Palladium-Catalysed Coupling Reactions En Route to Molecular Machines: Sterically Hindered Indenyl and Ferrocenyl Anthracenes and Triptycenes, and Biindenyls

Abstract
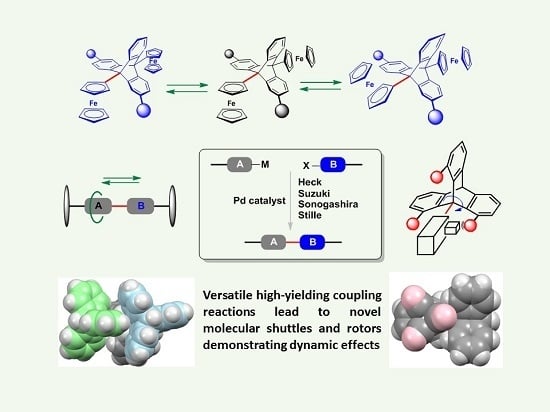
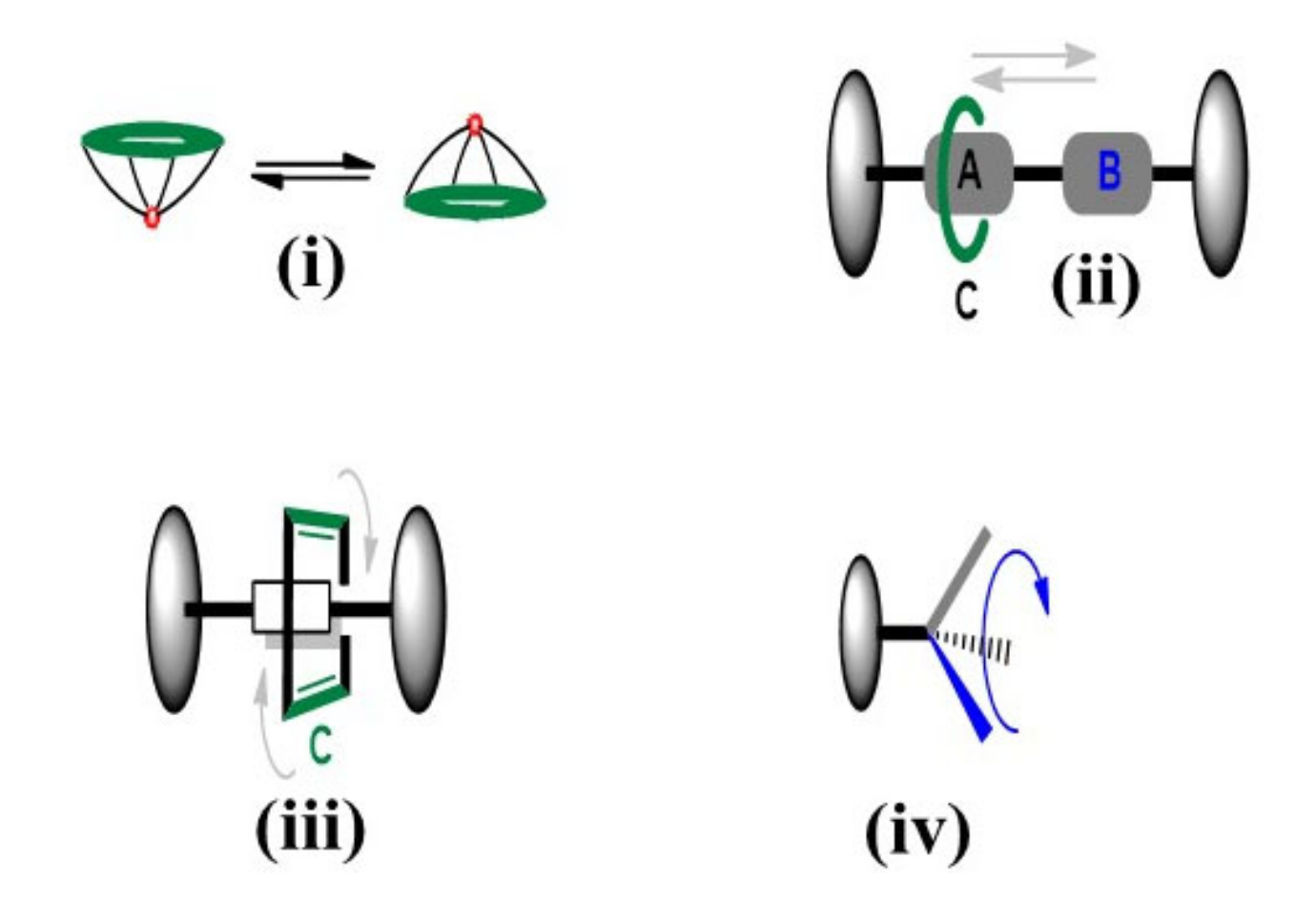
:
{kind=link}
{kind=link}
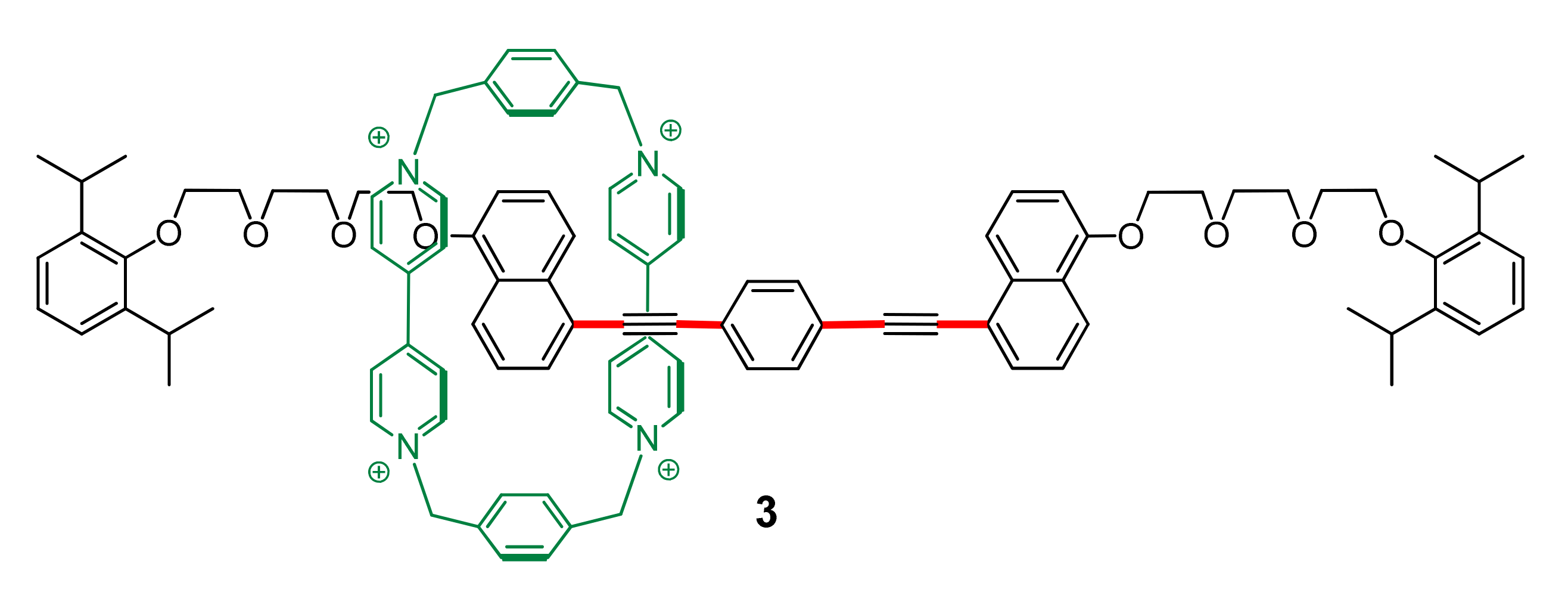{kind=link}
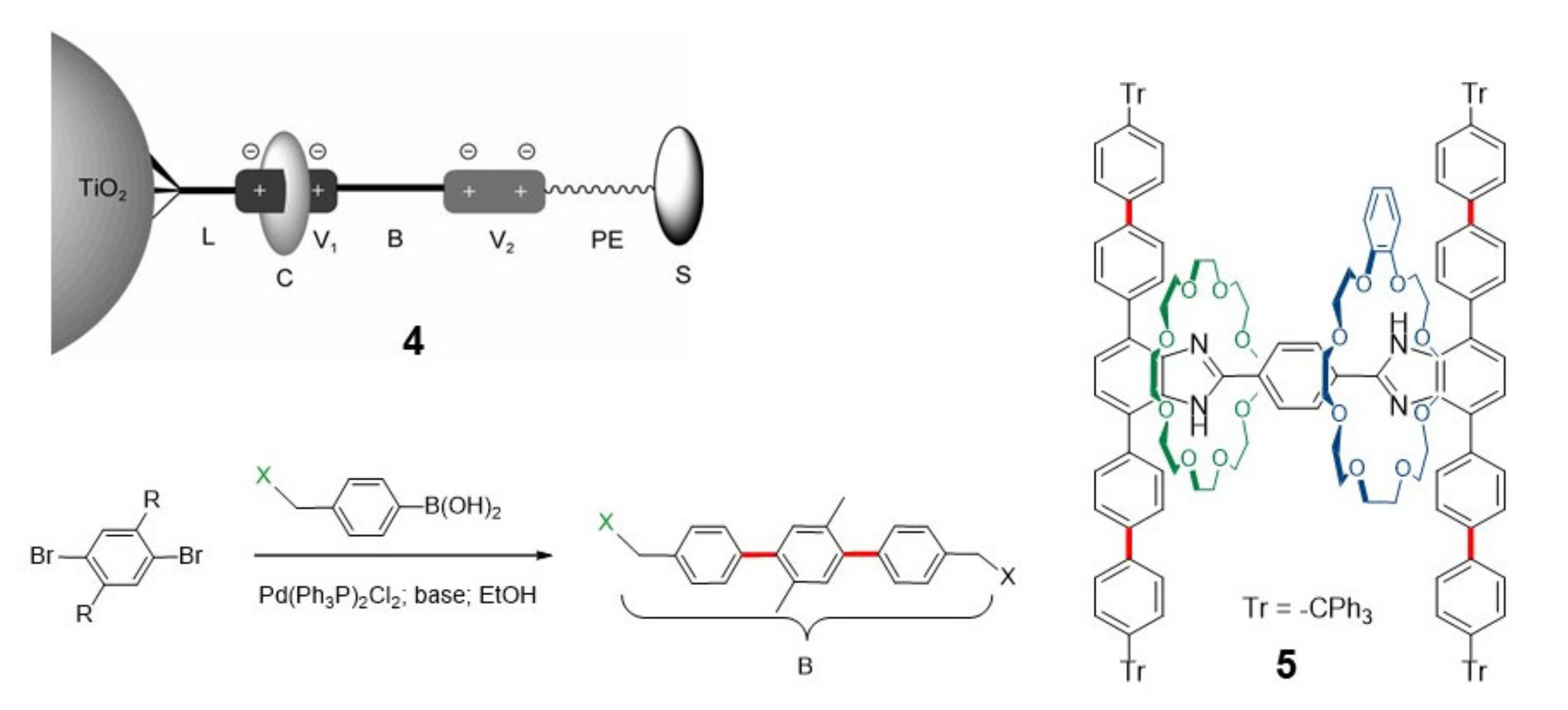{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
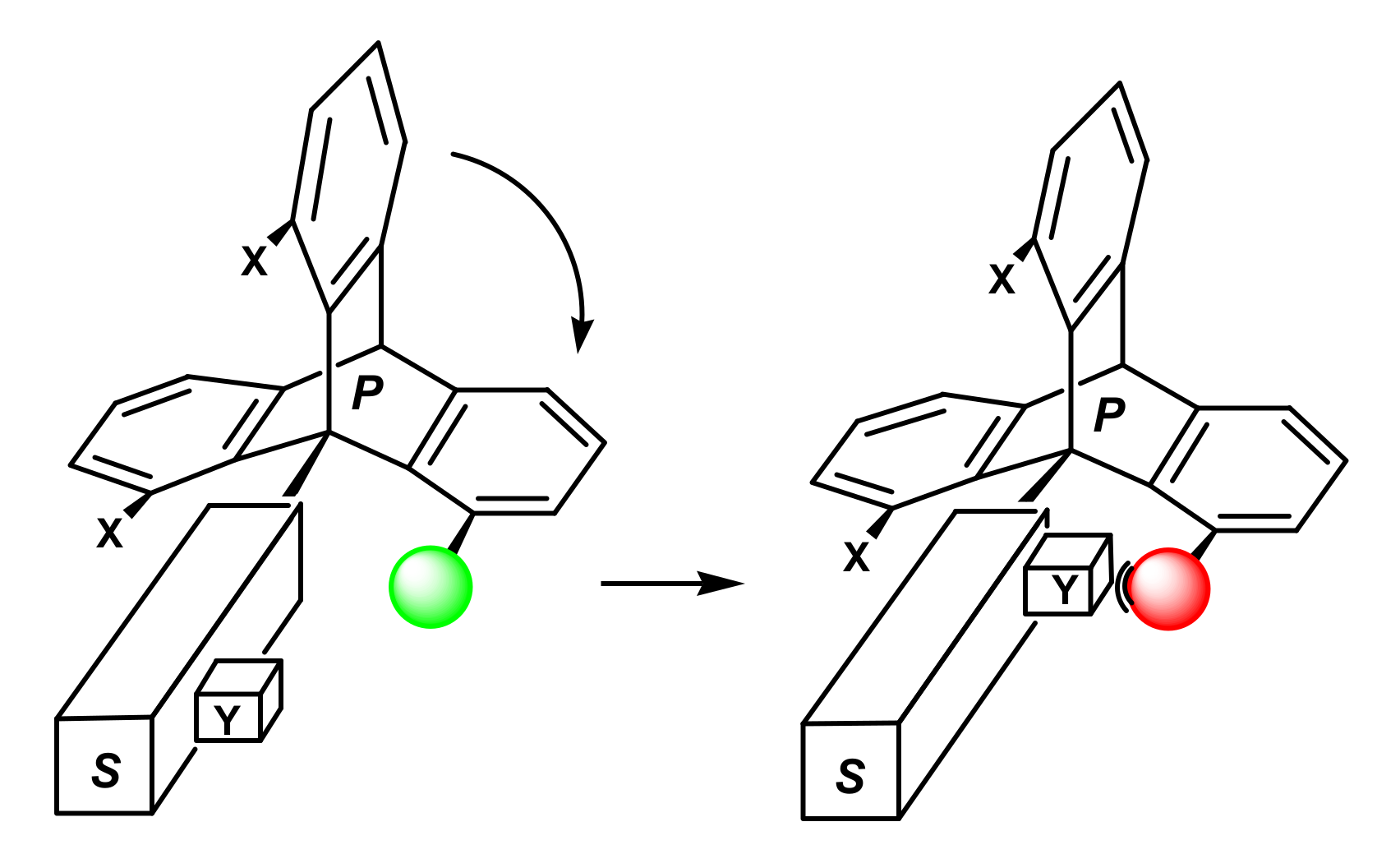
1. Introduction
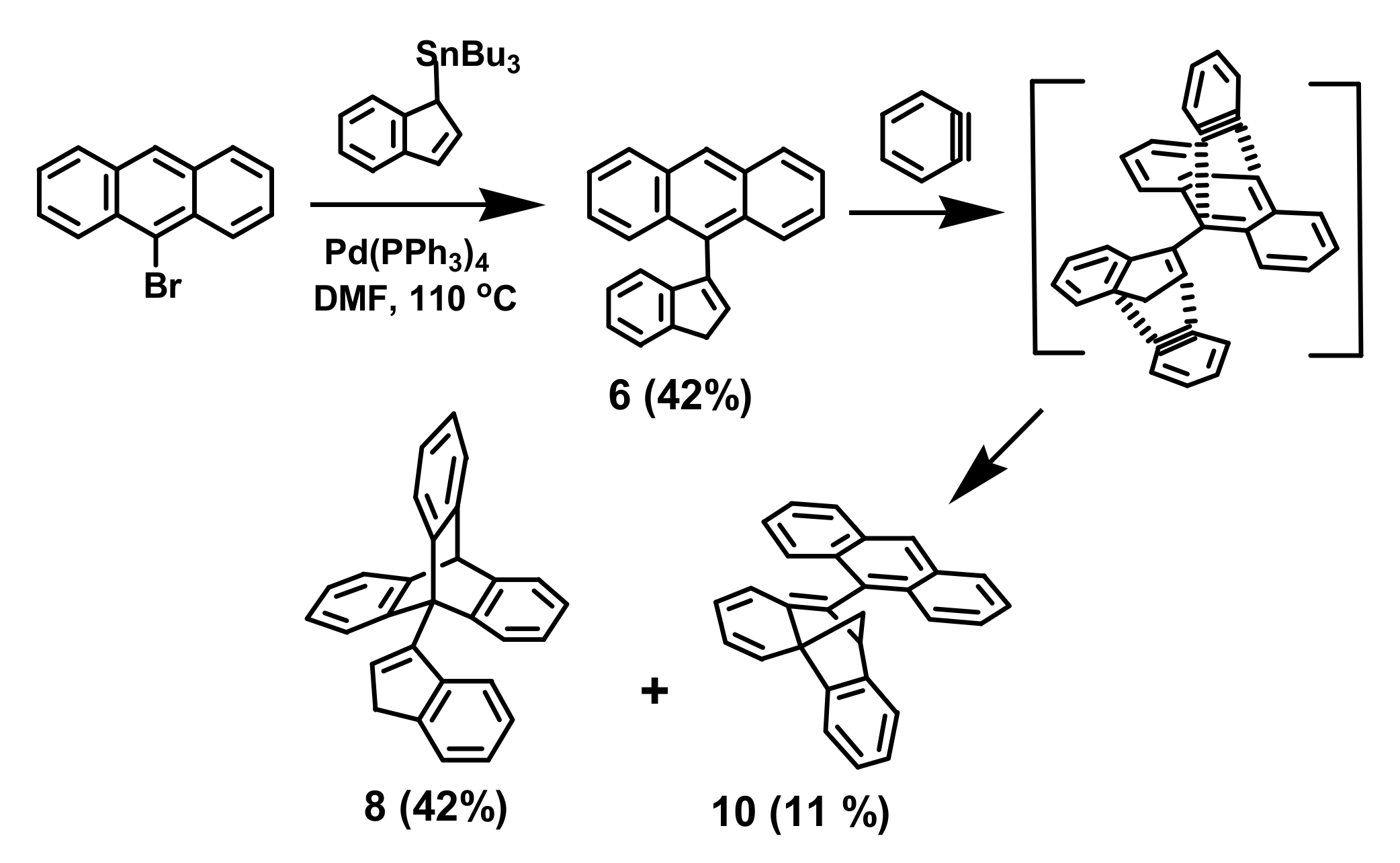
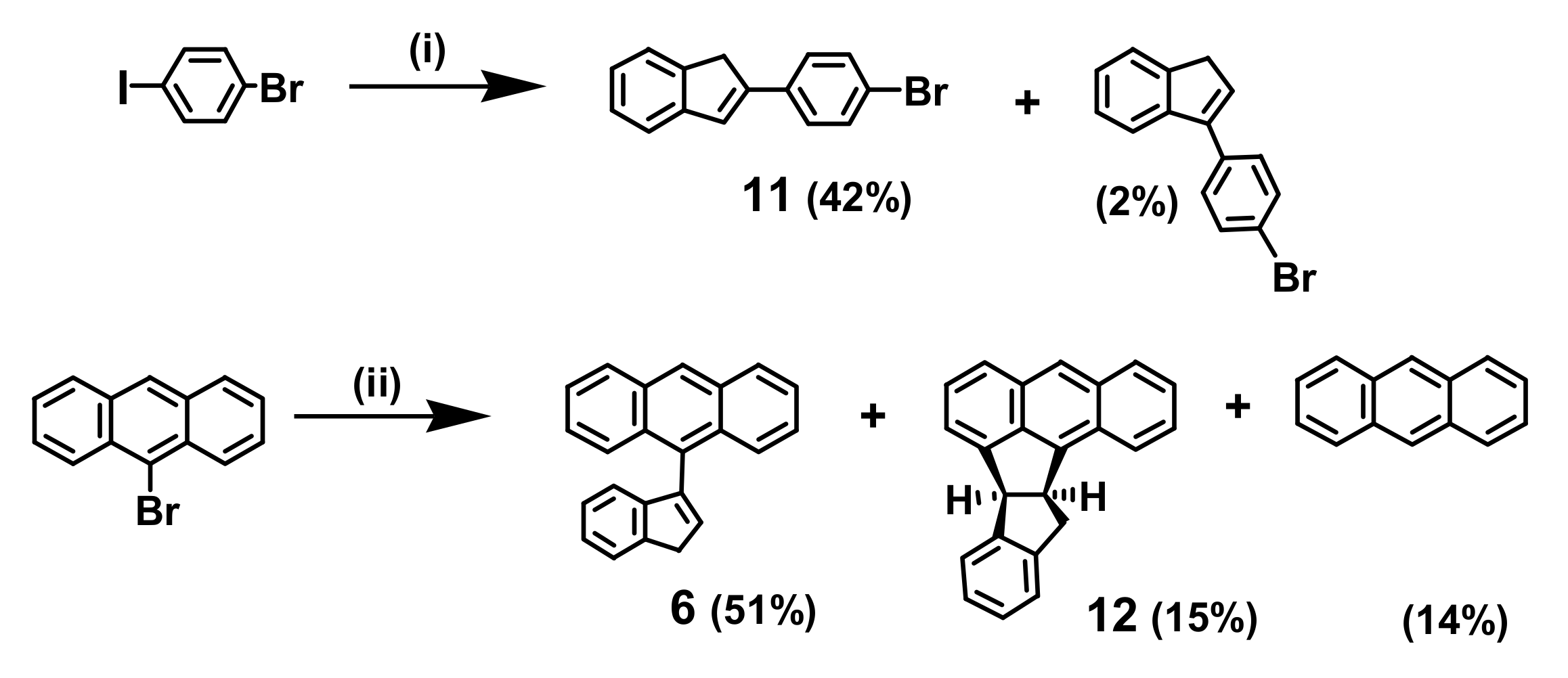
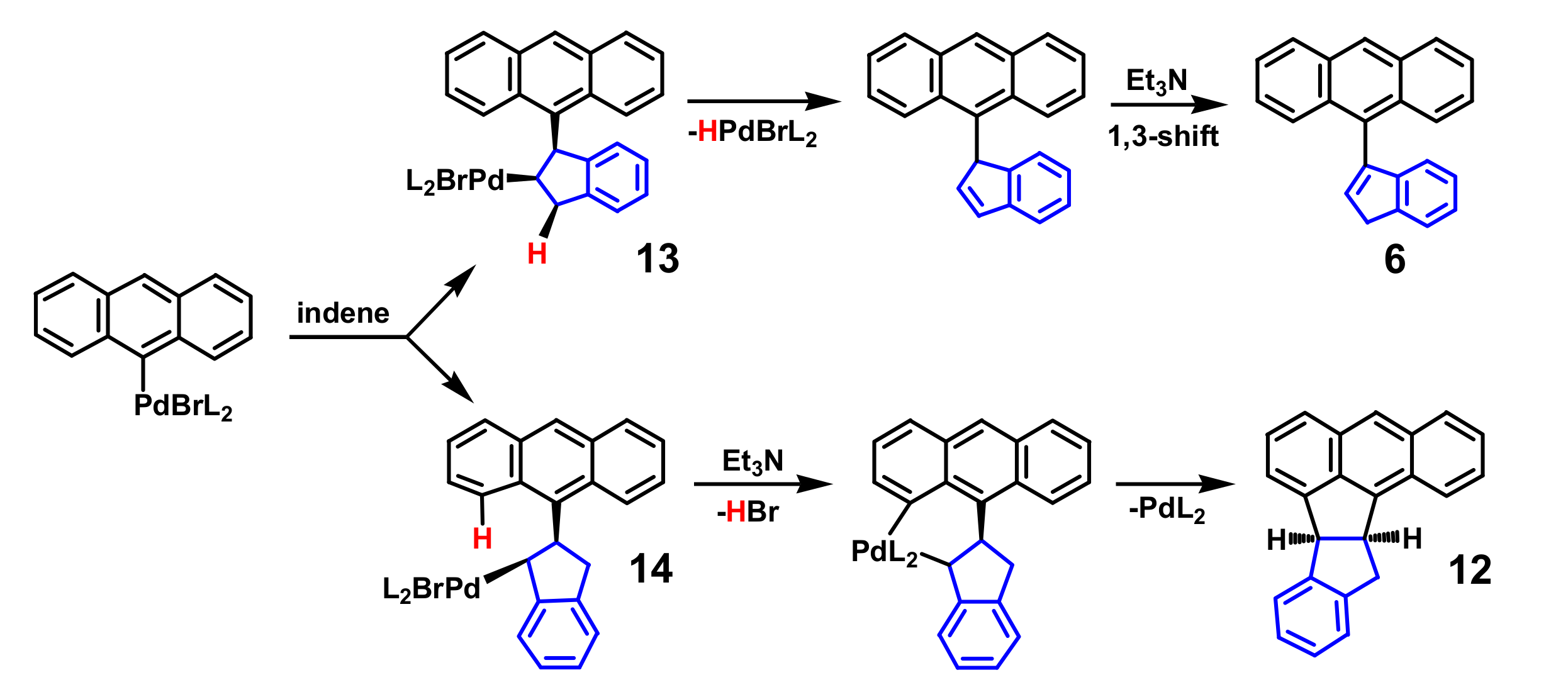
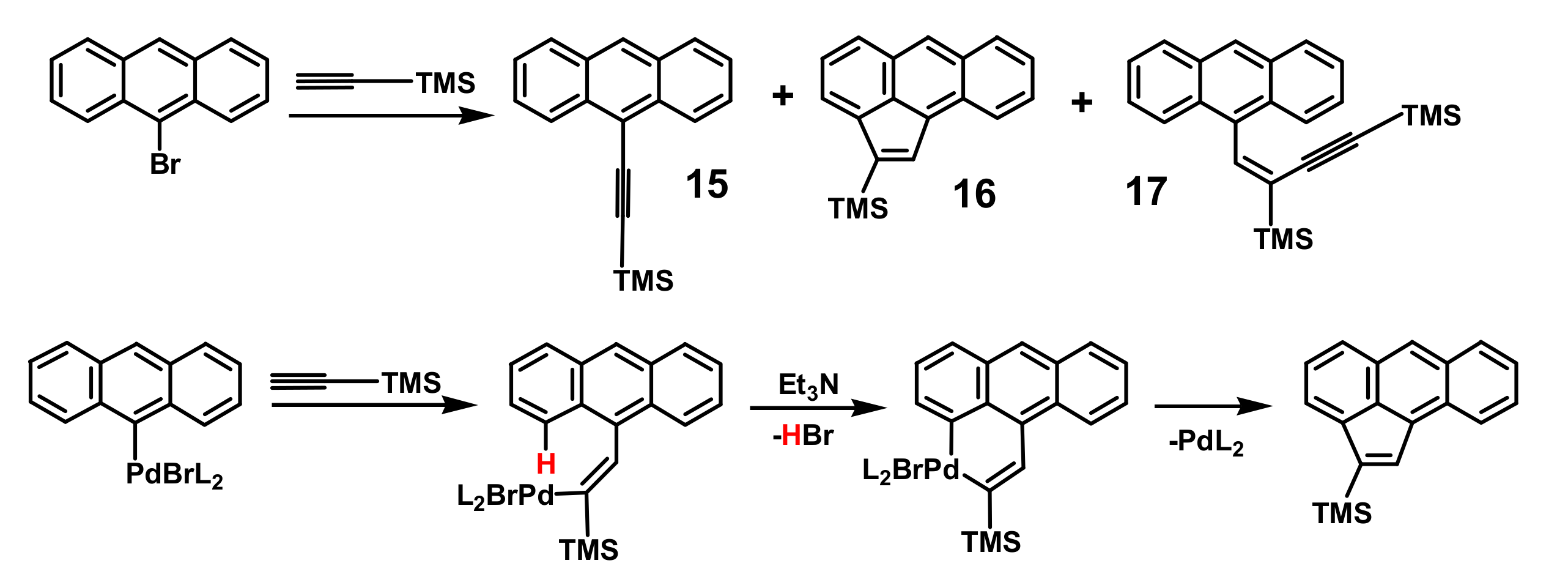
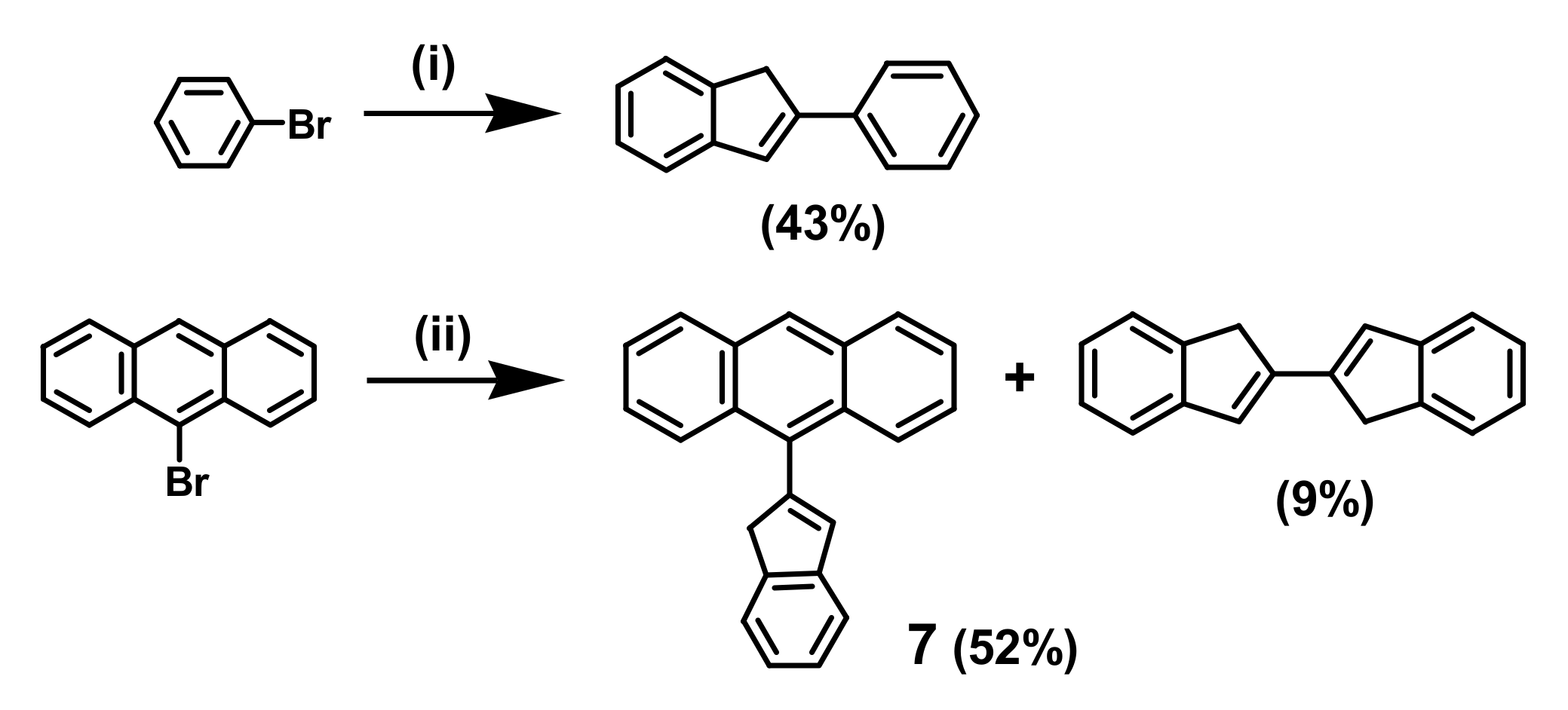
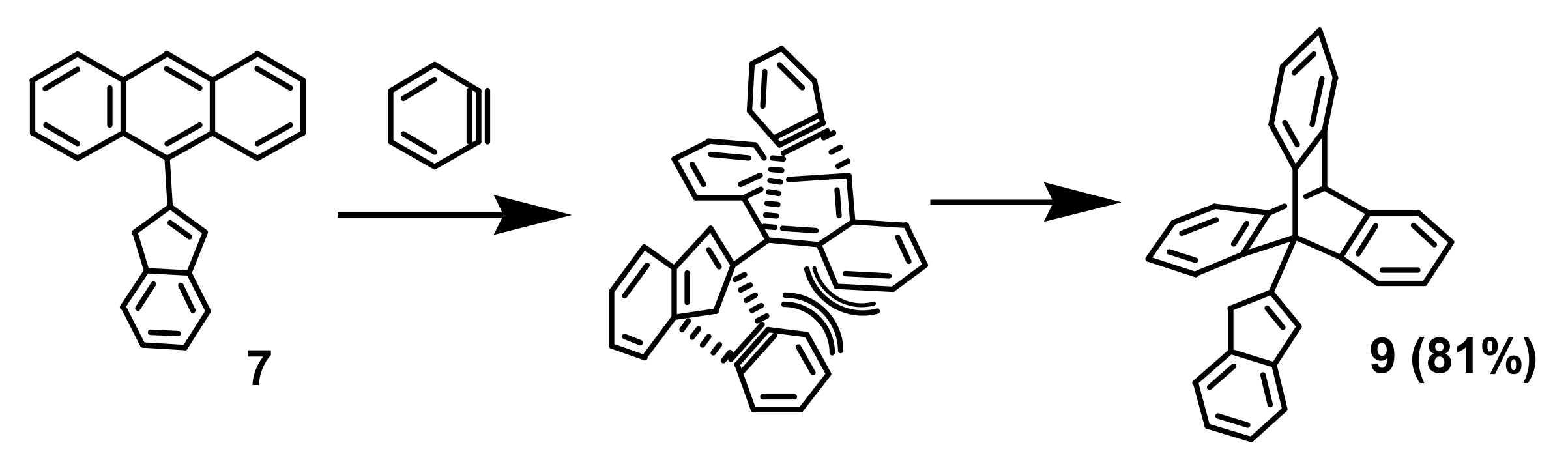
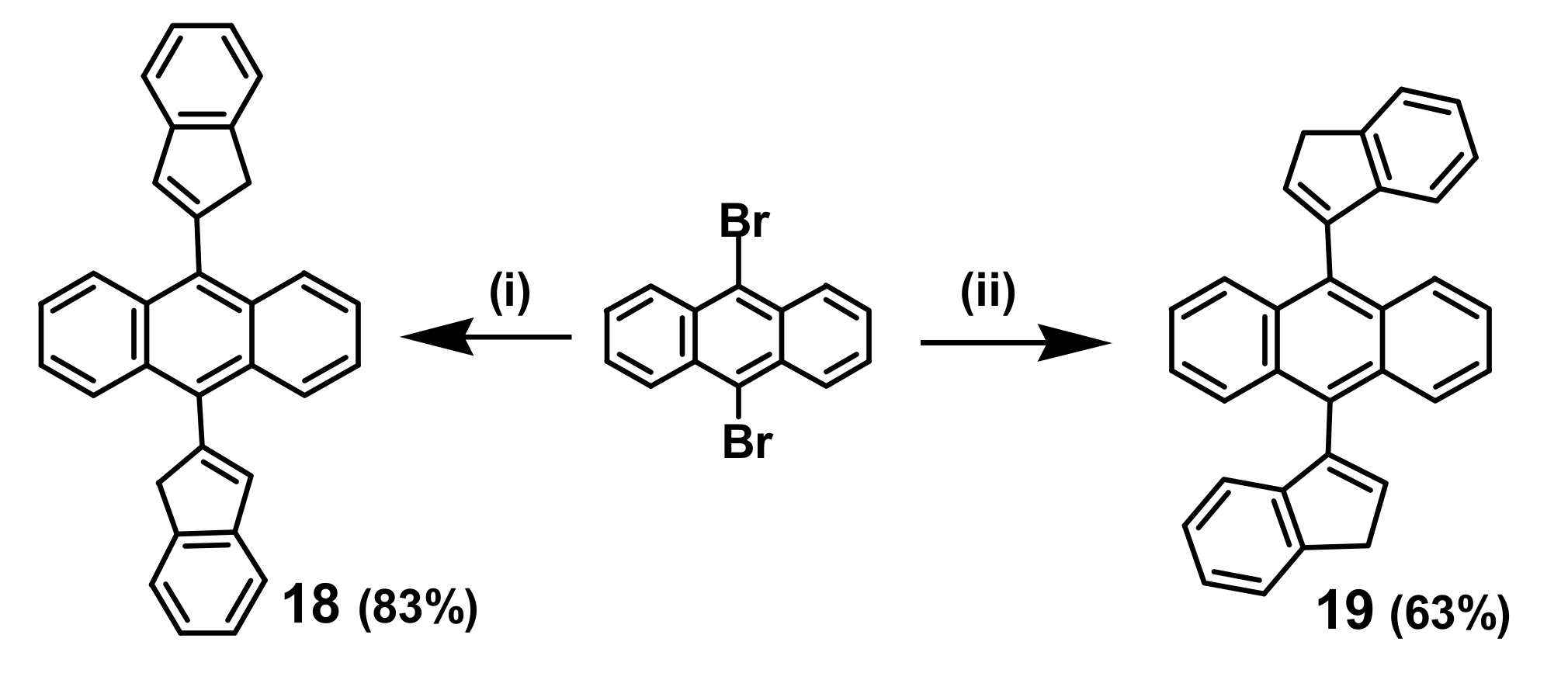
2. Indenyl Anthracenes and Triptycenes
2.1. Synthetic Aspects
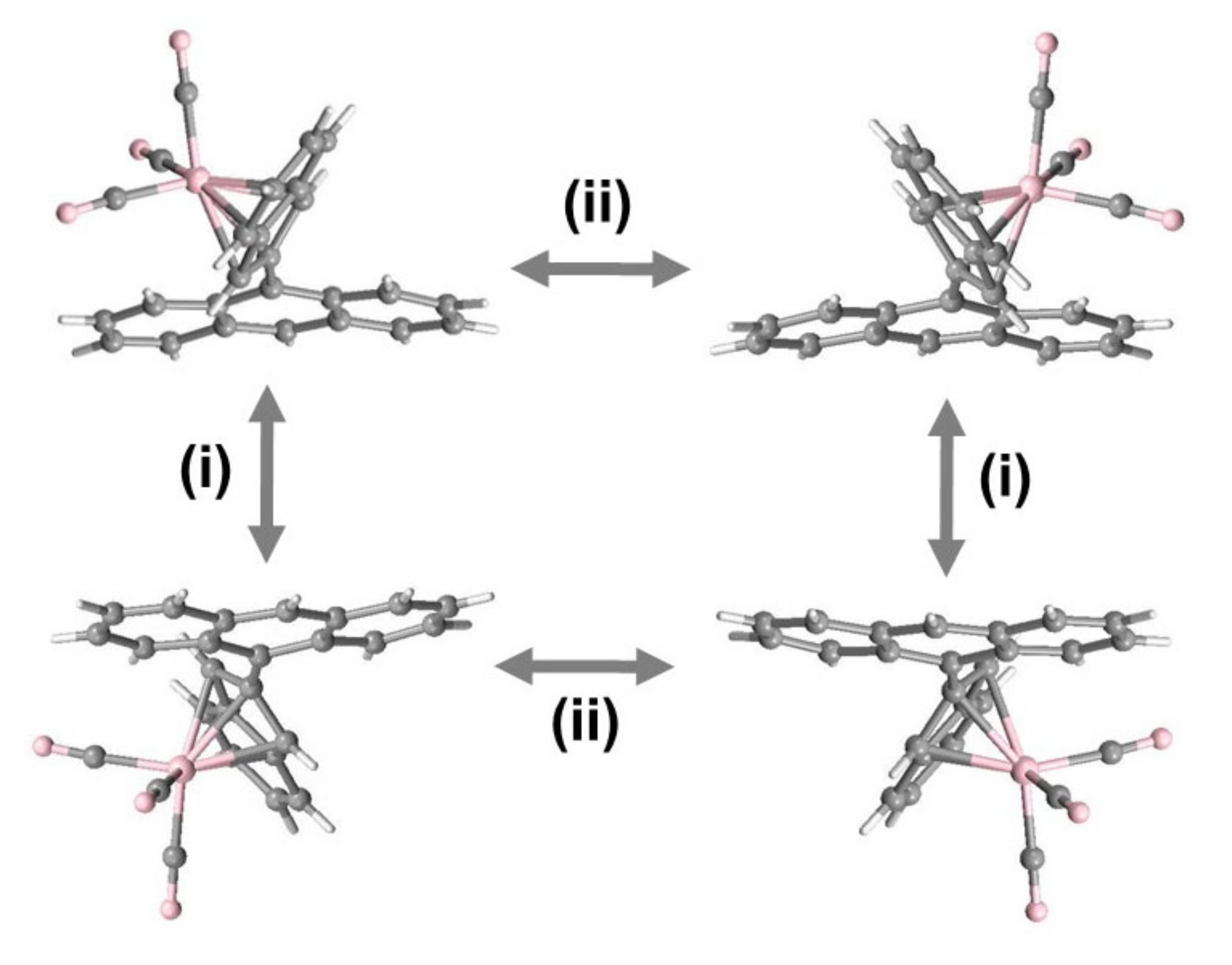
2.2. Rotational Barriers in di-Indenyl Anthracenes
2.3. Cycloadditions of Benzyne to 2-Phenylindene
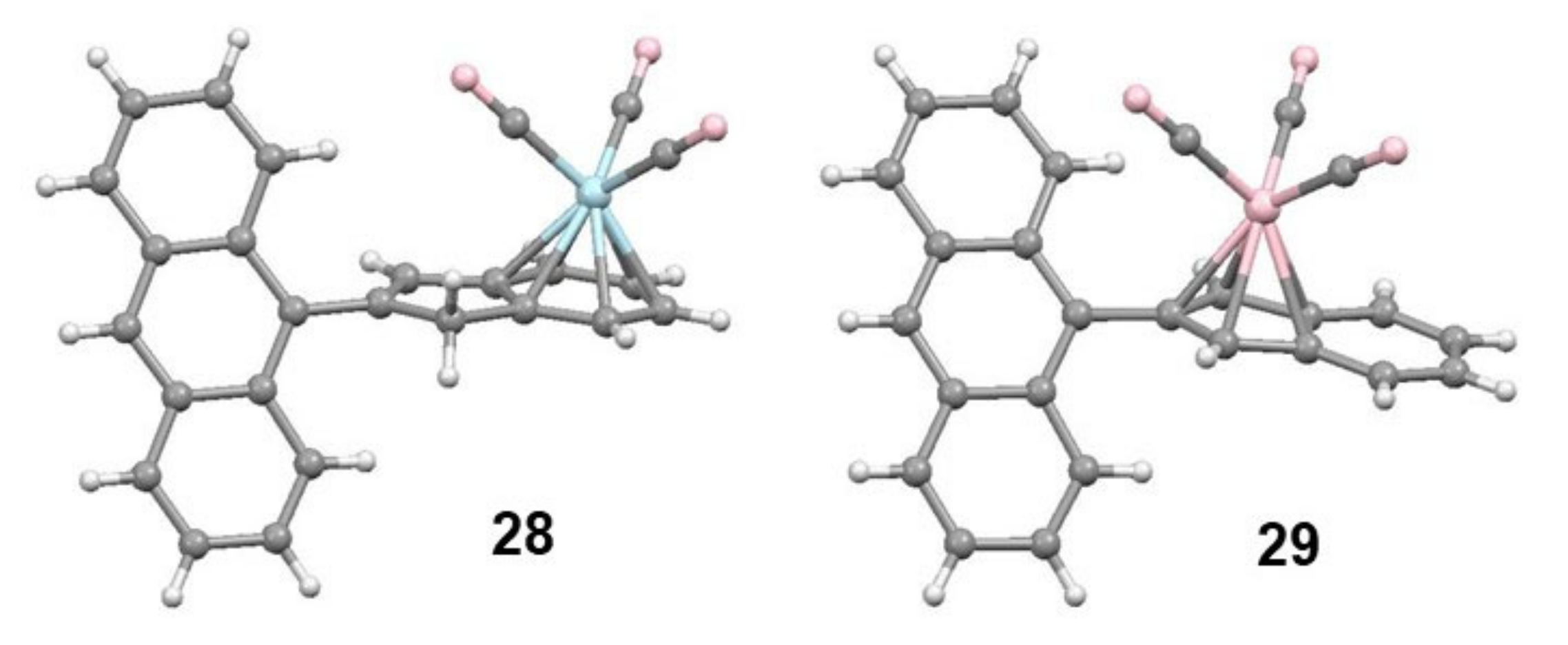
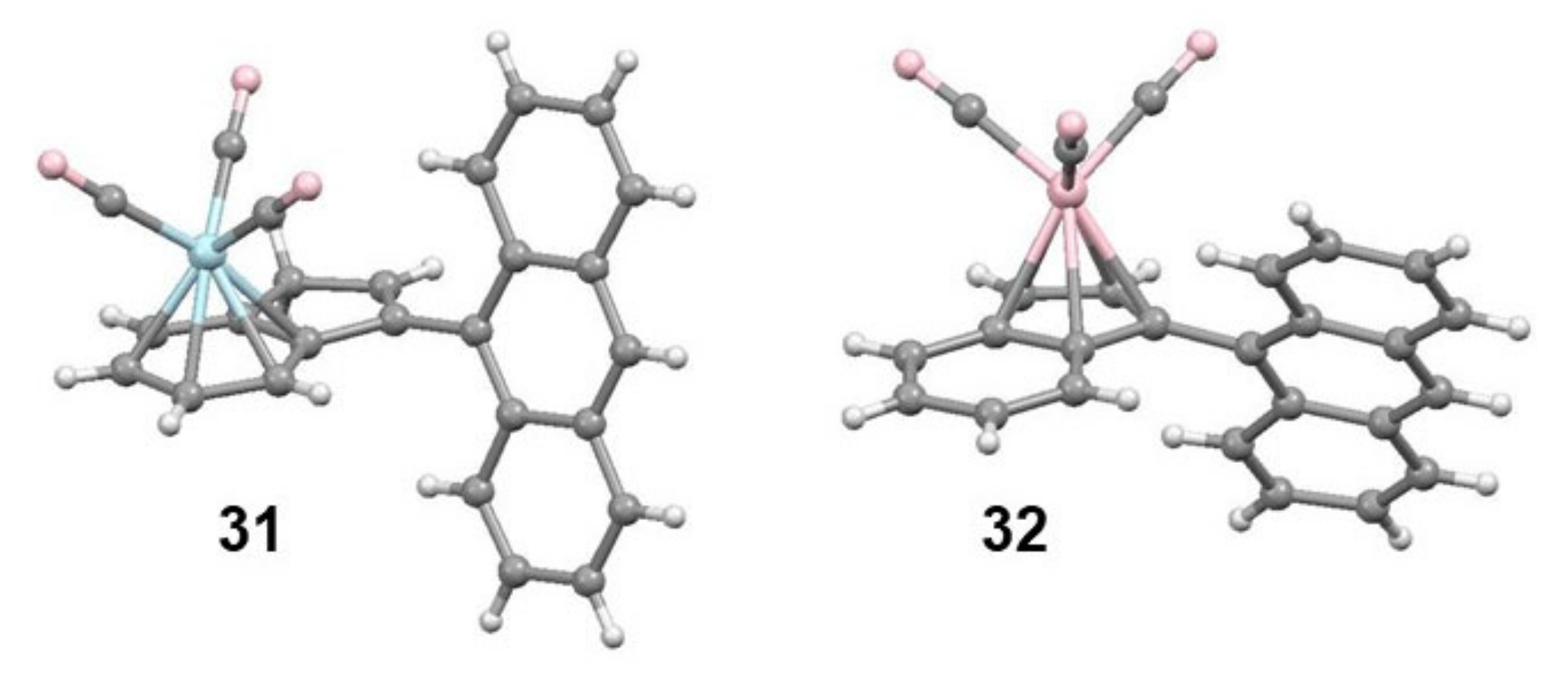
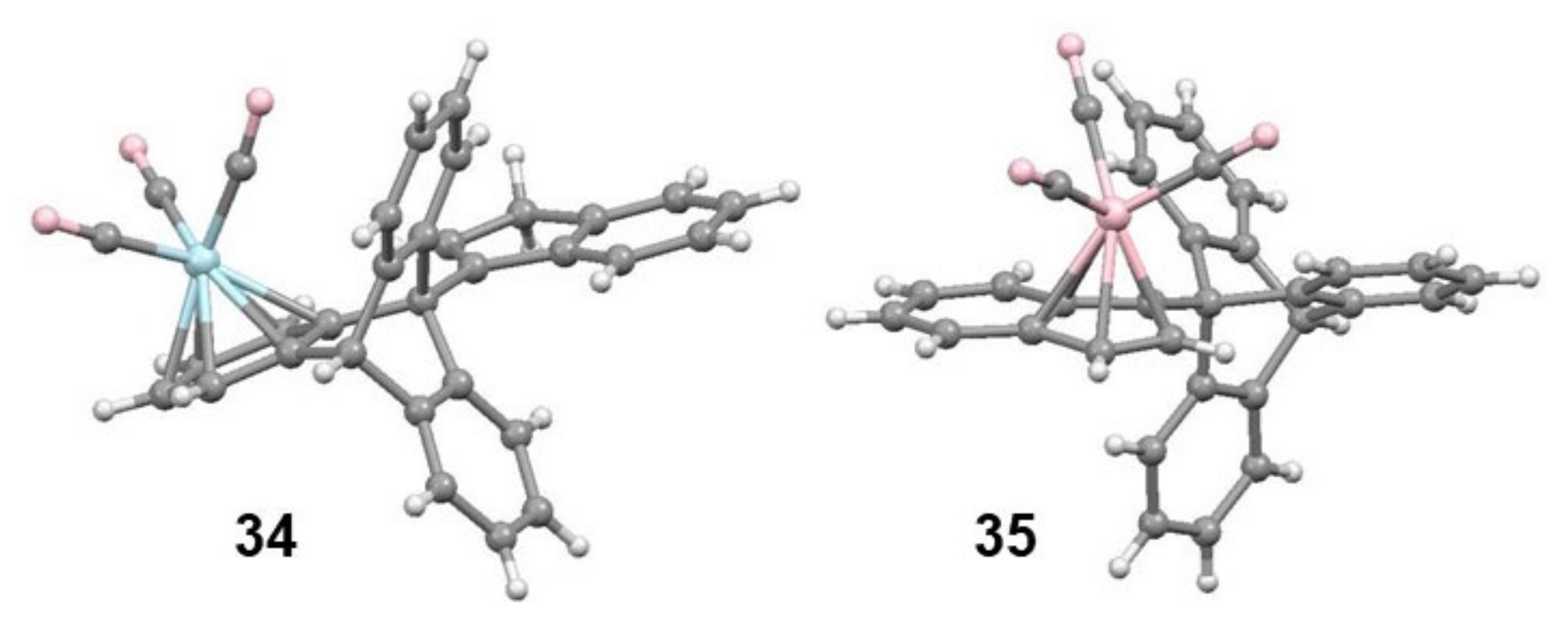
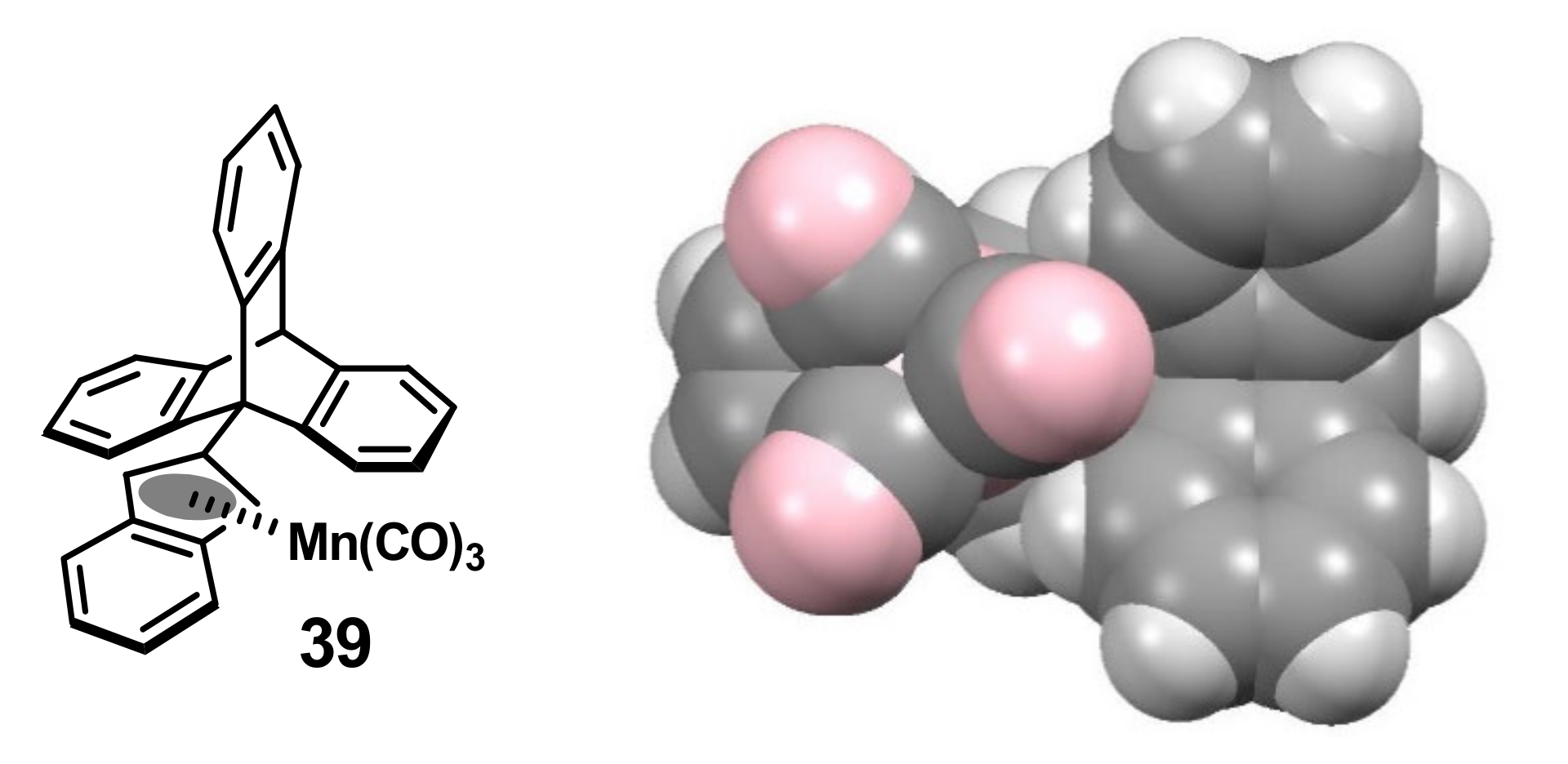
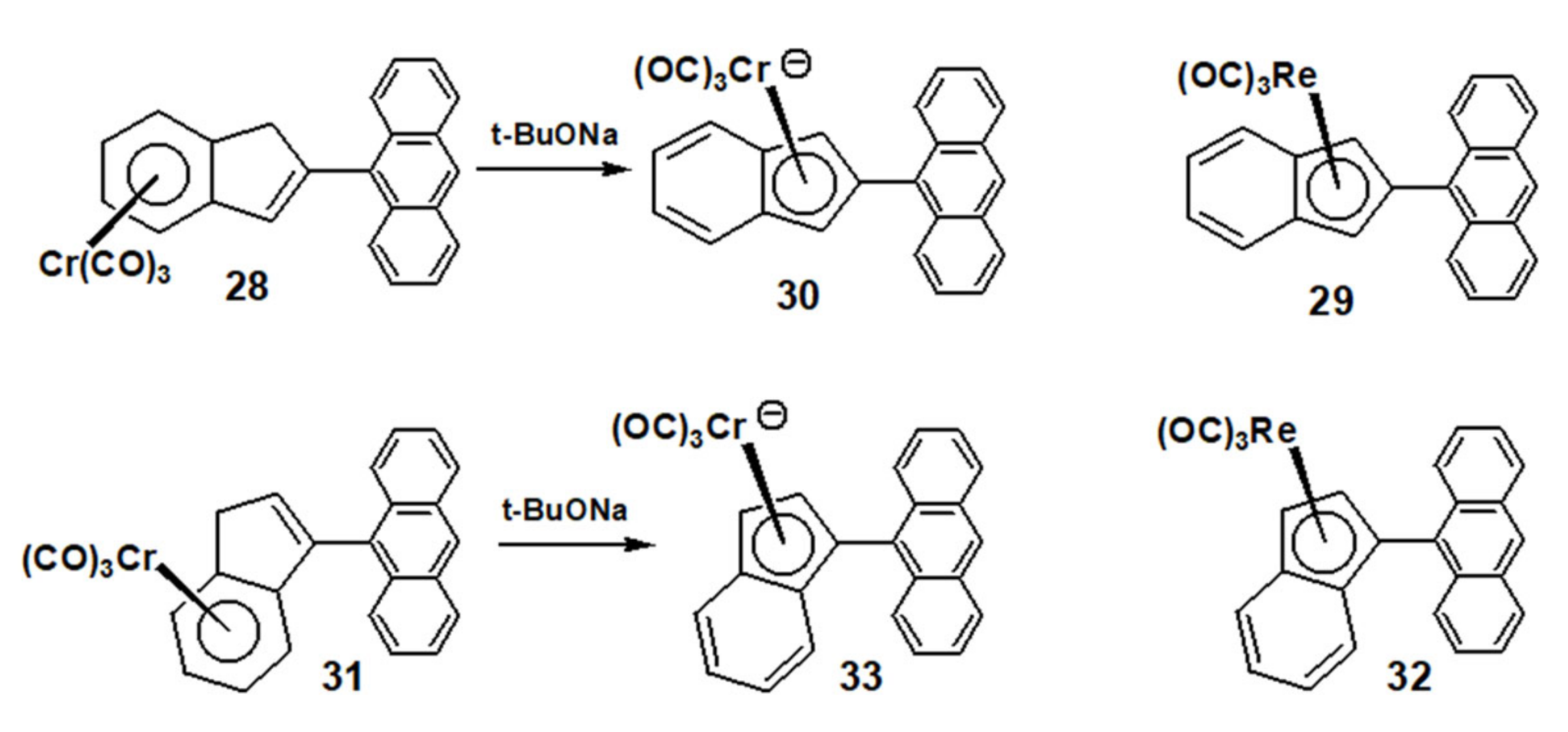
2.4. Organometallic Derivatives of Indenyl Anthracenes
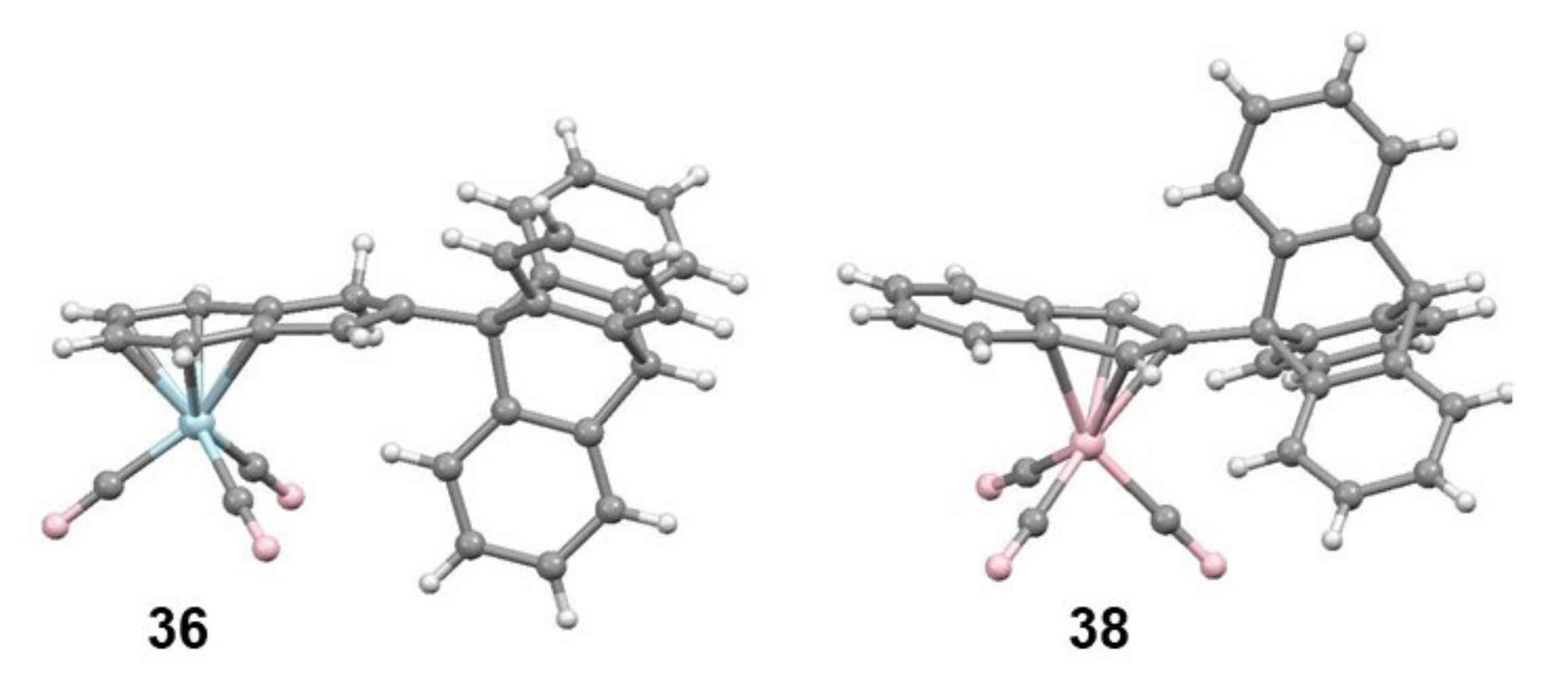
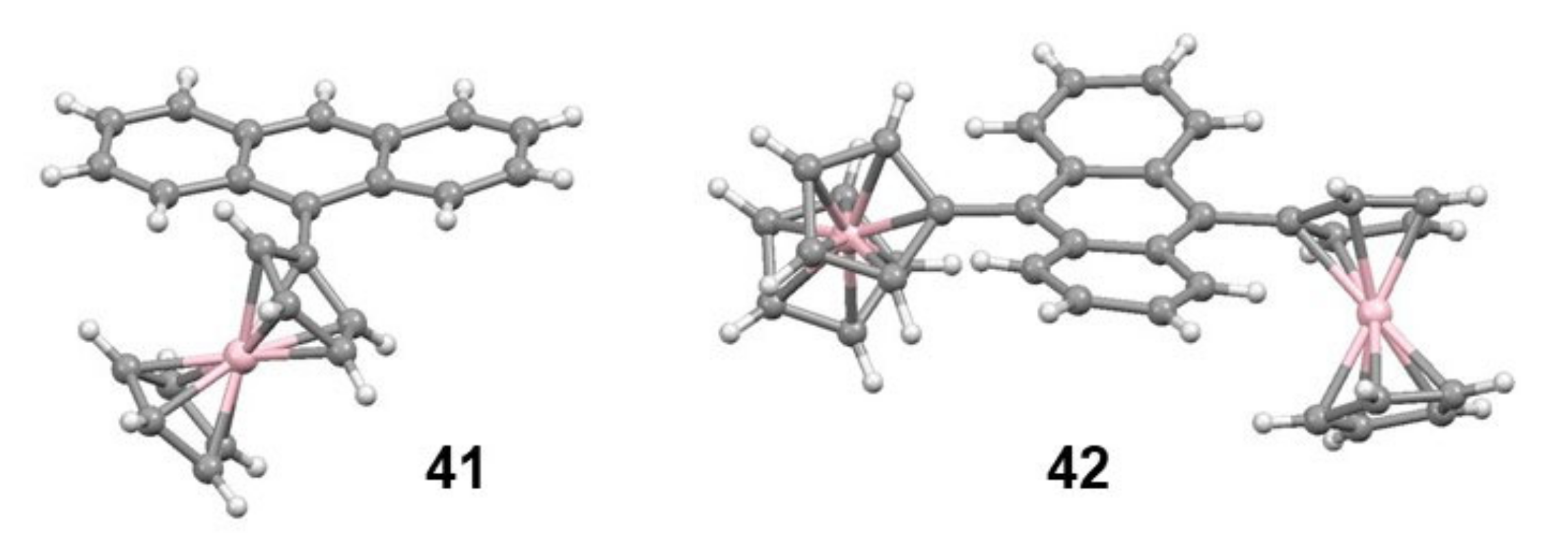
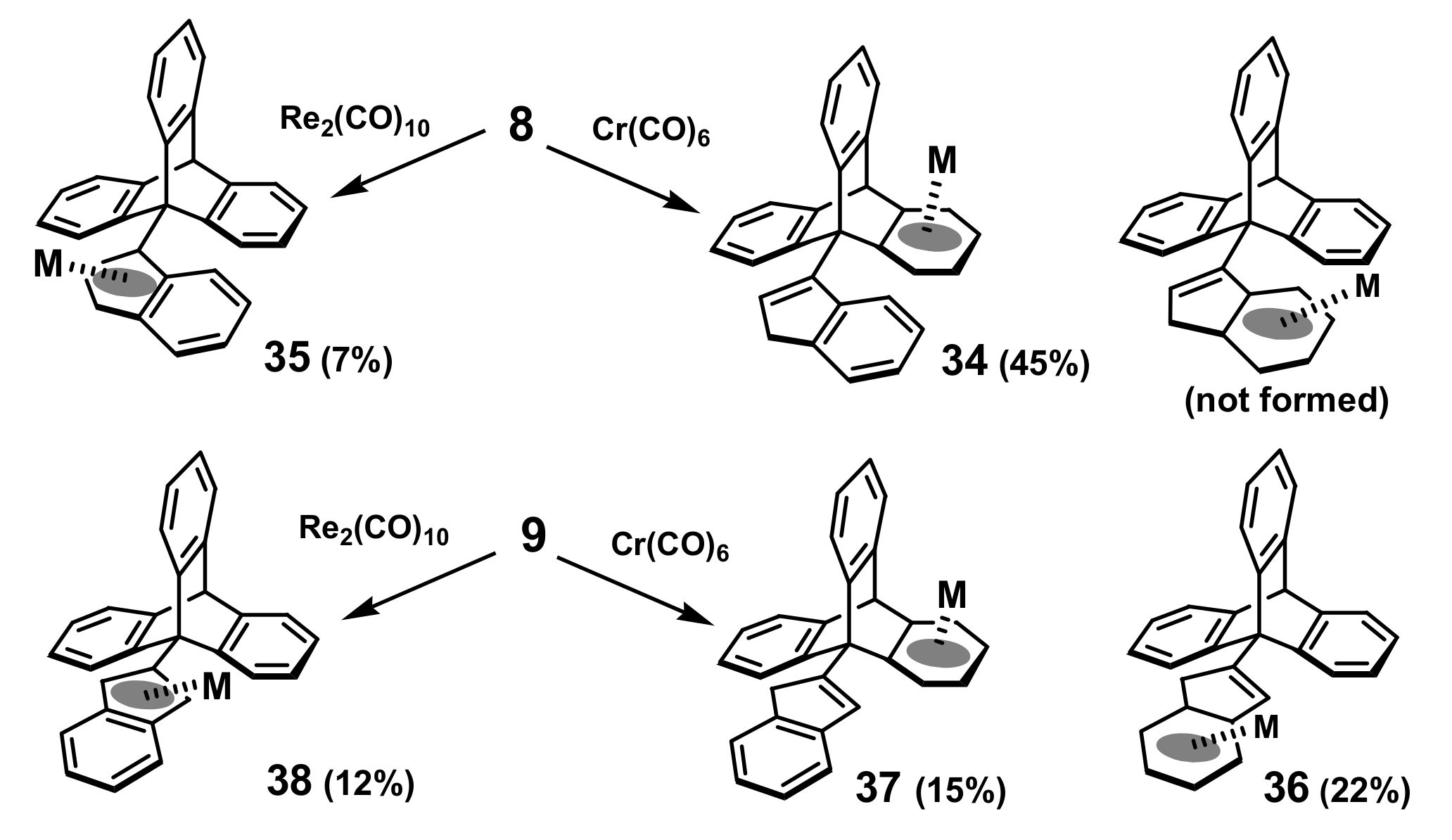
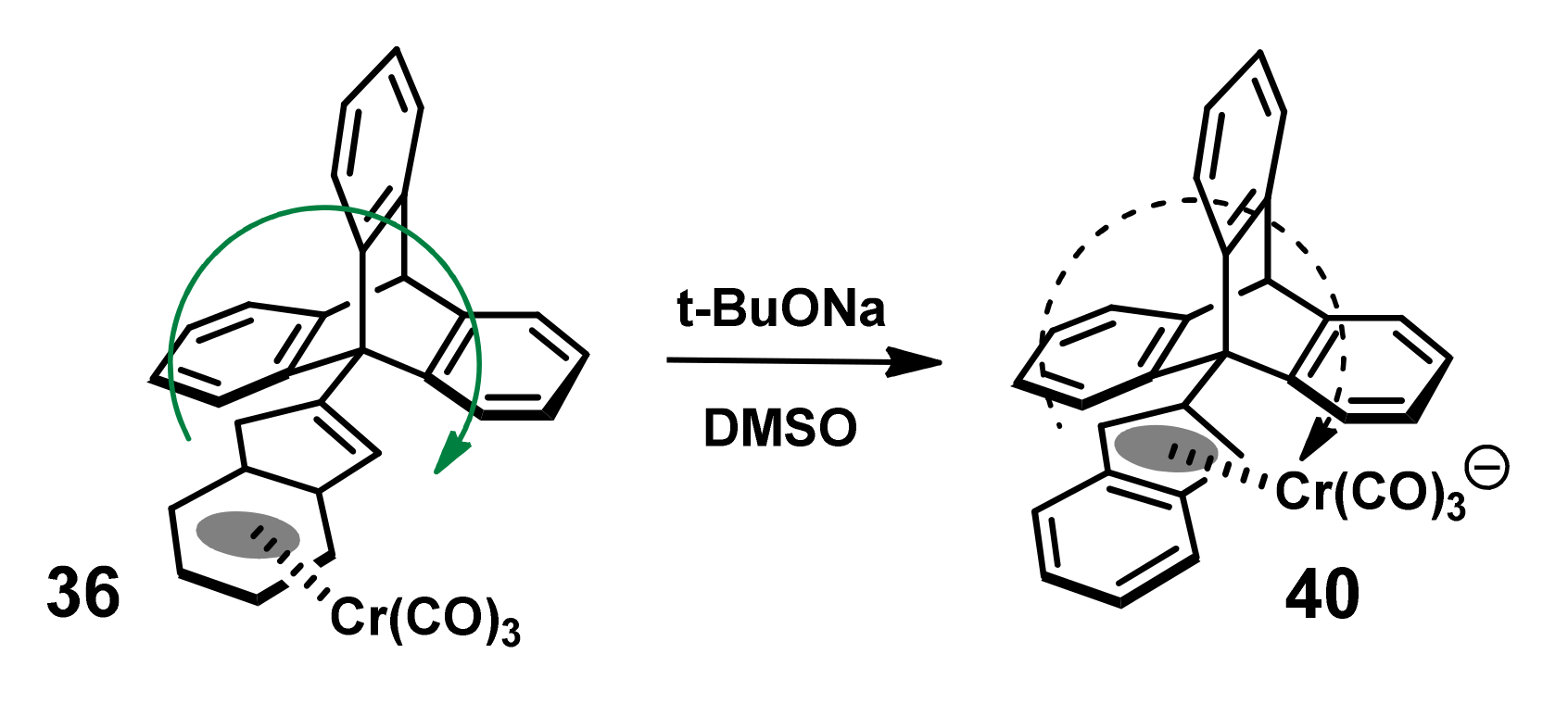
