
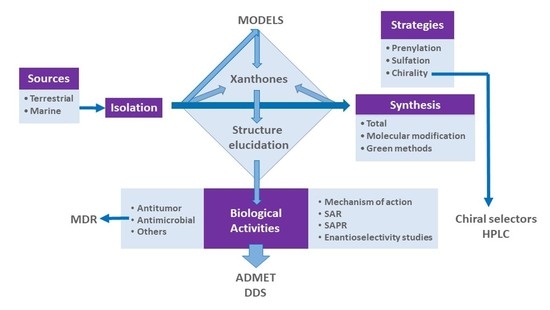
From Natural Products to New Synthetic Small Molecules: A Journey through the World of Xanthones

 , , ,
, , ,  , ,
, ,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
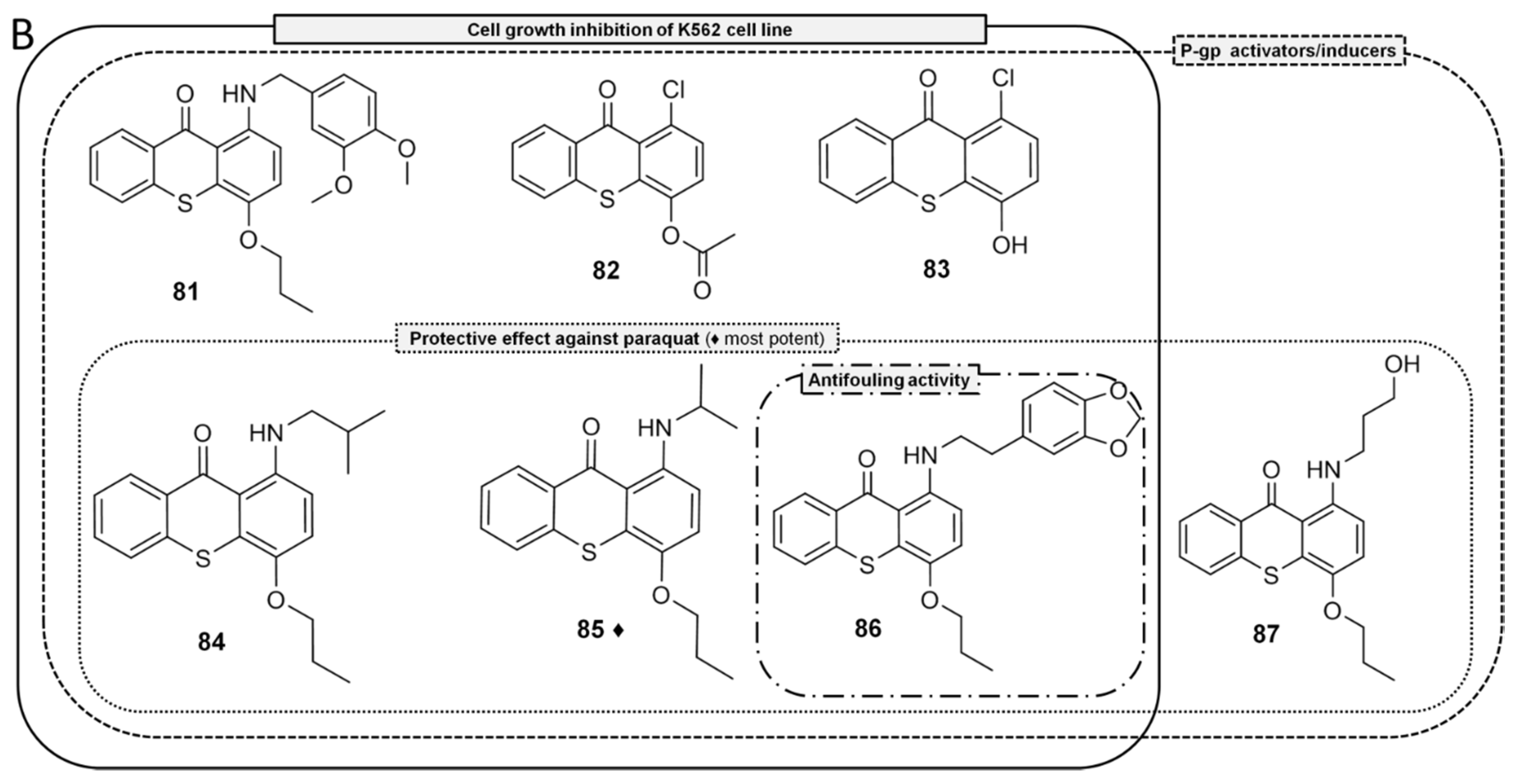{kind=link}
