
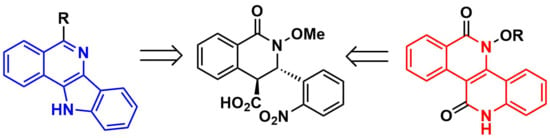
Two Annulated Azaheterocyclic Cores Readily Available from a Single Tetrahydroisoquinolonic Castagnoli–Cushman Precursor


Abstract
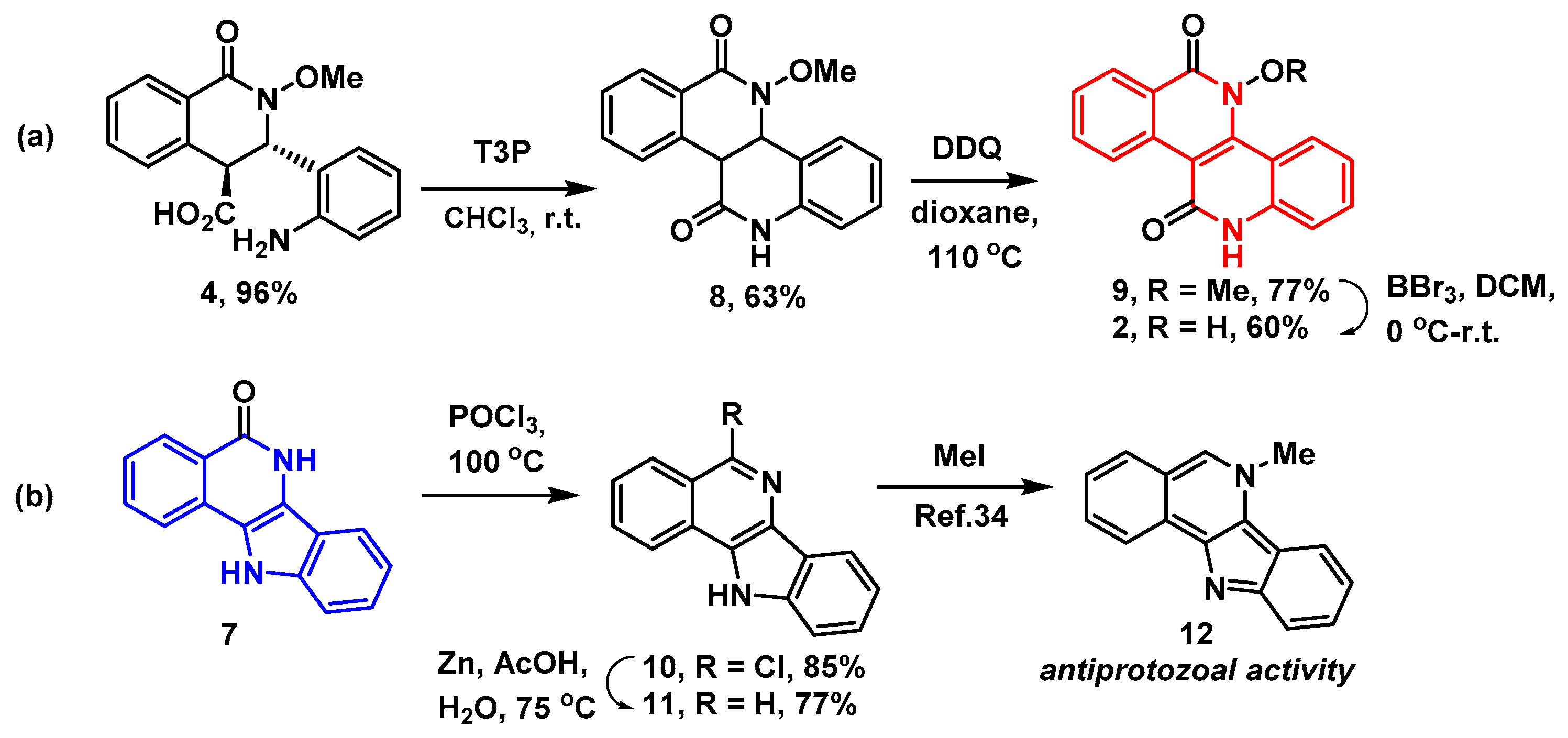
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
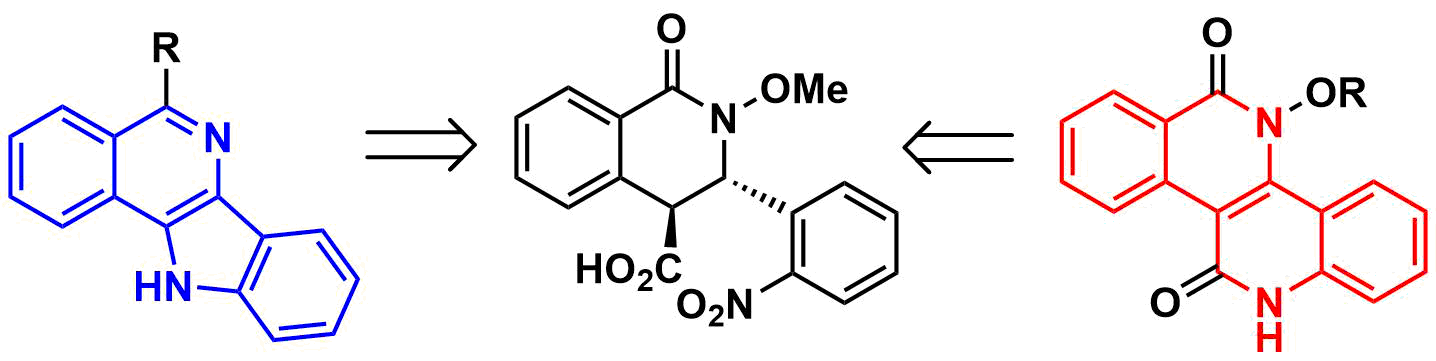
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis
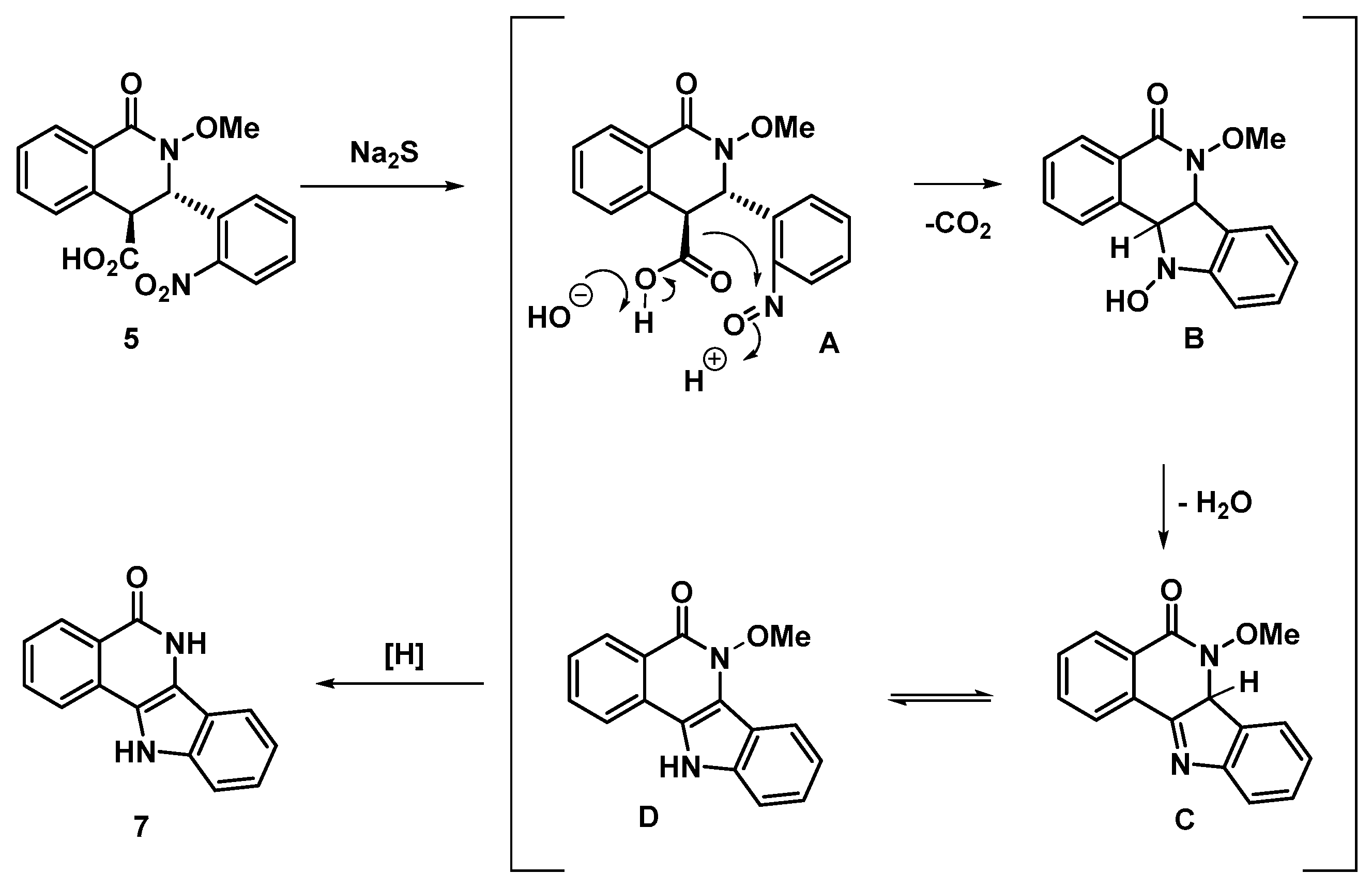
3.2.1. (±)-(3S,4S)-2-Methoxy-3-(2-nitrophenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (5)
3.2.2. 6,11-Dihydro-5H-indolo[3,2-c]isoquinolin-5-one (7)
3.2.3. (±)-(3S,4S)-2-Methoxy-3-(2-aminophenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (4)
3.2.4. 5-Methoxy-4b,12-dihydrodibenzo[c,h][1,6]naphthyridine-6,11(5H,10bH)-dione (8)
3.2.5. 5-Methoxydibenzo[c,h][1,6]naphthyridine-6,11(5H,12H)-dione (9)
3.2.6. 5-Hydroxydibenzo[c,h][1,6]naphthyridine-6,11(5H,12H)-dione (2)
3.2.7. 5-Chloro-11H-indolo[3,2-c]isoquinoline (10)
3.2.8. 11H-Indolo[3,2-c]isoquinoline (11)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gonzalez-Lopez, M.; Shaw, J.T. Cyclic anhydrides in formal cycloadditions and multicomponent reactions. Chem. Rev. 2009, 109, 164–189. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M.; Dar’in, D. Current diversity of cyclic anhydrides for the Castagnoli–Cushman-type formal cycloaddition reactions: Prospects and challenges. Tetrahedron Lett. 2016, 57, 1635–1640. [Google Scholar] [CrossRef]
- Cushman, M.; Dikshit, D.K. Formation of the 5-benzo[d]naphtho[2,3-b]pyran system during an attempted benzophenanthridine synthesis. J. Org. Chem. 1980, 45, 5064–5067. [Google Scholar] [CrossRef]
- Bakulina, O.; Bannykh, A.; Dar’in, D.; Krasavin, M. Cyclic Hydroxamic Acid Analogues of Bacterial Siderophores as Iron-Complexing Agents prepared through the Castagnoli-Cushman Reaction of Unprotected Oximes. Chem. Eur. J. 2017, 23, 17667–17673. [Google Scholar] [CrossRef]
- Abdein, M.A.; Abualreish, M.J.A. The Analytical Applications and Biological Activity of Hydroxamic acids. J. Adv. Chem. 2014, 10, 2118–2125. [Google Scholar]