2.5. Organometallic Derivatives of Indenyl Triptycenes
3. Ferrocenyl Anthracenes and Triptycenes
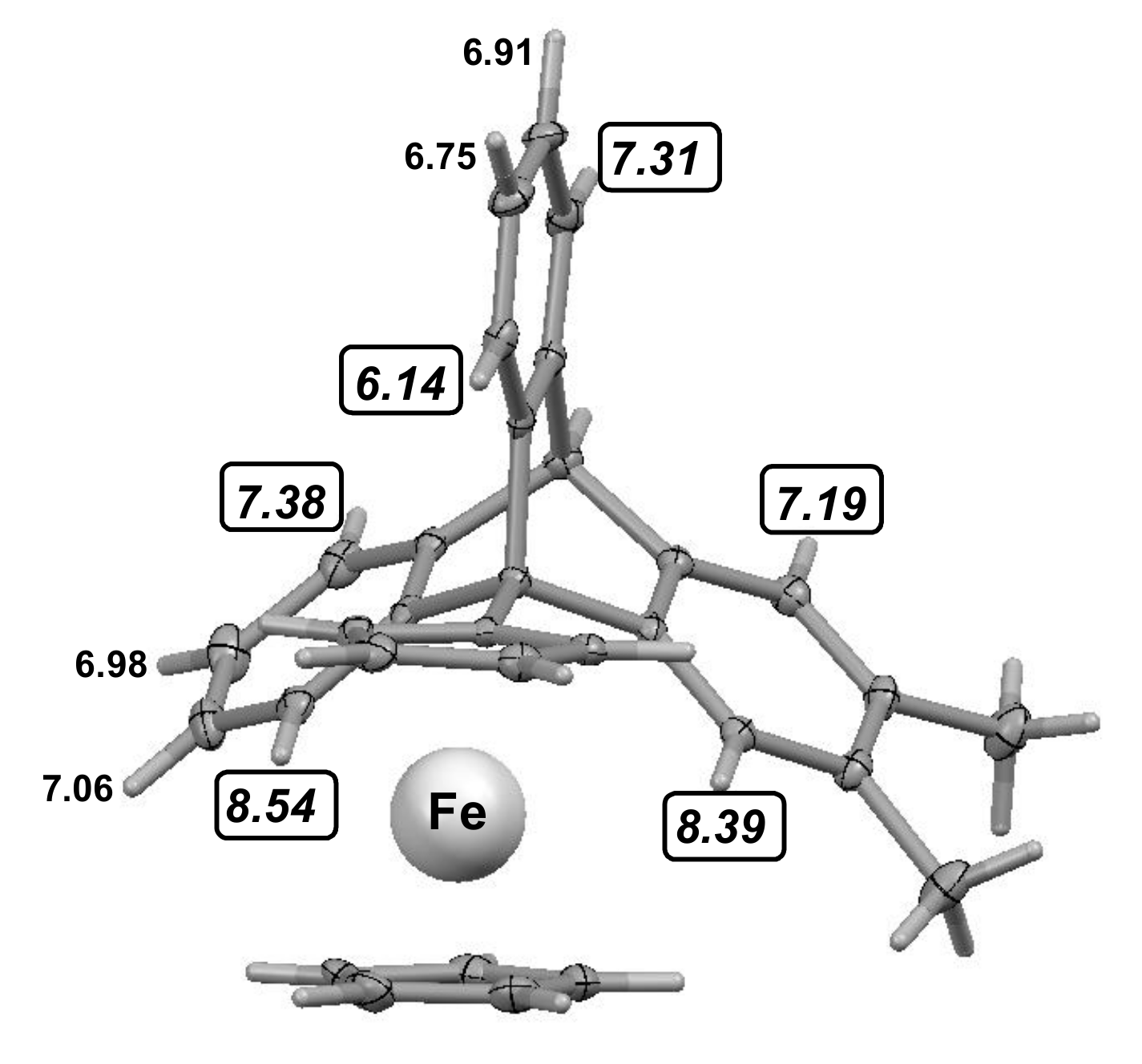
3.1. Mono- and di-Ferrocenyl Anthracenes
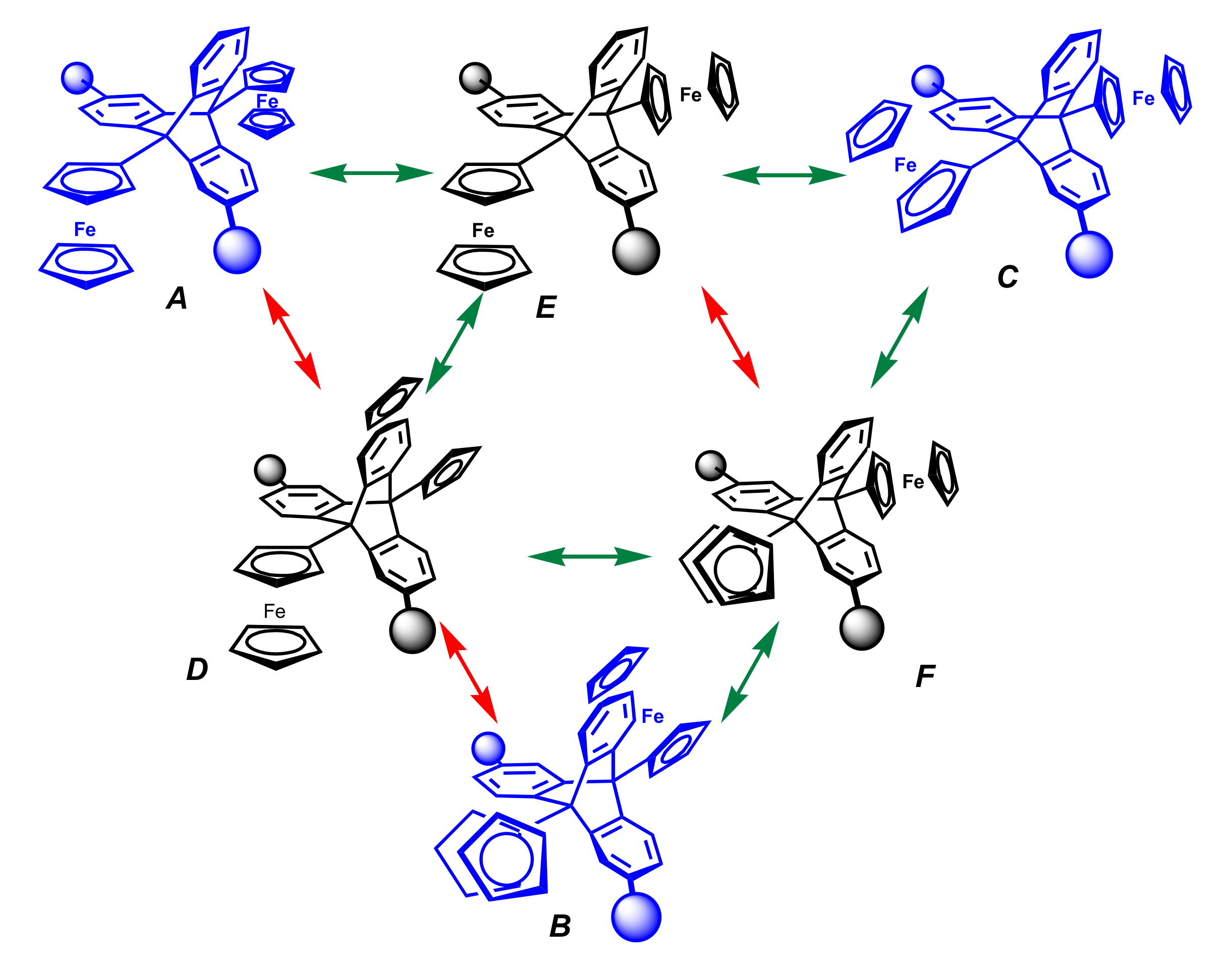
3.2. Mono- and di-Ferrocenyl Triptycenes
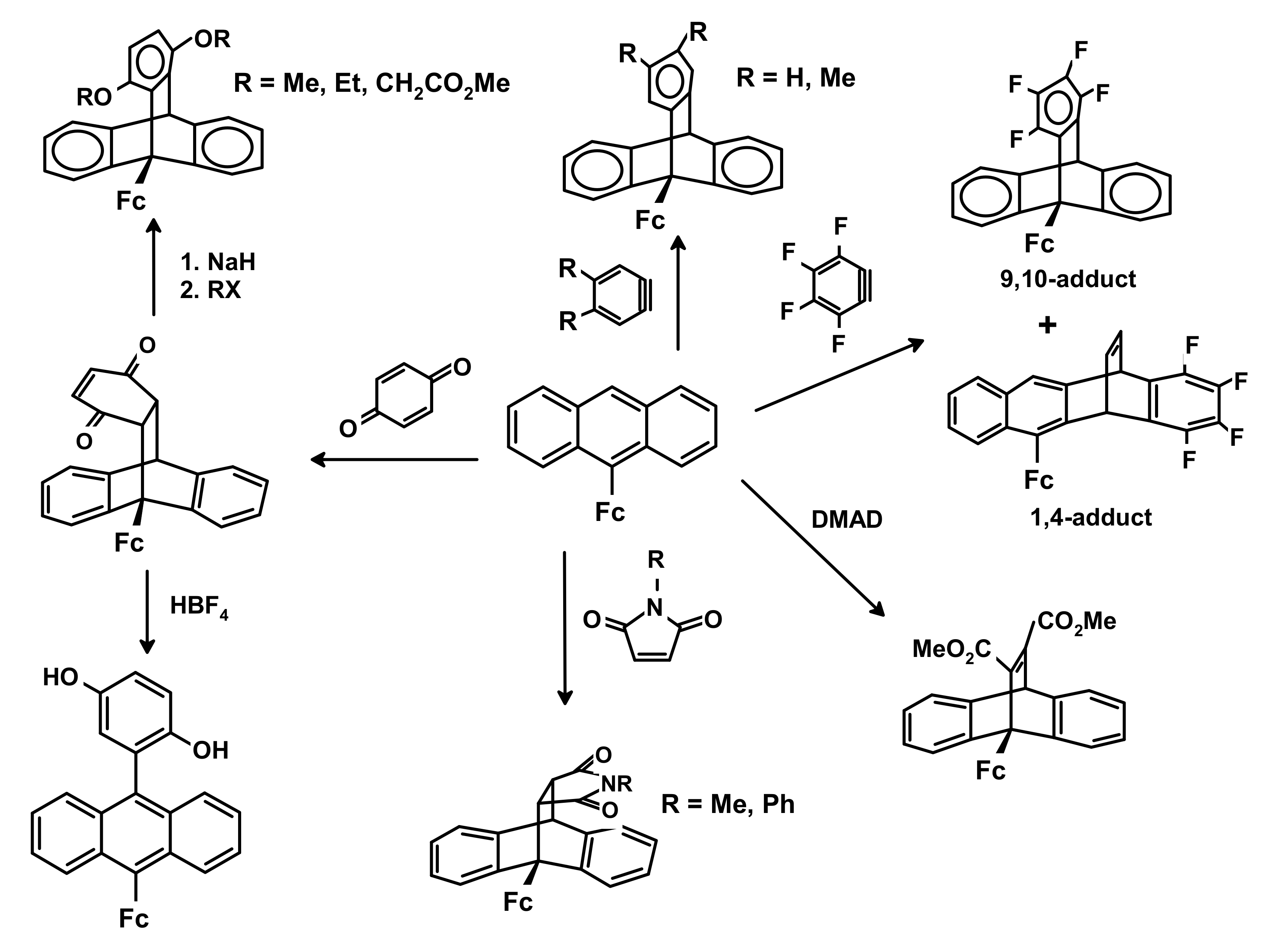
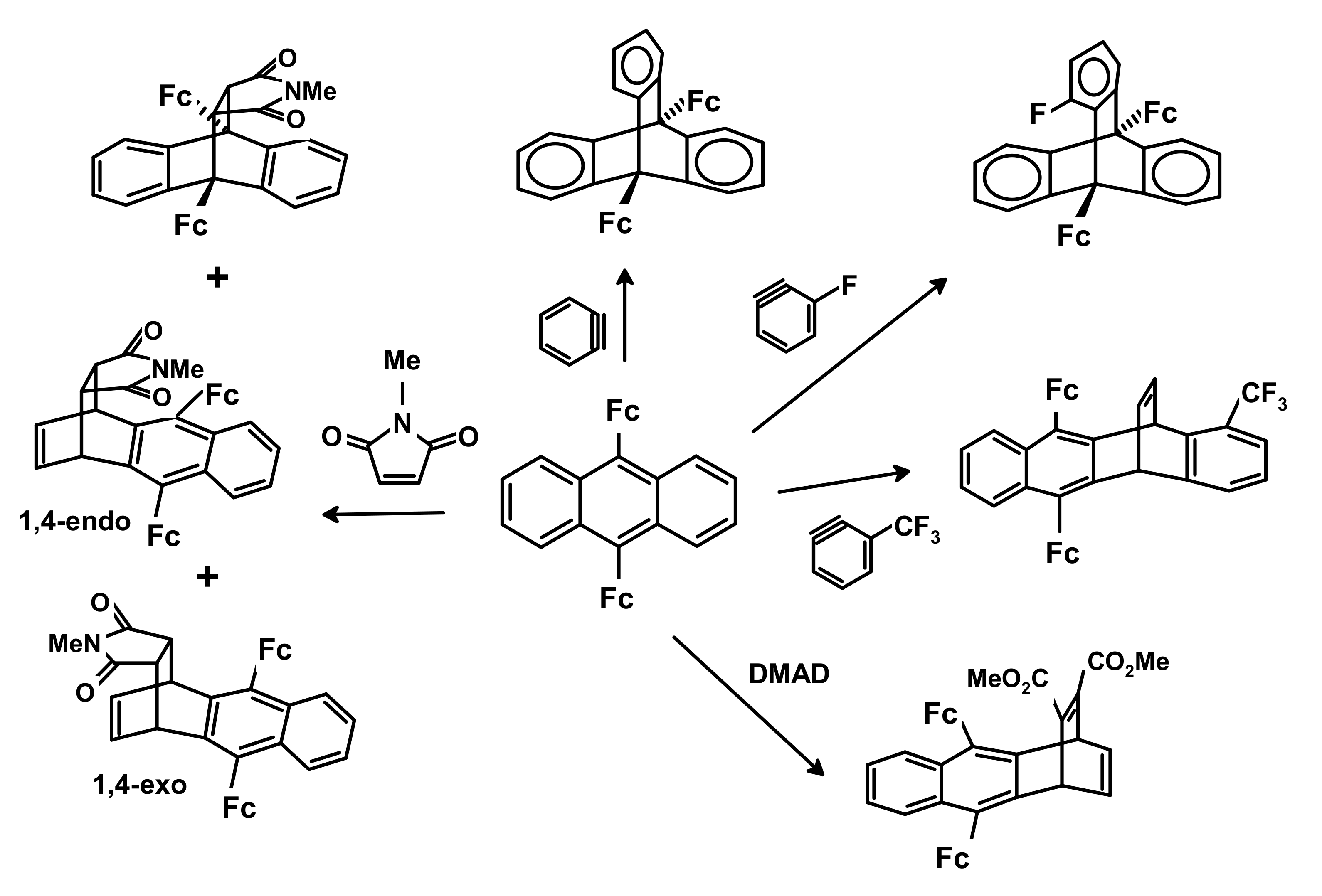
3.3. Cycloaddition Reactions of Mono- and di-Ferrocenyl Anthracenes
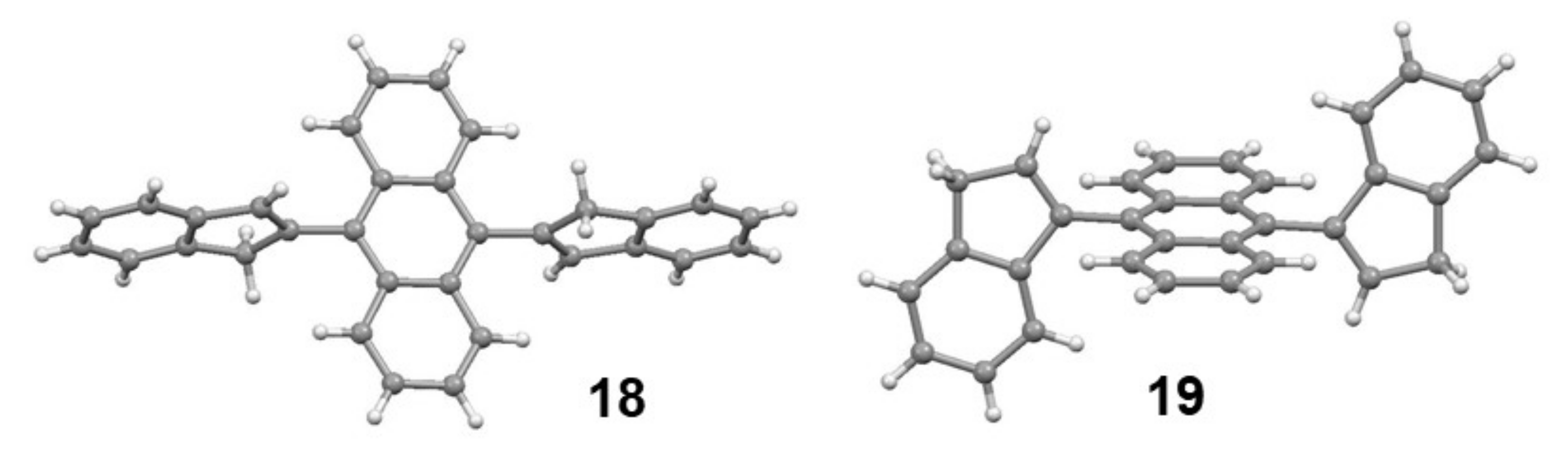
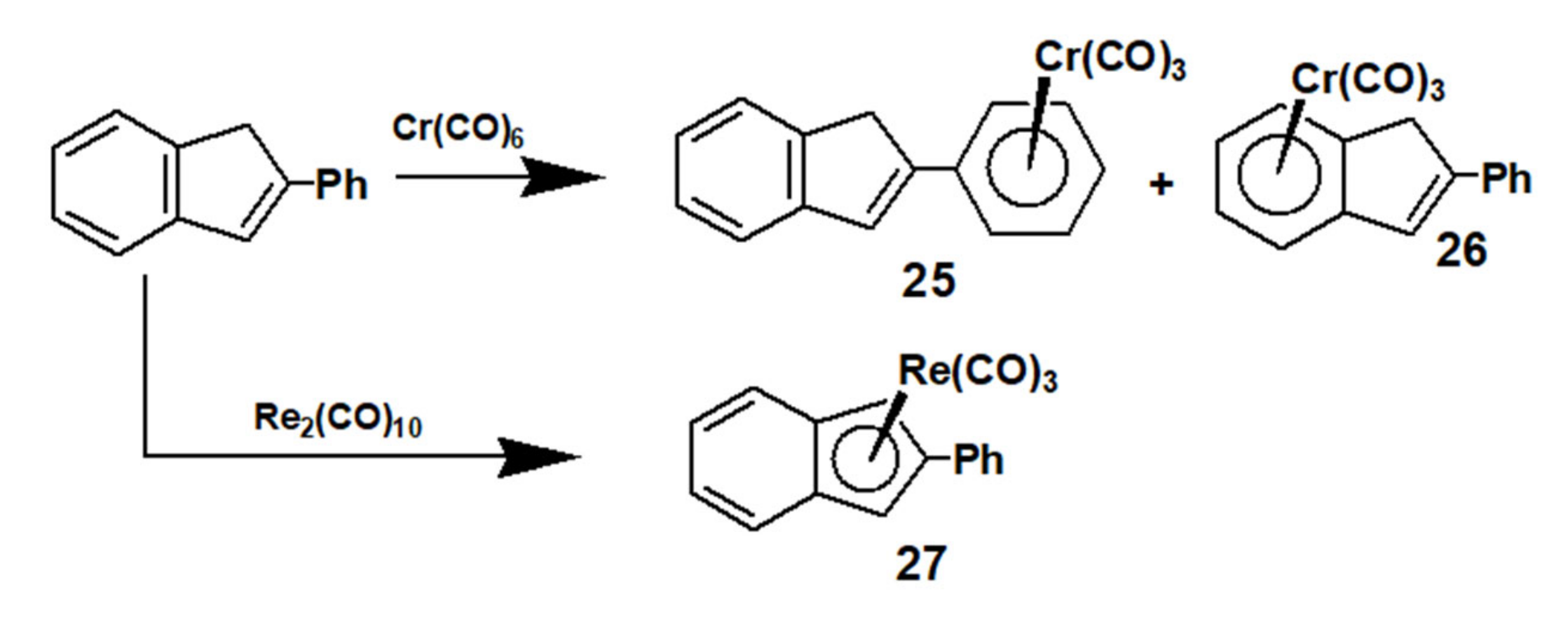
4. Syntheses and Dynamic Behaviour of Biindenyls
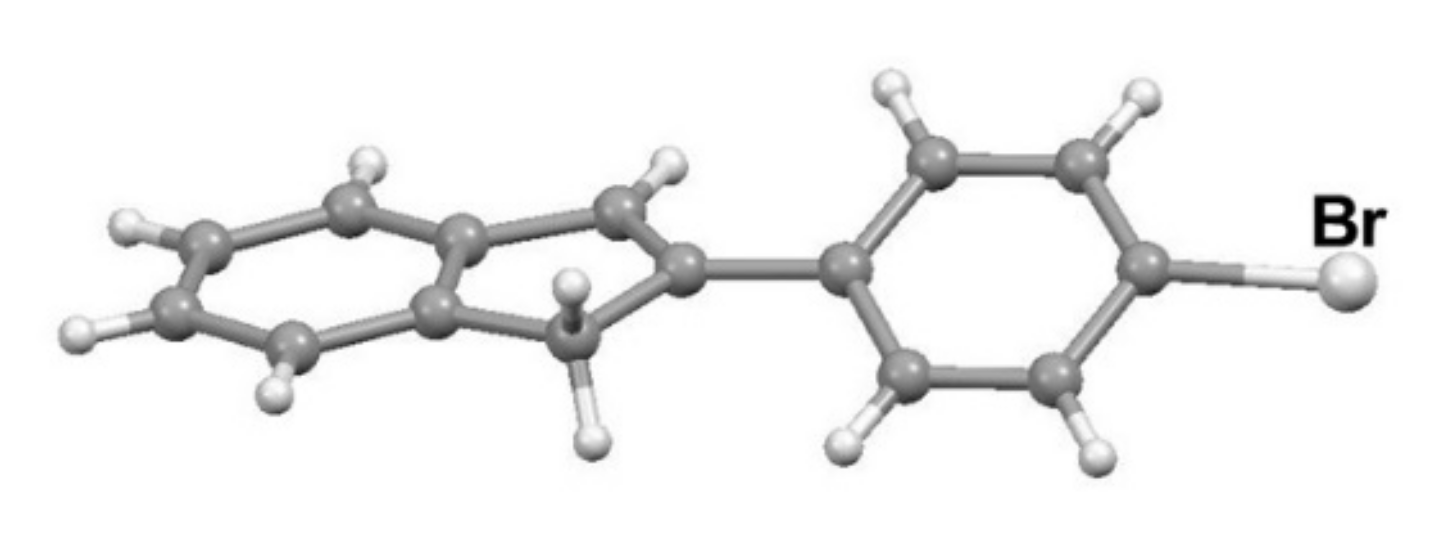
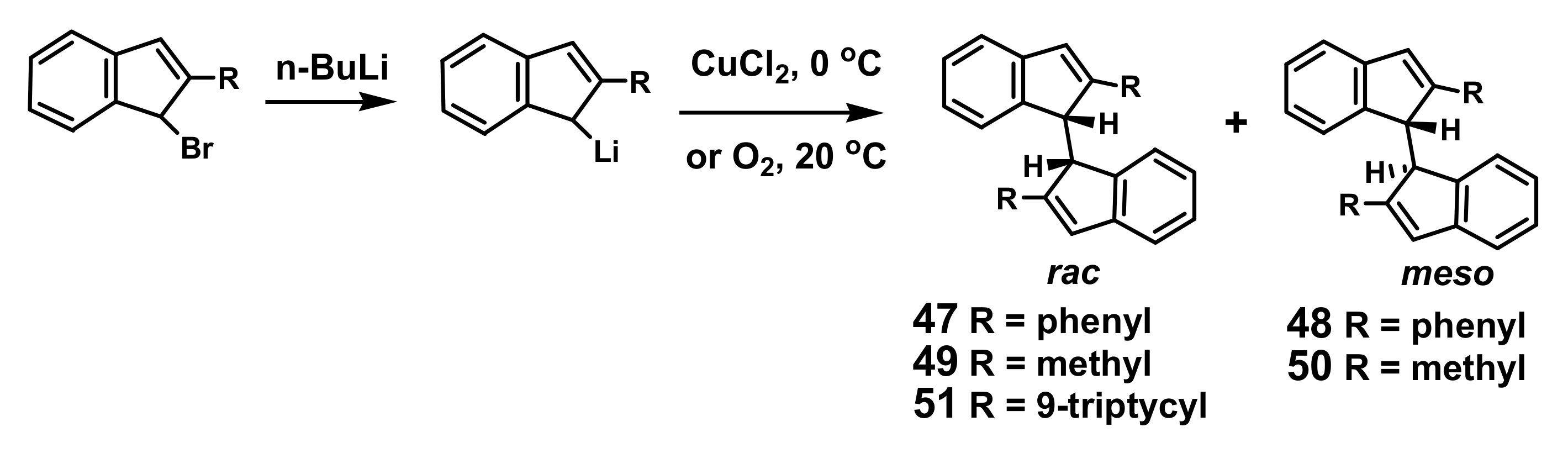
4.1. Cross-Coupling of 2-Phenyl- and 2-Methyl Indenes
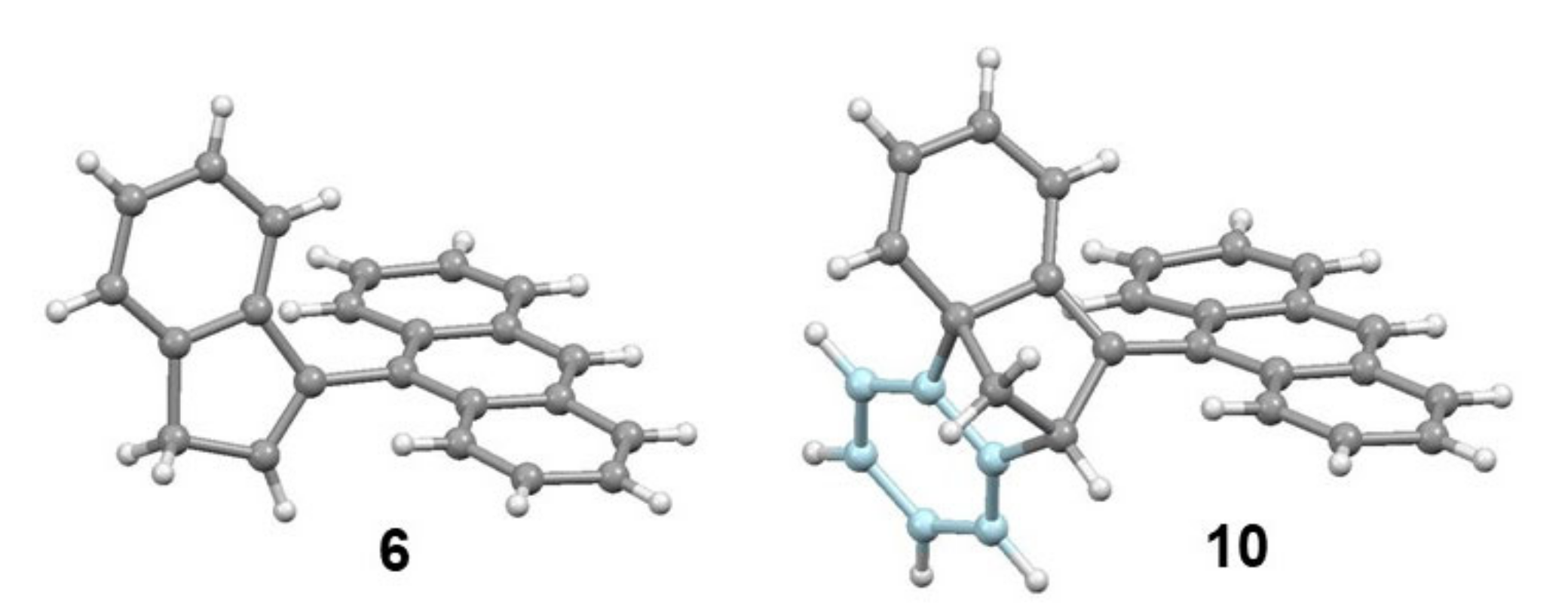
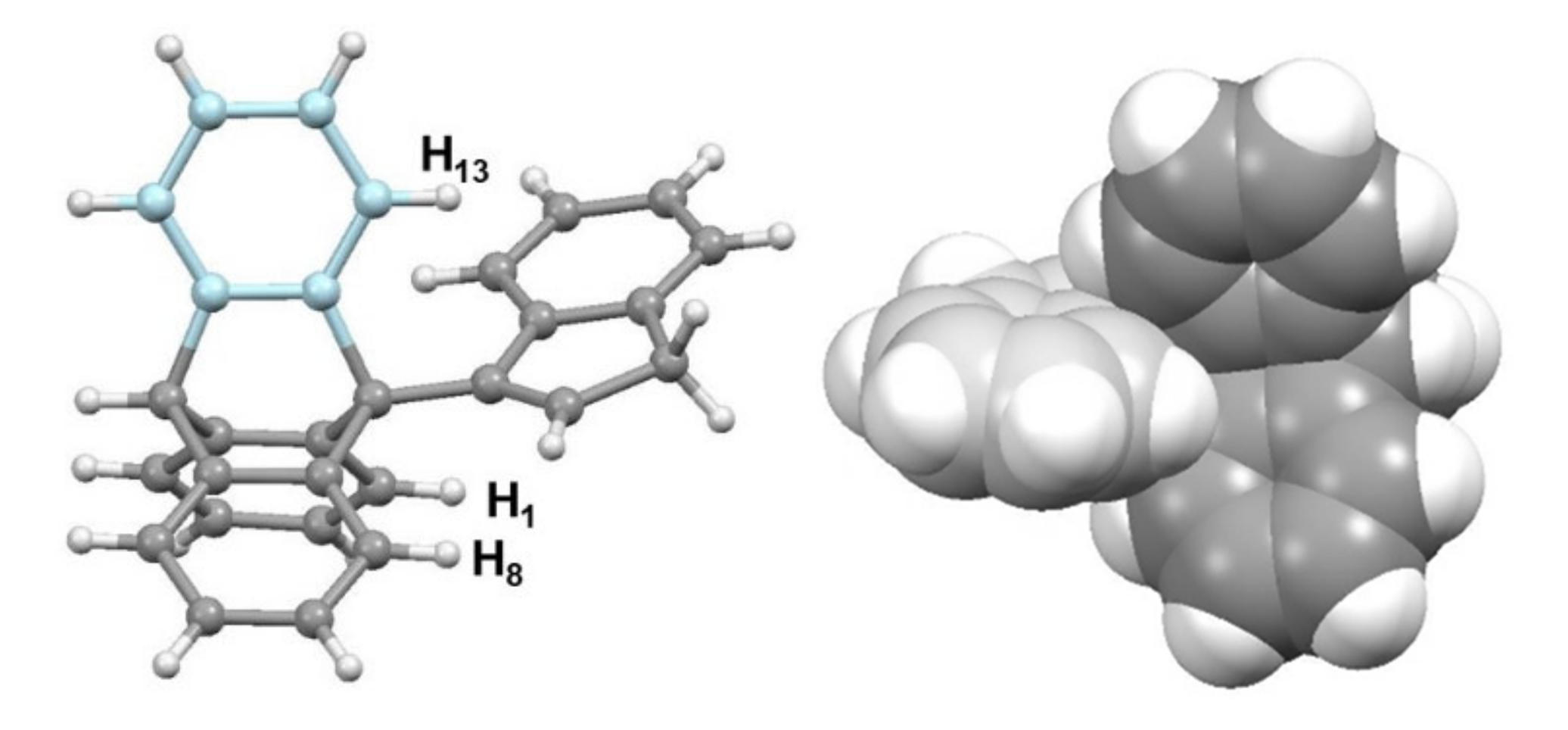
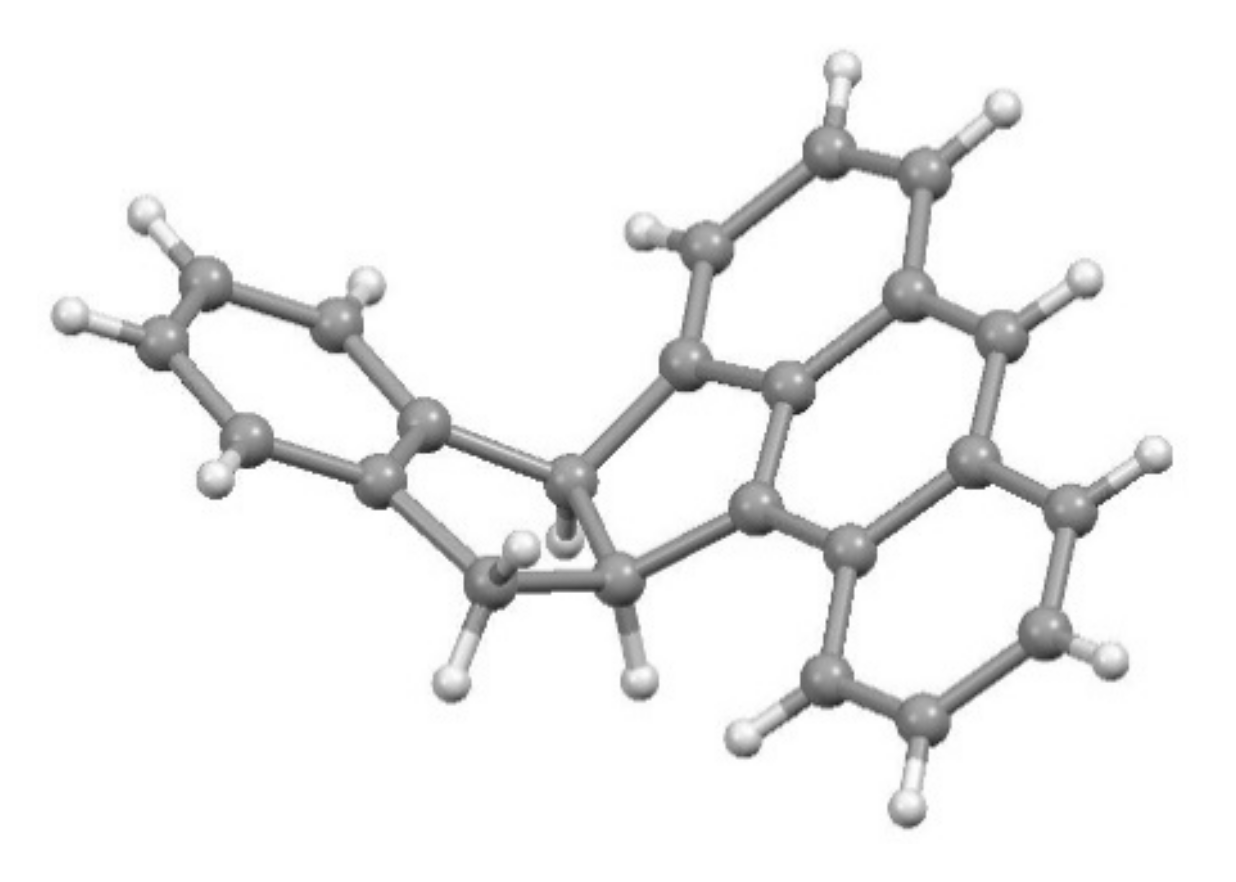
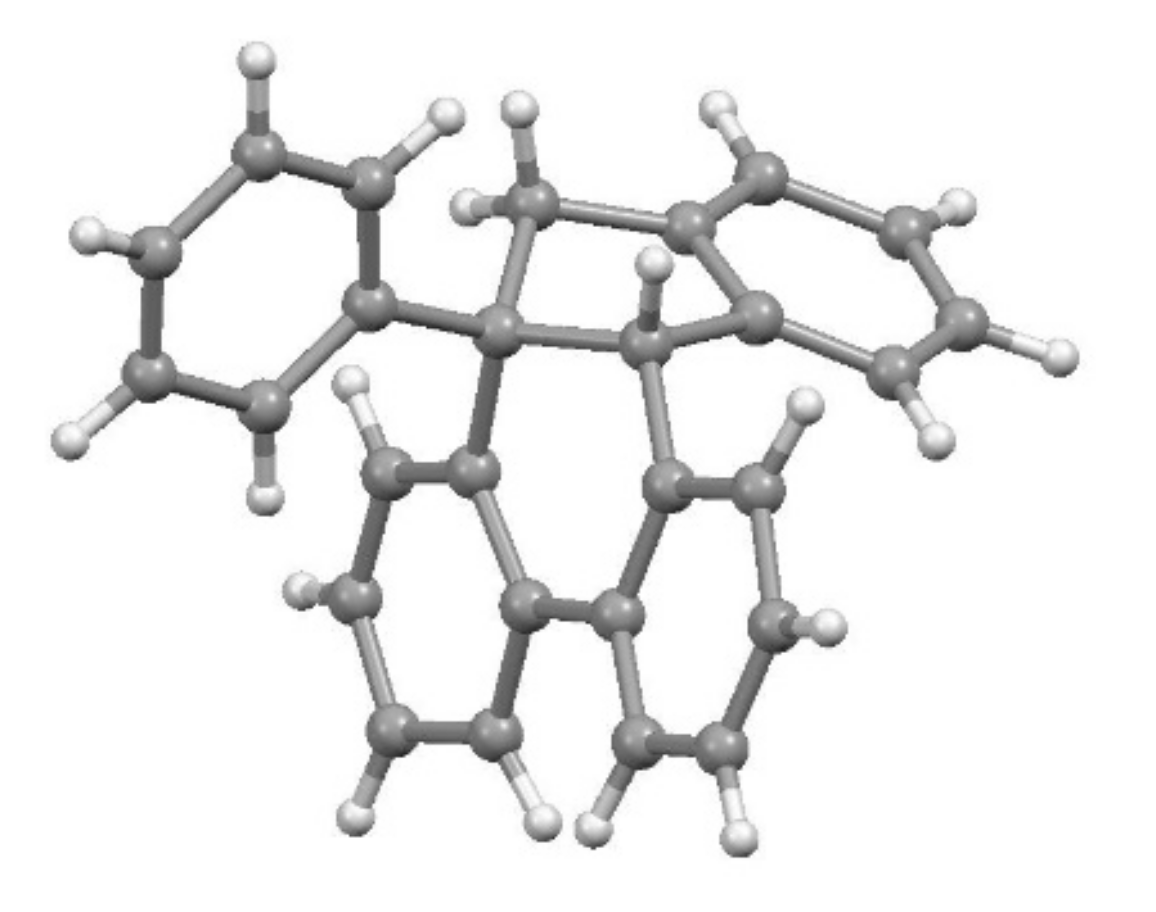
4.2. The Curious Case of the 2-Indenyltriptycene Dimer
5. Hindered Rotations in Phenyl-Anthracenes
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bruns, C.J.; Stoddart, J.F. The Nature of the Mechanical Bond; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Dattler, D.; Fuks, G.; Heiser, J.; Moulin, E.; Perrot, A.; Yao, X.; Giuseppone, N. Design of Collective Motions from Synthetic Molecular Switches, Rotors, and Motors. Chem. Rev. 2020, 120, 310–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negishi, E. Magical Power of Transition Metals: Past, Present, and Future (Nobel lecture). Angew. Chem. 2011, 50, 6738–6764. [Google Scholar] [CrossRef] [PubMed]
