
First Class of Phosphorus Dendritic Compounds Containing β-Cyclodextrin Units in the Periphery Prepared by CuAAC

 , , and
, , and 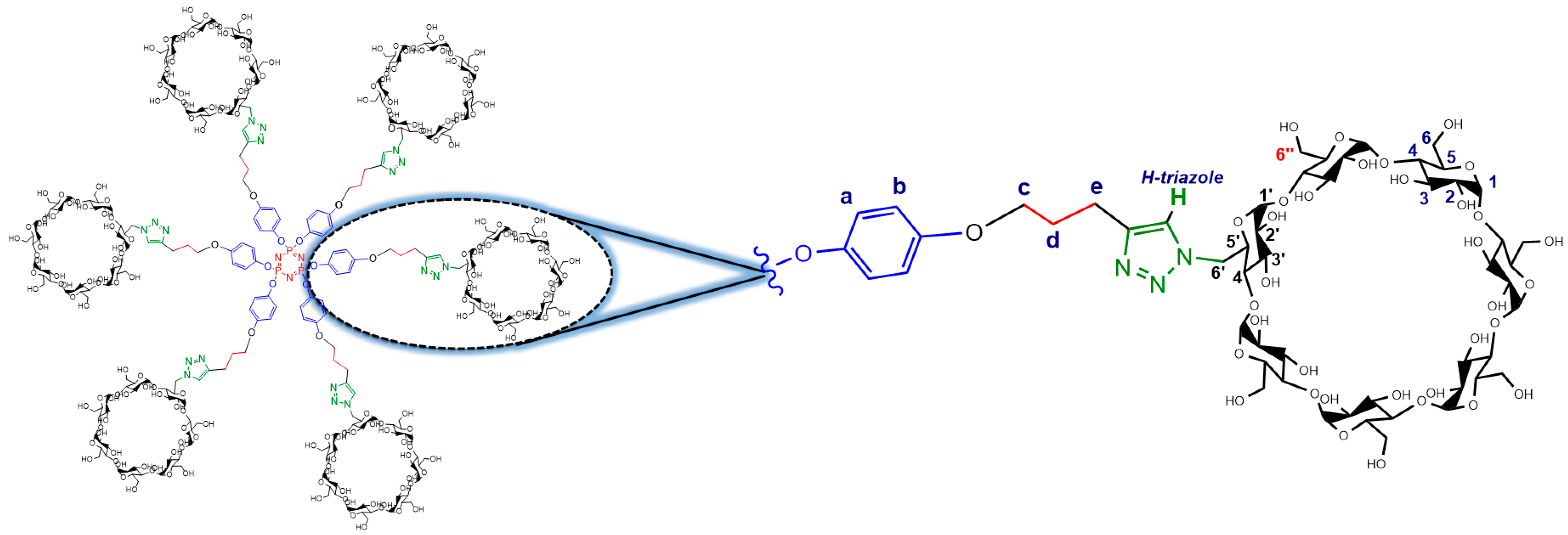{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
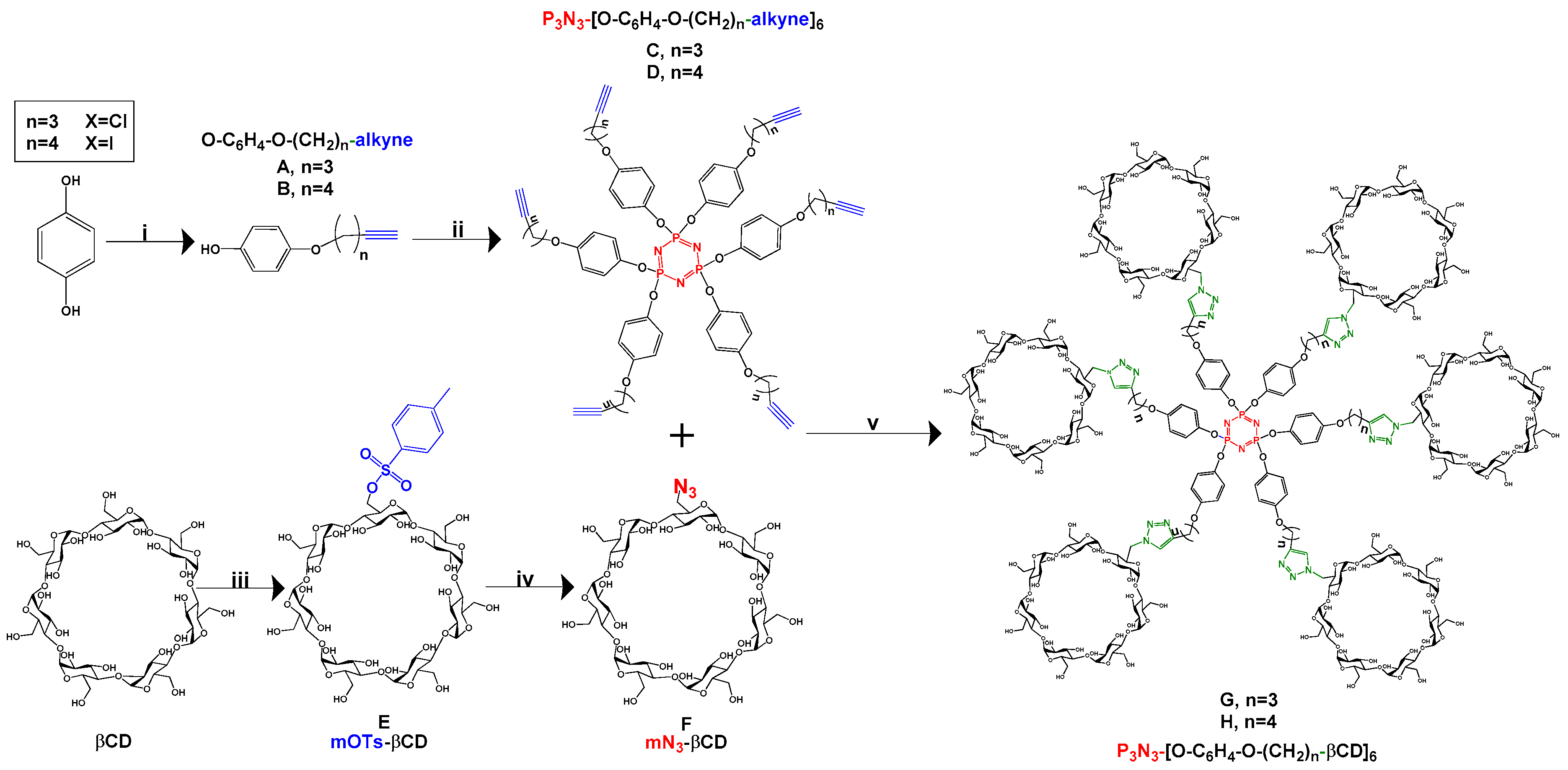
2.1. Synthesis
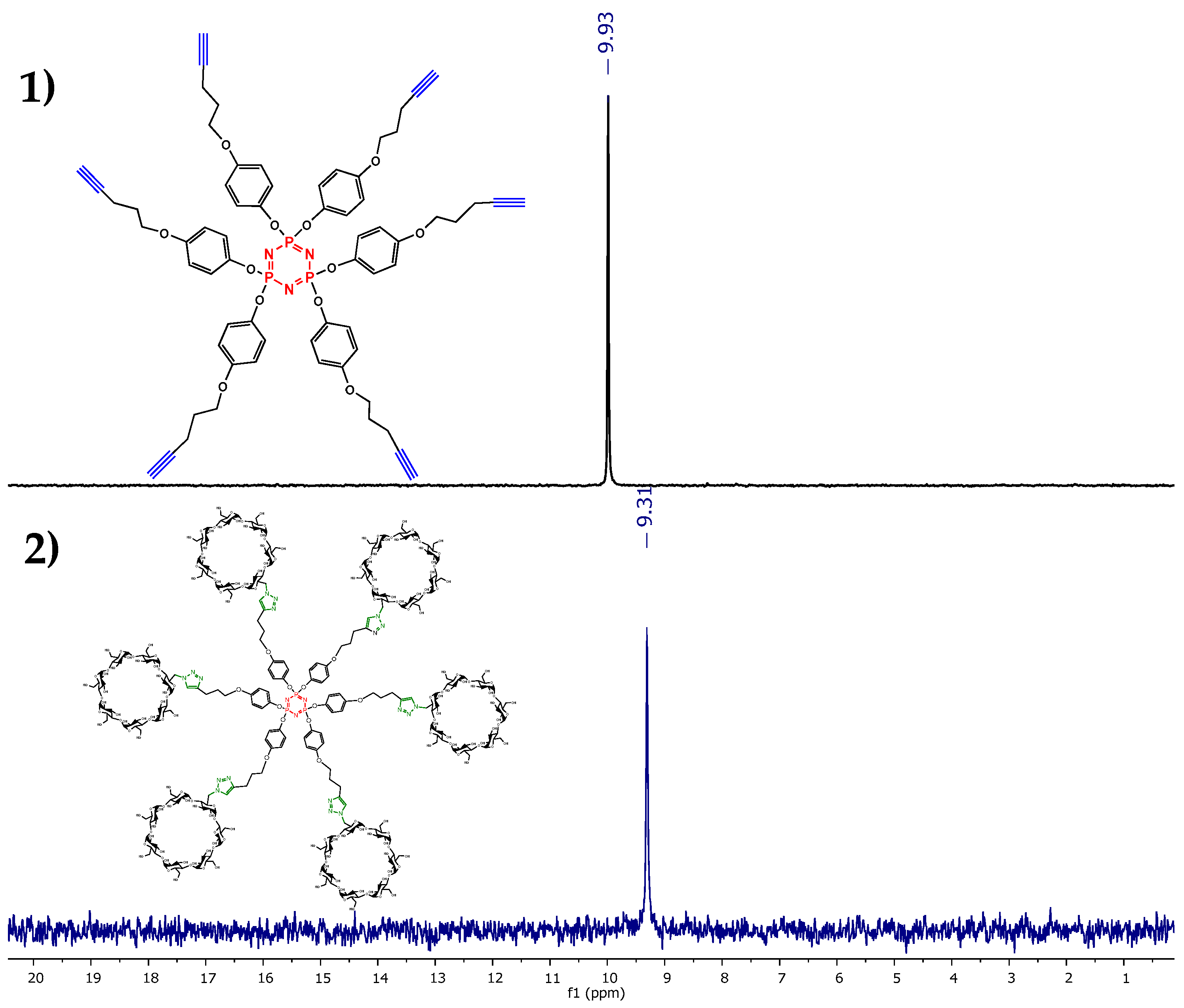
2.2. Characterization
2.3. Determination of Water Solubility for P3N3-(O-C6H4-O-(CH2)n-βCD)6 PDCs
3. Materials and Methods
3.1. General Notes
3.2. Synthetic Procedures
3.2.1. Synthesis of HO-C6H4-O-(CH2)n-alkyne (n = 3 and 4) A and B
3.2.2. Synthesis of P3N3-[O-C6H4-O-(CH2)n-alkyne]6 (n = 3 and 4) C and D
3.2.3. Synthesis of 6-O-monotosyl-β-cyclodextrin (mOTs-βCD) E
3.2.4. Synthesis of 6-O-monoazido-β-cyclodextrin (mN3-βCD) F
3.2.5. Synthesis of P3N3-[O-C6H4-O-(CH2)n-βCD]6 PDCs (n = 3 or 4) G and H
3.3. Characterization
3.4. Determination of Water Solubility for P3N3-[O-C6H4-O-(CH2)n-βCD]6 PDCs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Su, H.H.; Koo, J.M.M.; Cui, H. One-component nanomedicine. J. Control. Release. 2015, 219, 383–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, Y.; Kumar, A.; Tan, A.; Jin, S.; Mozhi, A.; Liang, X.J. pH-Sensitive nano-systems for drug delivery in cancer therapy. Biotechnol. Adv. 2014, 32, 693–710. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, P.; Jain, K.; Jain, N.K. Dendrimer as nanocarrier for drug delivery. Prog. Polym. Sci. 2014, 39, 268–307. [Google Scholar] [CrossRef]
- Tripathy, S.; Das, M.K. Dendrimers and their applications as novel drug delivery carriers. J. Appl. Pharm. Sci. 2013, 3, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Majoral, J.P.; Caminade, A.M.; Maraval, V. The specific contribution of phosphorus in dendrimer chemistry. Chem. Commun. 2002, 8, 2929–2942. [Google Scholar] [CrossRef]