{kind=link}
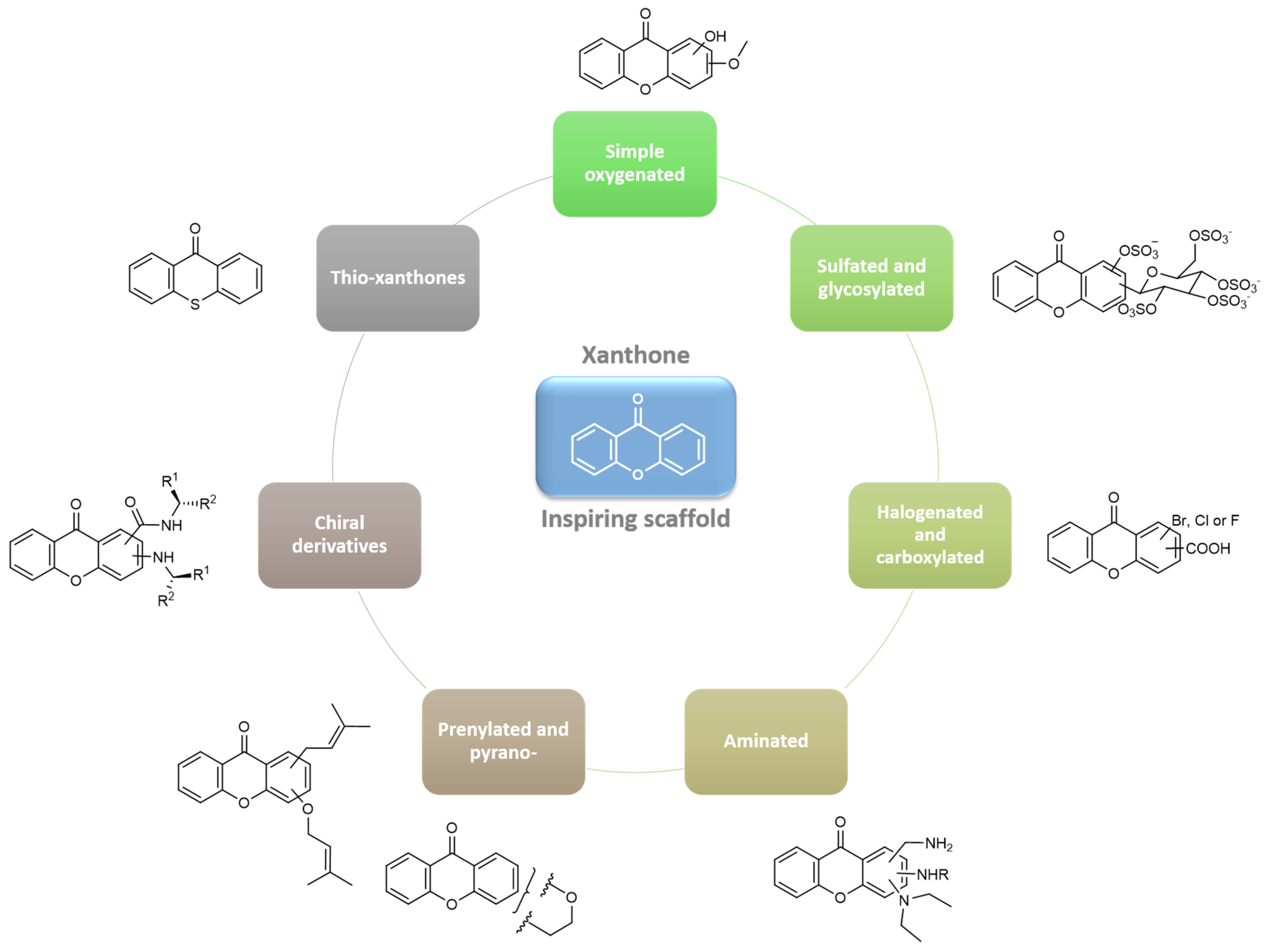{kind=link}
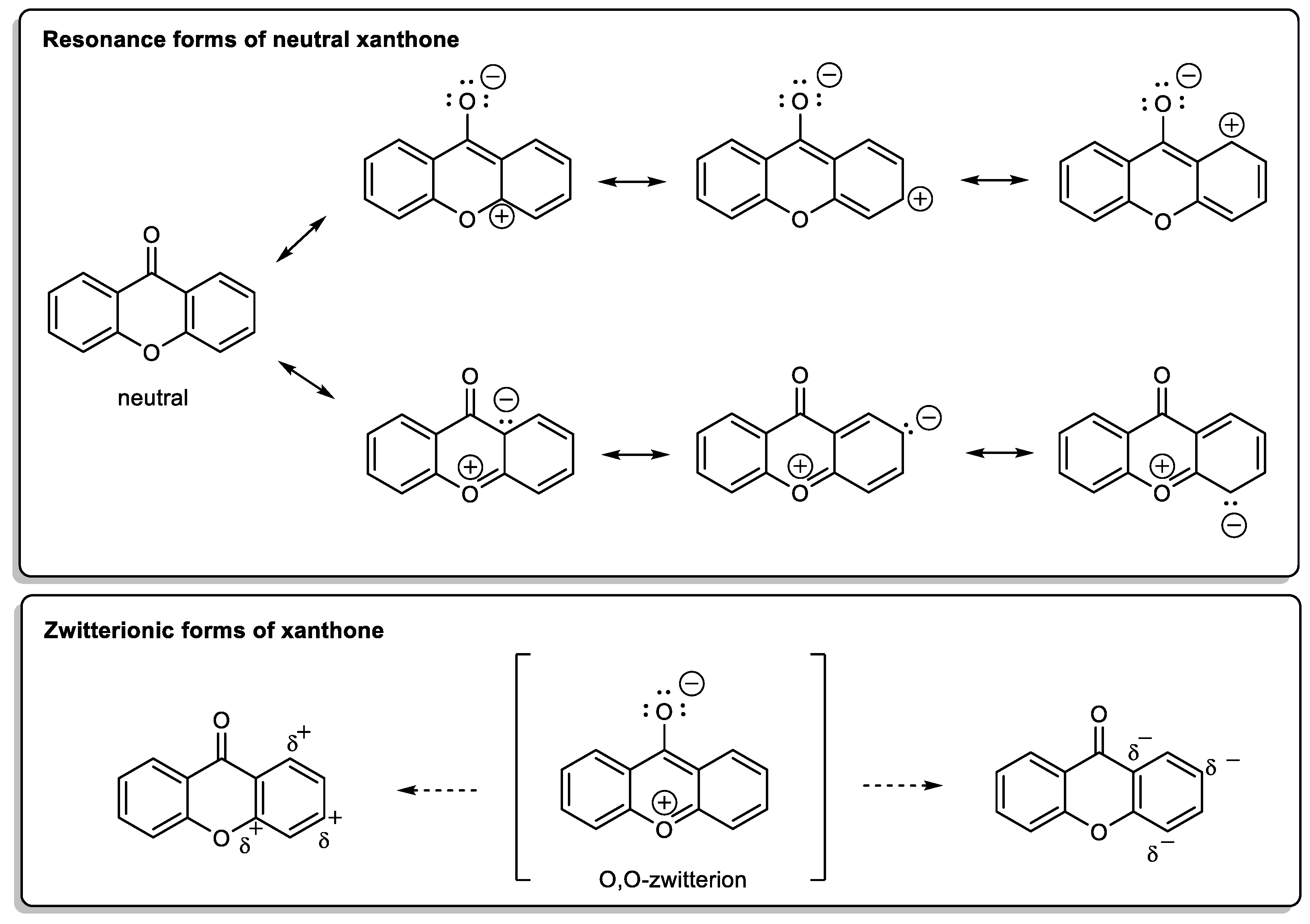{kind=link}
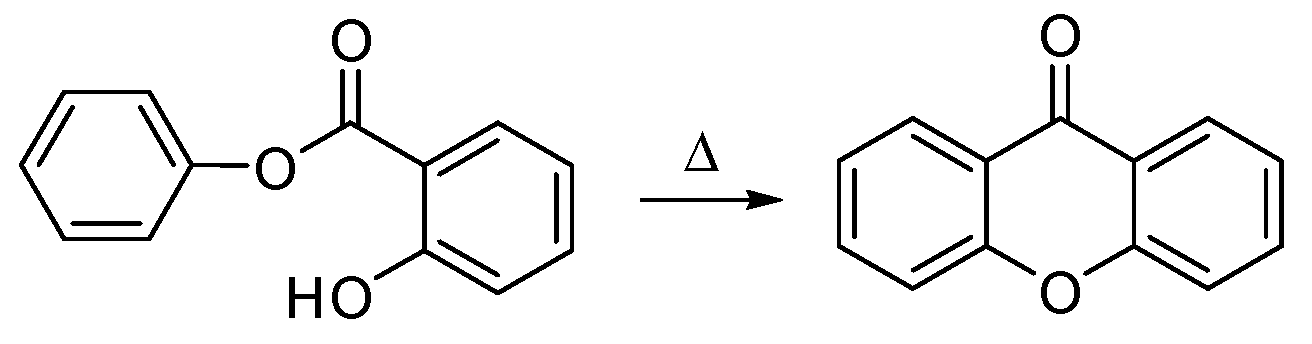{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Why Choose “Xanthone Derivatives”?