- Bandera, D.; Baldridge, K.K.; Linden, A.; Dorta, R.; Siegel, J.S. Stereoselective Coordination of C-5-Symmetric Corannulene Derivatives with an Enantiomerically Pure [Rh-I(nbd*)] Metal Complex. Angew. Chem. 2011, 50, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, H.; Hiraoka, Y.; Hiroto, S.; Sakamaki, D.; Seki, S.; Shinokubo, H. Nitrogen-embedded buckybowl and its assembly with C-60. Nat. Commun. 2015, 6, 8215–8224. [Google Scholar] [CrossRef] [Green Version]
- Yoon, I.; Benitez, D.; Zhao, Y.-L.; Miljanic, O.S.; Kim, S.-Y.; Tkatchouk, E.; Leung, K.C.-F.; Khan, S.I.; Goddard, W.A.; Stoddart, J.F. Functionally Rigid and Degenerate Molecular Shuttles Chem. Eur. J. 2009, 15, 1115–1122. [Google Scholar] [CrossRef]
- Lestini, E.; Nikitin, K.; Stolarczyk, J.; Fitzmaurice, D. Electron Transfer and Switching in Rigid [2]Rotaxanes Adsorbed on TiO2 Nanoparticles. Chem. Phys. Chem. 2012, 13, 797–810. [Google Scholar] [CrossRef]
- Nikitin, K.; Stolarczyk, J.; Lestini, E.; Müller-Bunz, H.; Fitzmaurice, D. Quantitative Conformational Study of Redox-Active [2]Rotaxanes, Part 2: Switching in Flexible and Rigid Bistable [2]Rotaxanes. Chem. Eur. J. 2008, 14, 1117–1128. [Google Scholar] [CrossRef]