- Sharma, A.K.; Gothwal, A.; Kesharwani, P.; Alsaab, H.; Iyer, A.K.; Gupta, U. Dendrimer nanoarchitectures for cancer diagnosis and anticancer drug delivery. Drug Discov. Today. 2017, 22, 314–326. [Google Scholar] [CrossRef]
- Cloninger, M.J. Biological applications of dendrimers. Curr. Opin. Chem. Biol. 2002, 6, 742–748. [Google Scholar] [CrossRef]
- Mignani, S.; El Kazzouli, S.; Bousmina, M.; Majoral, J.P. Dendrimer space concept for innovative nanomedicine: A futuristic vision for medicinal chemistry. Prog. Polym. Sci. 2013, 38, 993–1008. [Google Scholar] [CrossRef]
- Caminade, A.M.; Turrin, C.O.; Majoral, J.P. Biological properties of phosphorus dendrimers. New J. Chem. 2010, 34, 1512–1524. [Google Scholar] [CrossRef]
- Caminade, A.M. Phosphorus dendrimers for nanomedicine. Chem. Commun. 2017, 53, 9830–9838. [Google Scholar] [CrossRef] [PubMed]
- Launay, N.; Caminade, A.M.; Lahana, R.; Majoral, J.P. A General Synthetic Strategy for Neutral Phosphorus-Containing Dendrimers. Angew. Chem. Int. Ed. Engl. 1994, 33, 1589–1592. [Google Scholar] [CrossRef]
- Sebastián, R.M.; Magro, G.; Caminade, A.M.; Majoral, J.P. Dendrimers with N,N-disubstituted hydrazines as end groups, useful precursors for the synthesis of water-soluble dendrimers capped with carbohydrate, carboxylic or boronic acid derivatives. Tetrahedron 2000, 56, 6269–6277. [Google Scholar] [CrossRef]
- Hadad, C.; Majoral, J.P.; Muzart, J.; Caminade, A.M.; Bouquillon, S. First phosphorous D-xylose-derived glycodendrimers. Tetrahedron Lett. 2009, 50, 1902–1905. [Google Scholar] [CrossRef]