- Birkett, J.E.; Carrott, M.J.; Fox, O.D.; Jones, C.J.; Maher, C.J.; Roube, C.V.; Taylor, R.J.; Woodhead, D.A. Controlling Neptunium and Plutonium within Single Cycle Solvent Extraction Flowsheets for Advanced Fuel Cycles. J. Nucl. Sci. Technol. 2007, 44, 337–343. [Google Scholar] [CrossRef]
- Natarajan, R. Hydroxamic Acids as Chelating Mineral Collectors. In Hydroxamic Acids; Gupta, S.P., Ed.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2013; pp. 281–307. [Google Scholar] [CrossRef]
- Ezznaydy, G.; Shaban, A.; Telegdi, J.; Ouaki, B.; El Hajjaji, S. Inhibition of copper corrosion in saline solution by mono-hydroxamic acid. J. Mater. Environ. Sci. 2015, 6, 1819–1823. [Google Scholar]
- Schalk, I.J.; Hannauer, M.; Braud, A. New roles for bacterial siderophores in metal transport and tolerance. Environ. Microbiol. 2011, 13, 2844–2854. [Google Scholar] [CrossRef]
- Kapranov, L.E.; Reznikov, A.N.; Klimochkin, Y.N. The Main Structural Types of Inhibitors of Matrix Metalloproteinases. Pharm. Chem. J. 2017, 51, 175–181. [Google Scholar] [CrossRef]
- Manal, M.; Chandrasekar, M.J.; Gomathi Priya, J.; Nanjan, M.J. Inhibitors of histone deacetylase as antitumor agents: A critical review. Bioorg. Chem. 2016, 67, 18–42. [Google Scholar] [CrossRef]
- Plewe, M.B.; Butler, S.L.; Dress, K.R.; Hu, Q.; Johnson, T.W.; Kuehler, J.E.; Kuki, A.; Lam, H.; Liu, W.; Nowlin, D.; et al. Azaindole hydroxamic acids are potent HIV-1 integrase inhibitors. J. Med. Chem. 2009, 52, 7211–7219. [Google Scholar] [CrossRef] [PubMed]
- Bakulina, O.; Rashevskii, A.; Dar’in, D.; Halder, S.; Khagar, P.; Krasavin, M. Modular Assembly of Tunable Fluorescent Chemosensors Selective for Pb2+ and Cu2+ Metal Ions via the Multicomponent Castagnoli-Cushman Reaction. ChemistrySelect 2019, 4, 6066–6073. [Google Scholar] [CrossRef]
- Kiselev, E.; Dexheimer, T.S.; Pommier, Y.; Cushman, M. Design, synthesis, and evaluation of dibenzo[c,h][1,6]naphthyridines as topoisomerase I inhibitors and potential anticancer agents. J. Med. Chem. 2010, 53, 8716–8726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrkvicka, V.; Klasek, A.; Kimmel, R.; Pevee, A.; Kosmrlj, J. Thermal reaction of 3aH,5H-thiazolo[5,4-c]quinoline-2,4-diones–an easy pathway to 4-amino-1H-quinolin-2-ones and novel 6H-thiazolo[3,4-c]quinazoline-3,5-diones. Arkivoc 2008, 2008, 289–302. [Google Scholar]