1.1.1. Xanthone: The Molecule
1.1.2. A New Molecule Was Born from the Lab and from the Nature
1.1.3. Xanthone: A Privileged Scaffold
2. A Library of Natural Mimetic Xanthones Looking for Biological Diversity: From the Land and from the Sea
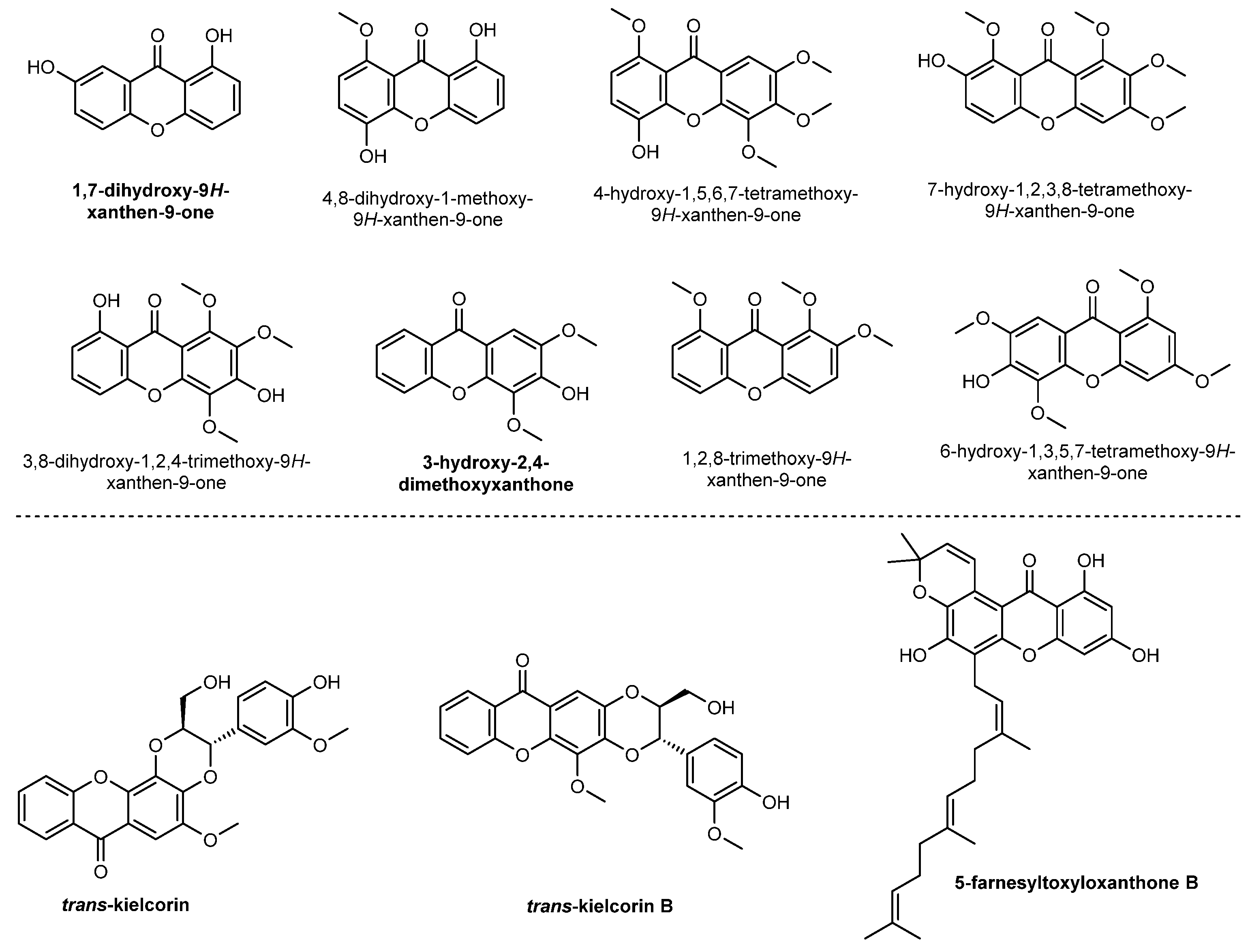
2.1. Simple Oxygenated Xanthones
2.2. Prenylated Xanthones
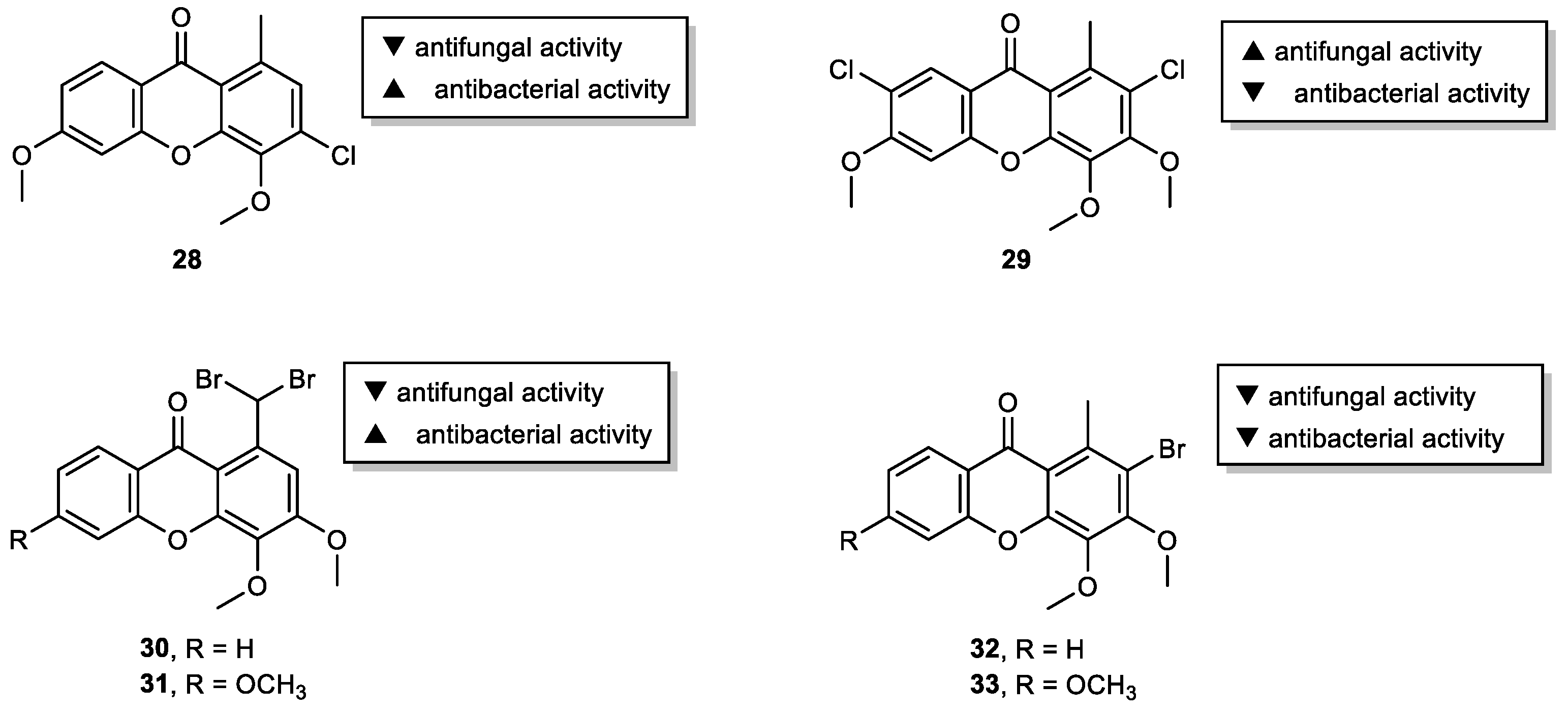
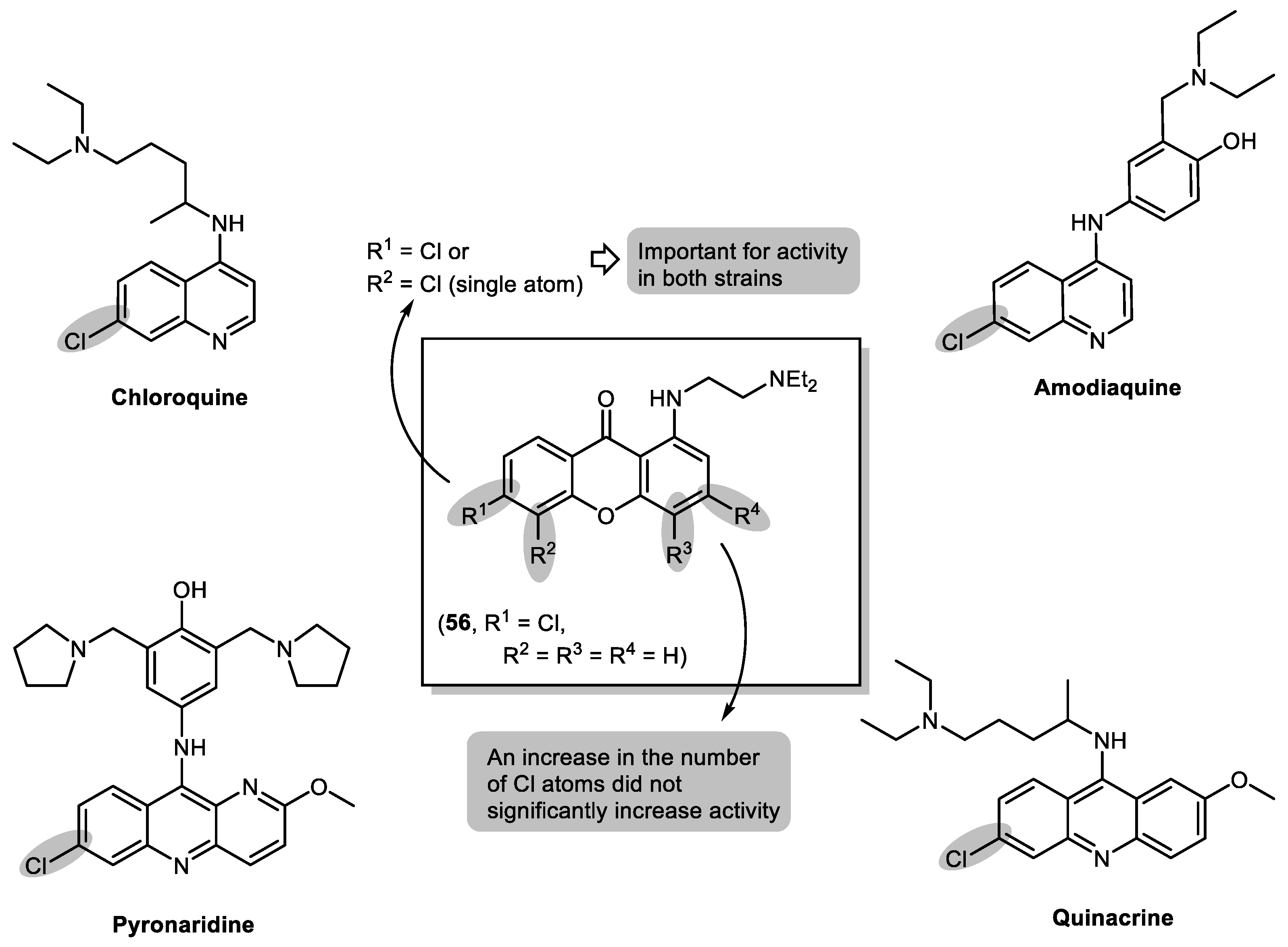
2.3. Halogenated and Carboxyxanthones
2.4. Aminated Xanthones
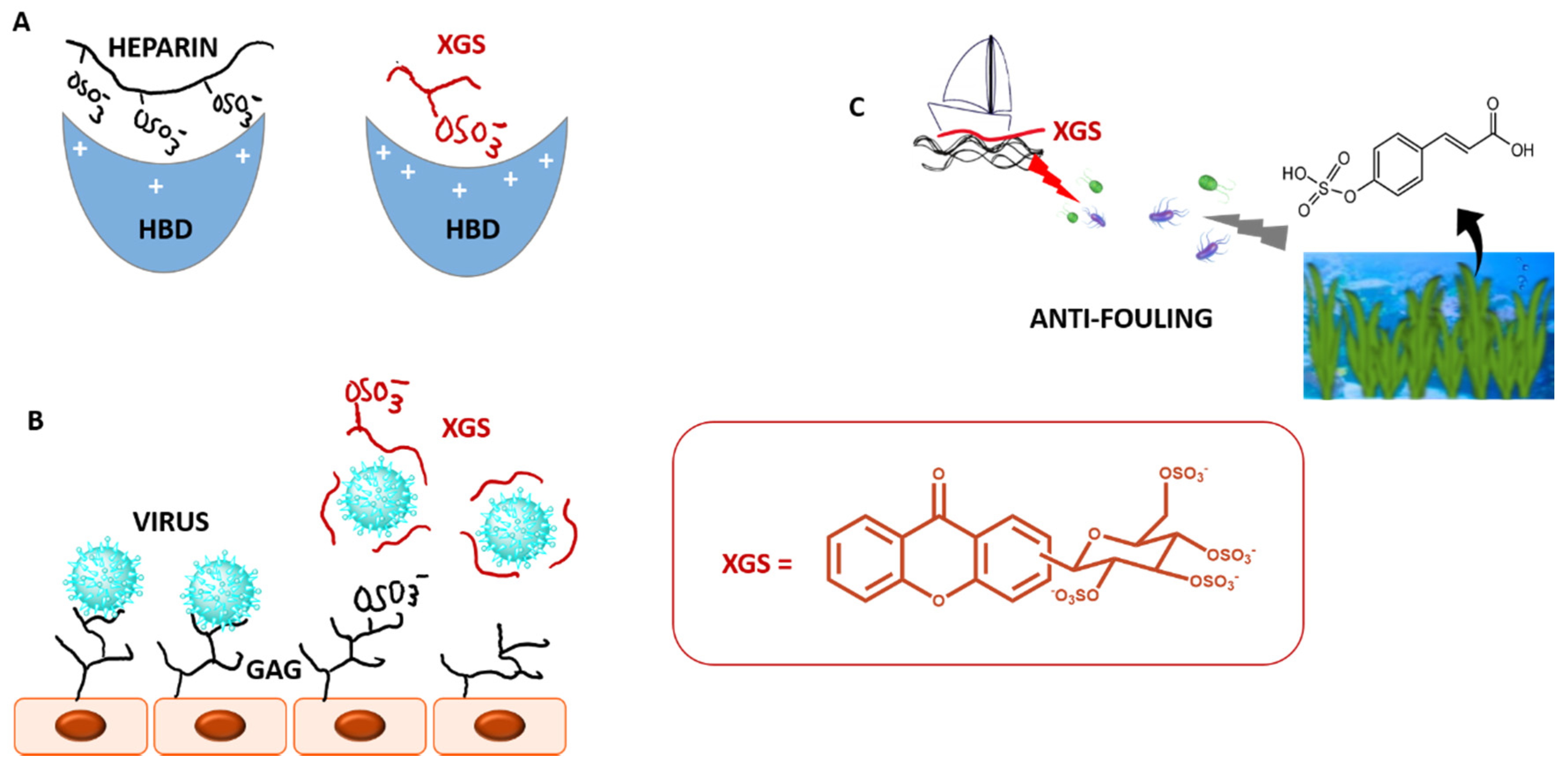