- Blattes, E.; Vercellone, A.; Eutamène, H.; Turrin, C.O.; Théodorou, V.; Majoral, J.P.; Caminade, A.M.; Prandi, J.; Nigou, J.; Puzo, G. Mannodendrimers prevent acute lung inflammation by inhibiting neutrophil recruitment. Proc. Natl. Acad. Sci. USA 2013, 110, 8795–8800. [Google Scholar] [CrossRef] [Green Version]
- Touaibia, M.; Roy, R. First synthesis of “majoral-type” glycodendrimers bearing covalently bound α-D-mannopyranoside residues onto a hexachlocyclotriphosphazene core. J. Org. Chem. 2008, 73, 9292–9302. [Google Scholar] [CrossRef]
- Blanzat, M.; Turrin, C.O.; Perez, E.; Rico-Lattes, I.; Caminade, A.M.; Majoral, J.P. Phosphorus-containing dendrimers bearing galactosylceramide analogs: Self-assembly properties. Chem. Commun. 2002, 1864–1865. [Google Scholar] [CrossRef]
- González-Méndez, I.; Hameau, A.; Laurent, R.; Bijani, C.; Bourdon, V.; Caminade, A.M.; Rivera, E.; Moineau-Chane Ching, K.I. β-Cyclodextrin PAMAM Dendrimer: How to Overcome the Tumbling Process for Getting Fully Available Host Cavities. Eur. J. Org. Chem. 2020, 1114–1121. [Google Scholar] [CrossRef]
- Arima, H.; Kihara, F.; Hirayama, F.; Uekama, K. Enhancement of gene expression by polyamidoamine dendrimer conjugates with α-, β-, and γ-cyclodextrins. Bioconjug. Chem. 2001, 12, 476–484. [Google Scholar] [CrossRef]
- Abdelwahab, A.F.; Ohyama, A.; Higashi, T.; Motoyama, K.; Khaled, K.A.; Sarhan, H.A.; Hussein, A.K.; Arima, H. Preparation and evaluation of polyamidoamine dendrimer conjugate with glucuronylglucosyl-β-cyclodextrin (G3) as a novel carrier for siRNA. J. Drug Target. 2014, 22, 927–934. [Google Scholar] [CrossRef]