- Zhu, K.; Baggi, G.; Loeb, S.J. Ring-through-ring molecular shuttling in a saturated [3]rotaxane. Nat. Chem. 2018, 10, 625–630. [Google Scholar] [CrossRef]
- Harrington, L.E.; Cahill, L.S.; McGlinchey, M.J. Toward an Organometallic Molecular Brake with a Metal Foot Pedal: Synthesis, Dynamic Behavior, and X-Ray Crystal Structure of [(9-Indenyl)-triptycene]chromium Tricarbonyl. Organometallics 2004, 23, 2884–2891. [Google Scholar] [CrossRef]
- Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M.J. Joining the rings: The preparation of 2- and 3-indenyl-triptycenes, and curious related processes. Org. Biomol. Chem. 2007, 5, 1952–1960. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Sitnikov, A.; Andriukhova, N.V.; Laishevtsev, I.P.; Luzikov, Y.N. A facile synthesis of 2-arylindenes by Pd-catalyzed direct arylation of indene with aryl iodides. Tetrahedron Lett. 2002, 43, 3213–3215. [Google Scholar] [CrossRef]
- Dang, H.; Garcia-Garibay, M.A. Palladium-Catalyzed Formation of Aceanthrylenes: A Simple Method for Cyclopentenelation of Aromatic Compounds. J. Am. Chem. Soc. 2001, 123, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.D.; Jellison, J.L.; Finke, A.D.; Wang, L.; Plunkett, K.N. Electron Acceptors Based on Functionalizable Cyclopenta[hi]aceanthrylenes and Dicyclopenta[de,mn]tetracenes. J. Am. Chem. Soc. 2012, 134, 15783–15789. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Plunkett, K.N. Orthogonal Functionalization of Cyclopenta[hi]aceanthrylenes. Org. Lett. 2013, 15, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Claus, T.K.; Telitel, S.; Welle, A.; Bastmeyer, M.; Vogt, A.P.; Delaittre, G.; Barner-Kowollik, C. Light-driven reversible surface functionalisation with anthracenes: Visible light writing and mild UV erasing. Chem. Commun. 2017, 53, 1599–1602. [Google Scholar] [CrossRef]
- Nikitin, K.; Müller-Bunz, H.; Guiry, P.J.; McGlinchey, M.J. A mechanistic rationale for the outcome of Sonogashira cross-coupling of 9-bromoanthracene and ethynyltrimethylsilane: An unexpected product 4-(9-anthracenyl)-1,3-bis(trimethylsilyl)-but-3-en-1-yne. J. Organometal. Chem. 2019, 880, 1–6. [Google Scholar] [CrossRef]
- Rogalski, S.; Kubicki, M.; Pietraszuk, G. Palladium catalysed regio and stereoselective synthesis of (E)-4-aryl-1,3-bis(trimethylsilyl)-but-3-en-1-yne. Tetrahedron 2018, 74, 6192–6198. [Google Scholar] [CrossRef]
- Nikitin, K.; Fleming, C.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M.J. Severe Energy Costs of Double Steric Interactions: Towards a Molecular Clamp. Eur. J. Org. Chem. 2010, 5203–5216. [Google Scholar] [CrossRef]
- Nikitin, K.; Bothe, C.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M.J. High and Low Rotational Barriers in Metal Tricarbonyl Complexes of 2- and 3-Indenyl Anthracenes and Triptycenes: Rational Design of Molecular Brakes. Organometallics 2012, 31, 6183–6198. [Google Scholar] [CrossRef]