- Stadlbauer, W.; Kappe, T. Synthese von Indolen und Isochinolonen aus Phenylmalonylheterocyclen. Monatsh. Chem. 1984, 115, 467–475. [Google Scholar] [CrossRef]
- Delvaux, E. Cryptolepine. J. Pharm. Belg. 1931, 13, 955. [Google Scholar]
- Lavrado, J.; Moreira, R.; Paulo, A. Indoloquinolines as scaffolds for drug discovery. Curr. Med. Chem. 2010, 17, 2348–2370. [Google Scholar] [CrossRef]
- Parvatkar, P.T.; Parameswaran, P.S.; Tilve, S.G. Isolation, biological activities, and synthesis of indoloquinoline alkaloids: Cryptolepine, isocryptolepine, and neocryptolepine. Curr. Org. Chem. 2011, 15, 1036–1057. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.W. Recent developments in naturally derived antimalarials: Cryptolepine analogues. J. Pharm. Pharmacol. 2007, 59, 899–904. [Google Scholar] [CrossRef]
- Fodor, L.; Csomós, P.; Csámpai, A.; Sohár, P. Novel indole syntheses by ring transformation of β-lactam-condensed 1,3-benzothiazines into indolo[2,3-b][1,4]benzothiazepines and indolo[3,2-c]isoquinolines. Tetrahedron 2012, 68, 851–856. [Google Scholar] [CrossRef]
- Hu, Z.; Tong, X.; Liu, G. Rhodium(III)-Catalyzed Cascade Cyclization/Electrophilic Amidation for the Synthesis of 3-Amidoindoles and 3-Amidofurans. Org. Lett. 2016, 18, 2058–2061. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Fettinger, J.C.; Haddadin, M.J.; Kurth, M.J. Expedient one-pot synthesis of indolo[3,2-c]isoquinolines via a base-promoted N-alkylation/tandem cyclization. Tetrahedron Lett. 2015, 56, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Jagtap, P.G.; Baloglu, E.; Southan, G.; Williams, W.; Roy, A.; Nivorozhkin, A.; Landrau, N.; Desisto, K.; Salzman, A.L.; Szabo, C. Facile and convenient syntheses of 6,11-dihydro-5H-indeno[1,2-c]isoquinolin- 5-ones and 6,11-dihydro-5H-indolo[3,2-c]isoquinolin-5-one. Org. Lett. 2005, 7, 1753–1756. [Google Scholar] [CrossRef]