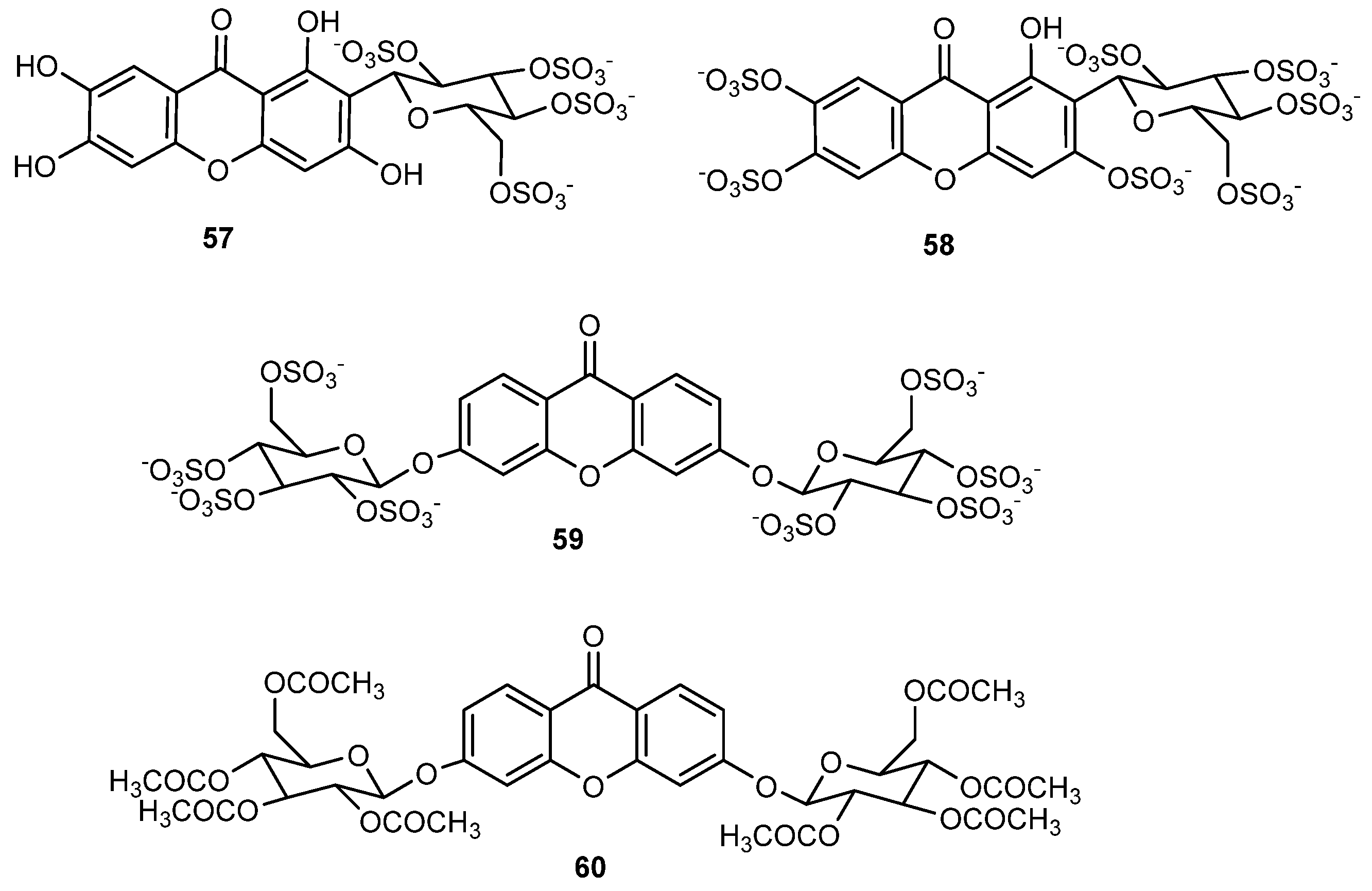
2.5. Sulfated and/or Glycosylated Xanthones
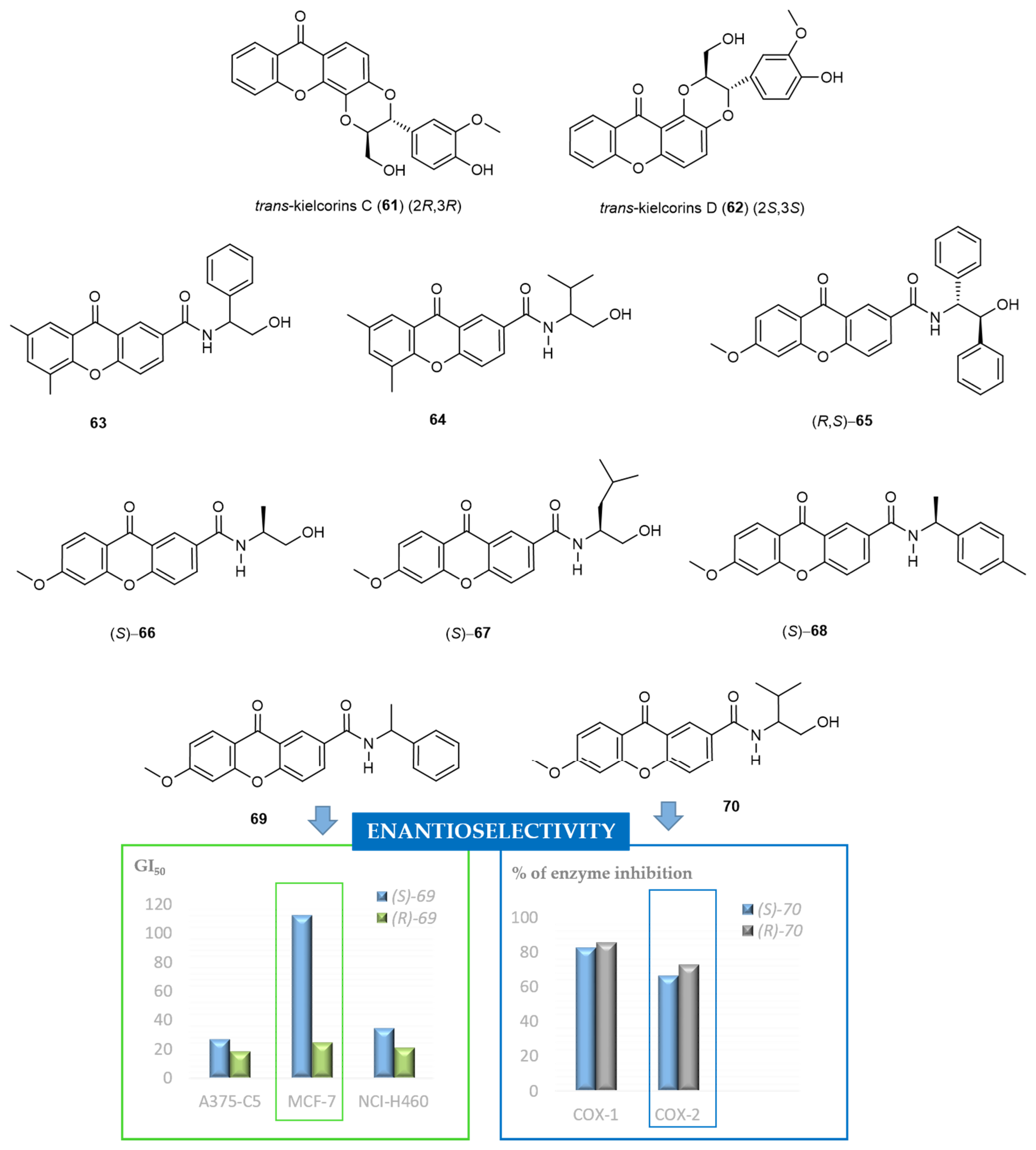
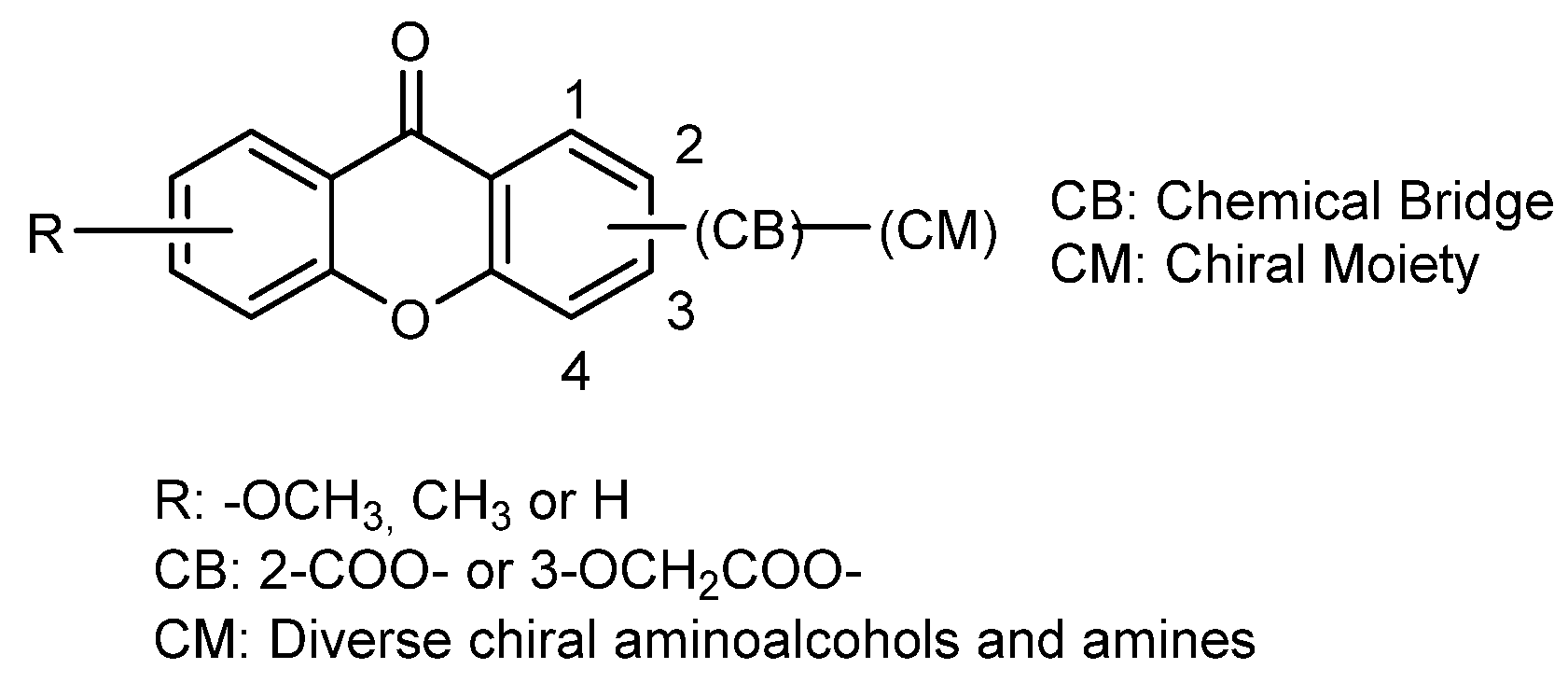
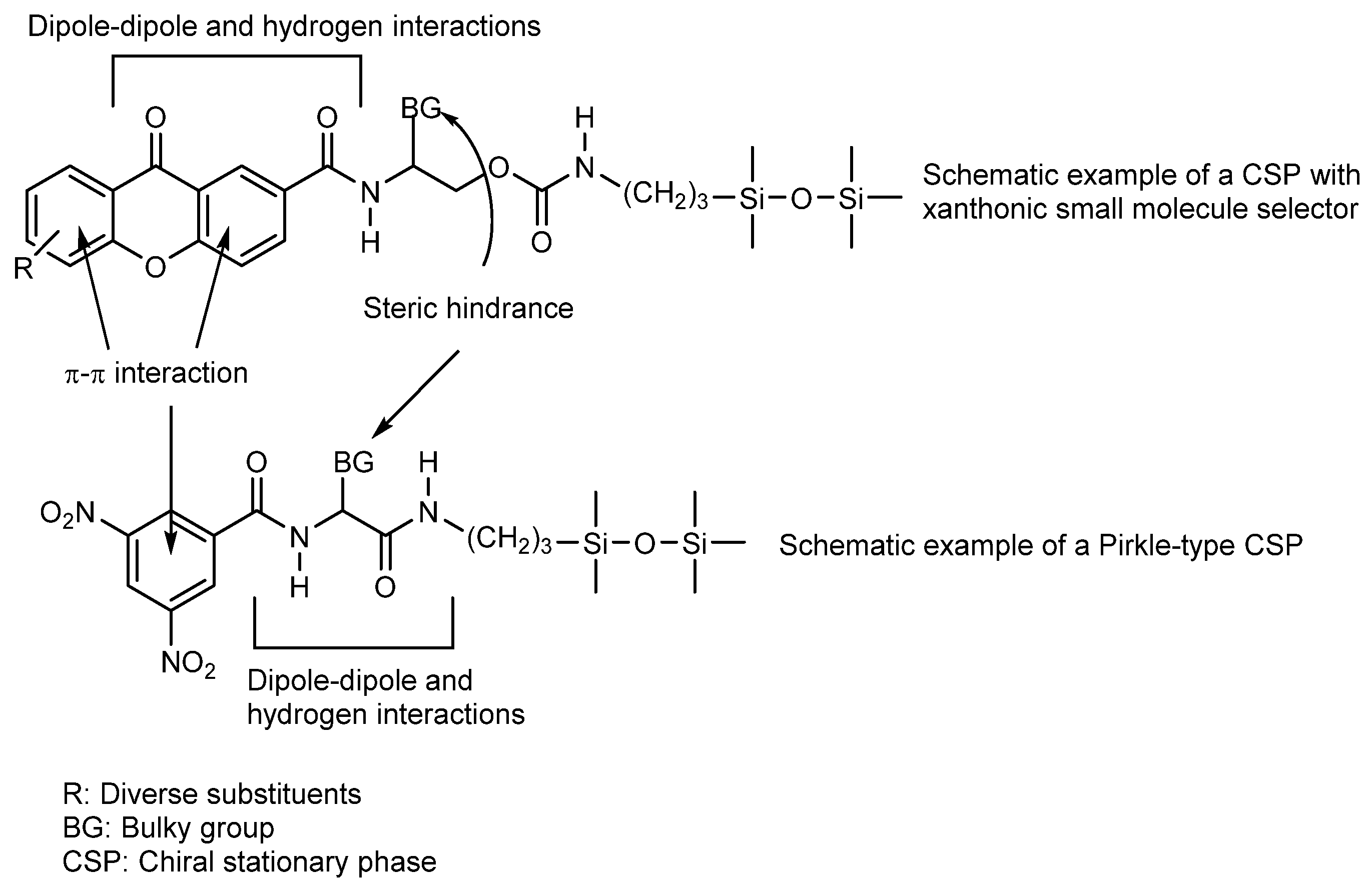
2.6. Chiral Derivatives of Xanthones