- Arima, H.; Motoyama, K.; Higashi, T. Sugar-appended polyamidoamine dendrimer conjugates with cyclodextrins as cell-specific non-viral vectors. Adv. Drug Deliv. Rev. 2013, 65, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Crini, G.; Fourmentin, S.; Fenyvesi, É.; Torri, G.; Fourmentin, M.; Morin-Crini, N. Fundamentals and Applications of Cyclodextrins. In Cyclodextrin Fundamentals. Reactivity and Analysis; Crini, G., Fourmentinn, S., Lichtfouse, E., Eds.; Springer: Cham, Switzerland, 2018; Volume 1, pp. 1–57. [Google Scholar] [CrossRef]
- Tian, B.; Liu, Y.; Liu, J. Cyclodextrin as a magic switch in covalent and non-covalent anticancer drug release systems. Carbohydr. Polym. 2020, 242, 116401. [Google Scholar] [CrossRef]
- Zhang, D.; Lv, P.; Zhou, C.; Zhao, Y.; Liao, X.; Yang, B. Cyclodextrin-based delivery systems for cancer treatment. Mater. Sci. Eng. C. 2019, 96, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.P.; da Silva Santos, S.; da Silva, J.V.; Parise-Filho, R.; Ferreira, E.I.; El Seoud, O.; Giarolla, J. Dendrimers in the context of nanomedicine. Int. J. Pharm. 2020, 573, 118814. [Google Scholar] [CrossRef] [PubMed]
- Aragão-Leoneti, V.; Campo, V.L.; Gomes, A.S.; Field, R.A.; Carvalho, I. Application of copper(I)-catalysed azide/alkyne cycloaddition (CuAAC) “click chemistry” in carbohydrate drug and neoglycopolymer synthesis. Tetrahedron 2010, 66, 9475–9492. [Google Scholar] [CrossRef]
- Ting, C.H.; Chen, J.T.; Hsu, C.S. Synthesis and thermal and photoluminescence properties of liquid crystalline polyacetylenes containing 4-alkanyloxyphenyl trans-4-alkylcyclohexanoate side groups. Macromolecules 2002, 35, 1180–1189. [Google Scholar] [CrossRef]
- Cavero, E.; Zablocka, M.; Caminade, A.M.; Majoral, J.P. Design of bisphosphonate-terminated dendrimers. Eur. J. Org. Chem. 2010, 14, 2759–2767. [Google Scholar] [CrossRef]