- Nikitin, K.; Bothe, C.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M.J. A Molecular Paddle-wheel with a Sliding Organometallic Latch: Syntheses, X-Ray Crystal Structures and Dynamic Behaviour of [Cr(CO)3(η6-2-(9-triptycyl)indenene)] and of [M(CO)3(η5-2-(9- triptycyl)indenyl)] (M = Mn, Re). Chem. Eur. J. 2009, 15, 1836–1843. [Google Scholar] [CrossRef]
- Butler, I.R.; Hobson, L.J.; Coles, S.J.; Hursthouse, M.B.; Abdul Malik, K.M. Ferrocenyl anthracenes: Synthesis and molecular structure. J. Organometal. Chem. 1997, 540, 27–40. [Google Scholar] [CrossRef]
- Vives, G.; Gonzalez, A.; Jaud, J.; Launay, J.; Rappenne, G. Synthesis of Molecular Motors Incorporating para-Phenylene-Conjugated or Bicyclo[2.2.2]octane-Insulated Electroactive Groups. Chem. Eur. J. 2007, 13, 5622–5631. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; Muldoon, J.; McGlinchey, M.J. Molecular Dials: Hindered Rotations in Mono and Diferrocenyl Anthracenes and Triptycenes. J. Am. Chem. Soc. 2010, 132, 17617–17622. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, M.J.; Nikitin, K. Direct measurement of the diamagnetic anisotropy of the ferrocenyl moiety: The origin of the unusual 1H chemical shifts in ferrocenyl-triptycenes and barrelenes. J. Organometal. Chem. 2014, 751, 809–814. [Google Scholar] [CrossRef]