- Li, L.; Chua, W.K.S. One-pot multistep synthesis of 3,4-fused isoquinolin-1(2H)-one analogs. Tetrahedron Lett. 2011, 52, 1574–1577. [Google Scholar] [CrossRef]
- Chupakhin, E.; Bakulina, O.; Dar’in, D.; Krasavin, M. Facile Access to Fe(III)-Complexing Cyclic Hydroxamic Acids in a Three-Component Format. Molecules 2019, 24, 864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiremath, S.P.; Saundane, A.R.; Mruthyunjayaswamy, B.H.M. A new method for the synthesis of 6H,11H-indolo[3,2-c]-isoquinolin-5-ones/thiones and their reactions. J. Heterocycl. Chem. 1993, 30, 603–609. [Google Scholar] [CrossRef]
- Cope, O.J.; Brown, R.K. The Reduction of Nitrobenzene by Sodium Sulphide in Aqueous Ethanol. Can. J. Chem. 1961, 39, 1695–1710. [Google Scholar] [CrossRef]
- Kiselev, E.; Empey, N.; Agama, K.; Pommier, Y.; Cushman, M. Dibenzo[c,h][1,5]naphthyridinediones as topoisomerase I inhibitors: Design, synthesis, and biological evaluation. J. Org. Chem. 2012, 77, 5167–5172. [Google Scholar] [CrossRef] [Green Version]
- Nohira, H.; Sato, K.; Mukaiyama, T. The Reactions of Nitrosobenzene and Some Methylene Compounds. B. Chem. Soc. Jpn. 1963, 36, 870–872. [Google Scholar] [CrossRef]
- Payette, J.N.; Yamamoto, H. Nitrosobenzene-mediated C-C bond cleavage reactions and spectral observation of an oxazetidin-4-one ring system. J. Am. Chem. Soc. 2008, 130, 12276–12278. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhao, X.; Zhang-Negrerie, D.; Du, Y. Reductive cleavage of the N–O bond: Elemental sulfur-mediated conversion of N-alkoxyamides to amides. Org. Chem. Front. 2019, 6, 347–351. [Google Scholar] [CrossRef]
- Van Baelen, G.; Hostyn, S.; Dhooghe, L.; Tapolcsanyi, P.; Matyus, P.; Lemiere, G.; Dommisse, R.; Kaiser, M.; Brun, R.; Cos, P.; et al. Structure-activity relationship of antiparasitic and cytotoxic indoloquinoline alkaloids, and their tricyclic and bicyclic analogues. Bioorg. Med. Chem. 2009, 17, 7209–7217. [Google Scholar] [CrossRef]