- Zhong, N.; Byun, H.S.; Bittman, R. An improved synthesis of 6-O-monotosyl-6-deoxy-β-cyclodextrin. Tetrahedron Lett. 1998, 39, 2919–2920. [Google Scholar] [CrossRef]
- Tripodo, G.; Wischke, C.; Neffe, A.T.; Lendlein, A. Efficient synthesis of pure monotosylated beta-cyclodextrin and its dimers. Carbohydr. Res. 2013, 381, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Yousef, T.; Hassan, N.; Akbar, E.A. Synthesis of the dendritic type β-cyclodextrin on primary face via click reaction applicable as drug nanocarrier. Carbohydr. Polym. 2015, 132, 205–213. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.; Hu, J.; Li, C.; Liu, S. Multi-responsive supramolecular double hydrophilic diblock copolymer driven by host-guest inclusion complexation between β-cyclodextrin and adamantyl moieties. Macromol. Chem. Phys. 2009, 210, 2125–2137. [Google Scholar] [CrossRef]
- Caminade, A.M.; Laurent, R.; Turrin, C.O.; Rebout, C.; Delavaux-Nicot, B.; Ouali, A.; Zablocka, M.; Majoral, J.P. Phosphorus dendrimers as viewed by 31P NMR spectroscopy; Synthesis and characterization. C. R. Chim. 2010, 13, 1006–1027. [Google Scholar] [CrossRef]
- Blais, J.C.; Turrin, C.O.; Caminade, A.M.; Majoral, J.P. MALDI TOF mass spectrometry for the characterization of phosphorus-containing dendrimers. Scope and limitations. Anal. Chem. 2000, 72, 5097–5105. [Google Scholar] [CrossRef] [PubMed]
- Turrin, C.O.; Maraval, V.; Leclaire, J.; Dantras, E.; Lacabanne, C.; Caminade, A.M.; Majoral, J.P. Surface, core, and structure modifications of phosphorus-containing dendrimers. Influence on the thermal stability. Tetrahedron 2003, 59, 3965–3973. [Google Scholar] [CrossRef] [Green Version]
- Caminade, A.M.; Majoral, J.P. Water-soluble phosphorus-containing dendrimers. Prog. Polym. Sci. 2005, 30, 491–505. [Google Scholar] [CrossRef]