- Nikitin, K.; Muldoon, J.; Müller-Bunz, H.; McGlinchey, M.J. A Ferrocenyl Kaleidoscope: Slow Interconversion of Six Diastereomers of 2,6-Di-tert-butyl-9,10-diferrocenyltriptycene. Chem. Eur. J. 2015, 21, 4664–4670. [Google Scholar] [CrossRef] [PubMed]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe VII. Justus Liebigs Ann. Chem. 1931, 486, 191–202. [Google Scholar] [CrossRef]
- Klanderman, B.H.; Criswell, T.R. Reactivity of benzyne towards anthracene systems. J. Org. Chem. 1969, 34, 2430–3426. [Google Scholar] [CrossRef]
- Nikitin, K.; Müller-Bunz, H.; McGlinchey, M.J. Diels-Alder Reactions of 9-Ferrocenyl and 9,10-Diferrocenylanthracene: Steric control of 9,10- versus 1,4-Cycloaddition. Organometallics 2013, 32, 6118–6129. [Google Scholar] [CrossRef]
- Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M.J. Different Rearrangement Behaviour of the Cation or Anion Derived from the Diels-Alder Adduct of 9-Ferrocenylanthracene and 1,4-Benzoquinone: Ring-Opening or Paddlewheel Formation. Chem. Eur. J. 2011, 17, 14241–14247. [Google Scholar] [CrossRef]
- Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; Risse, W.; McGlinchey, M.J. Twin Triptycyl Spinning Tops: A Simple Case of Molecular Gearing with Dynamic C2 Symmetry. Eur. J. Org. Chem. 2008, 3079–3084. [Google Scholar] [CrossRef]