- Van Baelen, G.; Meyers, C.; Lemière, G.L.F.; Hostyn, S.; Dommisse, R.; Maes, L.; Augustyns, K.; Haemers, A.; Pieters, L.; Maes, B.U.W. Synthesis of 6-methyl-6H-indolo[3,2-c]isoquinoline and 6-methyl-6H-indolo[2,3-c]isoquinoline: Two new unnatural isoquinoline isomers of the cryptolepine series. Tetrahedron 2008, 64, 11802–11809. [Google Scholar] [CrossRef]
- Martin, M.J.; Trudell, M.L.; Diaz Arauzo, H.; Allen, M.S.; LaLoggia, A.J.; Deng, L.; Schultz, C.A.; Tan, Y.C.; Bi, Y.; Narayanan, K.; et al. Molecular yardsticks. Rigid probes to define the spatial dimensions of the benzodiazepine receptor binding site. J. Med. Chem. 1992, 35, 4105–4117. [Google Scholar] [CrossRef] [PubMed]
- Béres, M.; Timári, G.; Hajós, G. Straightforward synthesis of 11H-indolo[3,2-c]isoquinoline and benzofuro[3,2-c]isoquinoline by ring transformation. Tetrahedron Lett. 2002, 43, 6035–6038. [Google Scholar] [CrossRef]
- Hayler, J.D.; Leahy, D.K.; Simmons, E.M. A Pharmaceutical Industry Perspective on Sustainable Metal Catalysis. Organometallics 2018, 38, 36–46. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Salerno, C.P.; Resat, M.; Magde, D.; Kraut, J. Synthesis of Caged NAD(P)+Coenzymes: Photorelease of NADP+. J. Am. Chem. Soc. 1997, 119, 3403–3404. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 4, 5 and 7 are available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karchuganova, E.; Bakulina, O.; Dar’in, D.; Krasavin, M. Two Annulated Azaheterocyclic Cores Readily Available from a Single Tetrahydroisoquinolonic Castagnoli–Cushman Precursor. Molecules 2020, 25, 2049. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092049
Karchuganova E, Bakulina O, Dar’in D, Krasavin M. Two Annulated Azaheterocyclic Cores Readily Available from a Single Tetrahydroisoquinolonic Castagnoli–Cushman Precursor. Molecules. 2020; 25(9):2049. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092049
Chicago/Turabian StyleKarchuganova, Elizaveta, Olga Bakulina, Dmitry Dar’in, and Mikhail Krasavin. 2020. "Two Annulated Azaheterocyclic Cores Readily Available from a Single Tetrahydroisoquinolonic Castagnoli–Cushman Precursor" Molecules 25, no. 9: 2049. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092049