- Jozwiakowski, M.J.; Connors, K.A. Aqueous solubility behavior of three cyclodextrins. Carbohydr. Res. 1985, 143, 51–59. [Google Scholar] [CrossRef]
- Loftsson, T.; Jarho, P.; Másson, M.; Järvinen, T. Cyclodextrins in drug delivery. Expert Opin. Drug Deliv. 2005, 2, 335–351. [Google Scholar] [CrossRef]
- Folgado, E.; Guerre, M.; Bijani, C.; Ladmiral, V.; Caminade, A.M.; Ameduri, B.; Ouali, A. Well-defined poly(vinylidene fluoride) (PVDF) based-dendrimers synthesized by click chemistry: Enhanced crystallinity of PVDF and increased hydrophobicity of PVDF films. Polym. Chem. 2016, 7, 5625–5629. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorroza-Martínez, K.; González-Méndez, I.; Vonlanthen, M.; Moineau-Chane Ching, K.I.; Caminade, A.-M.; Illescas, J.; Rivera, E. First Class of Phosphorus Dendritic Compounds Containing β-Cyclodextrin Units in the Periphery Prepared by CuAAC. Molecules 2020, 25, 4034. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25184034
Sorroza-Martínez K, González-Méndez I, Vonlanthen M, Moineau-Chane Ching KI, Caminade A-M, Illescas J, Rivera E. First Class of Phosphorus Dendritic Compounds Containing β-Cyclodextrin Units in the Periphery Prepared by CuAAC. Molecules. 2020; 25(18):4034. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25184034
Chicago/Turabian StyleSorroza-Martínez, Kendra, Israel González-Méndez, Mireille Vonlanthen, Kathleen I. Moineau-Chane Ching, Anne-Marie Caminade, Javier Illescas, and Ernesto Rivera. 2020. "First Class of Phosphorus Dendritic Compounds Containing β-Cyclodextrin Units in the Periphery Prepared by CuAAC" Molecules 25, no. 18: 4034. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25184034