- Nori-shargha, D.; Asadzadeh, S.; Ghanizadeh, F.R.; Deyhimic, F.; Aminic, M.M.; Jameh-Bozorghi, S. Ab initio study of the structures and dynamic stereochemistry of biaryls. J. Mol. Struct. 2005, 717, 41–51. [Google Scholar] [CrossRef]
- Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; Muldoon, J.; McGlinchey, M.J. Restricted Rotation in 9-Phenylanthracenes: A Prediction Fulfilled. Org. Lett. 2011, 13, 256–259. [Google Scholar] [CrossRef] [PubMed]











































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGlinchey, M.J.; Nikitin, K. Palladium-Catalysed Coupling Reactions En Route to Molecular Machines: Sterically Hindered Indenyl and Ferrocenyl Anthracenes and Triptycenes, and Biindenyls. Molecules 2020, 25, 1950. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25081950
McGlinchey MJ, Nikitin K. Palladium-Catalysed Coupling Reactions En Route to Molecular Machines: Sterically Hindered Indenyl and Ferrocenyl Anthracenes and Triptycenes, and Biindenyls. Molecules. 2020; 25(8):1950. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25081950
Chicago/Turabian StyleMcGlinchey, Michael J., and Kirill Nikitin. 2020. "Palladium-Catalysed Coupling Reactions En Route to Molecular Machines: Sterically Hindered Indenyl and Ferrocenyl Anthracenes and Triptycenes, and Biindenyls" Molecules 25, no. 8: 1950. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25081950