.17 | 0.50 | P | 14.6 | 90 | 0.42 | ||
| 1.0 | 2.30 | 0.52 | ||||||||
| B1B | 1.2 | 2.04 | 0.47 | 15.5 | 101 | 0.41 | 13 | |||
| −1.2 | 2.18 | 0.52 | ||||||||
| 8 | B1 | 5.8 | 2.69 | 0.24 | ||||||
| 3.8 | 2.78 | 0.17 | ||||||||
| B2 | 0.7 | 2.13 | 0.05 | P | 3.9 | 43 | 0.95 | 42 | ||
| −0.8 | 2.23 | 0.10 | −2.8 | 56 | 0.38 | 41 | ||||
| 9 | B1 | −3.7 | 1.70 | 0.64 | P2 | 20.0 | 78.4 | 0.96 | 38 | |
| −5.0 | 1.79 | 0.57 | 24.5 | 70.0 | 0.86 | 33 | 89 | |||
| B2 | −5.4 | 1.79 | 0.62 | P1 | 17.7 | 123.8 | 0.68 | 40 | ||
| −4.0 | 1.90 | 0.72 | 24.7 | 128.0 | 0.74 | 28 | 89 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knitsch, R.; Brinkkötter, M.; Wiegand, T.; Kehr, G.; Erker, G.; Hansen, M.R.; Eckert, H. Solid-State NMR Techniques for the Structural Characterization of Cyclic Aggregates Based on Borane–Phosphane Frustrated Lewis Pairs. Molecules 2020, 25, 1400. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25061400
Knitsch R, Brinkkötter M, Wiegand T, Kehr G, Erker G, Hansen MR, Eckert H. Solid-State NMR Techniques for the Structural Characterization of Cyclic Aggregates Based on Borane–Phosphane Frustrated Lewis Pairs. Molecules. 2020; 25(6):1400. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25061400
Chicago/Turabian StyleKnitsch, Robert, Melanie Brinkkötter, Thomas Wiegand, Gerald Kehr, Gerhard Erker, Michael Ryan Hansen, and Hellmut Eckert. 2020. "Solid-State NMR Techniques for the Structural Characterization of Cyclic Aggregates Based on Borane–Phosphane Frustrated Lewis Pairs" Molecules 25, no. 6: 1400. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25061400