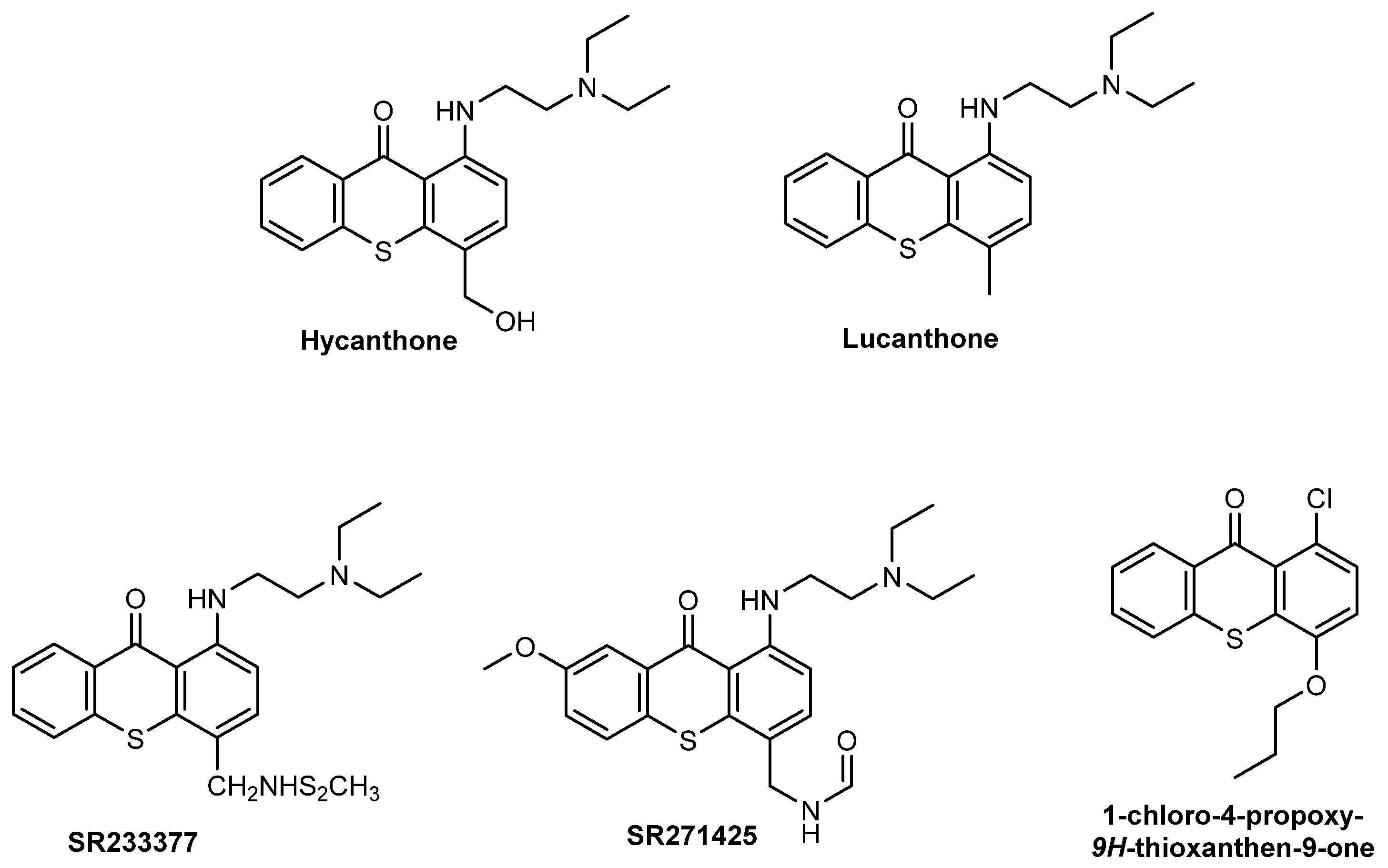
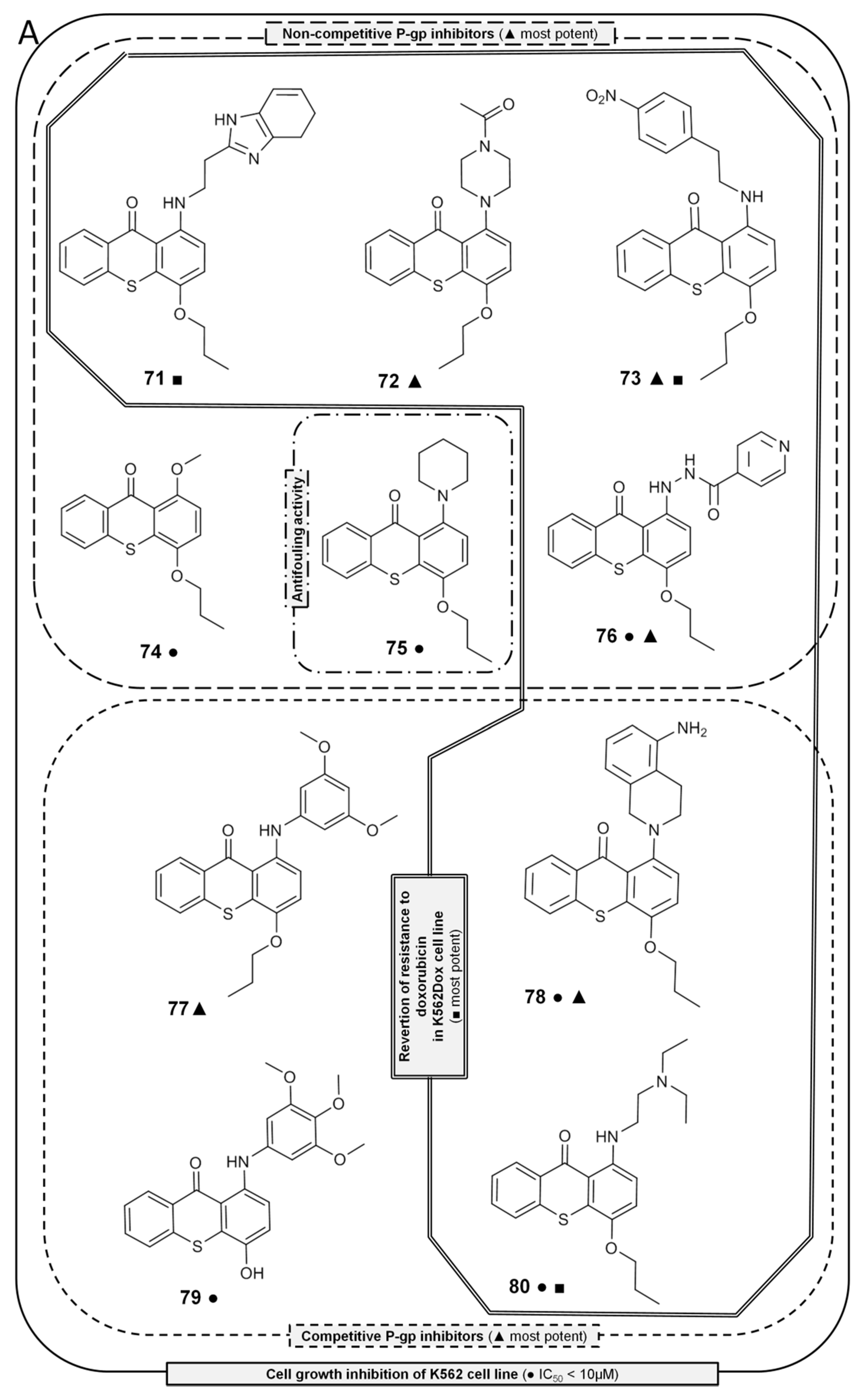
2.7. Thioxanthones
3. Conclusions
- (1)
- isolated new compounds from terrestrial and marine sources and/or analyzed species not yet studied with regard to their secondary metabolites;
- (2)
- used Nature-based strategies to guide the synthetic ways whenever possible;
- (3)
- synthesized these compounds for structure elucidation purposes to obtain them through “greener” methods and in quantities suitable for biological/pharmacological assays, as well as to obtain nature-based analogues with improved druglike properties;
- (4)
- contributed with new data in the area of NMR and X-ray crystallography;
- (5)
- evaluated several biological activities for different chemical families of xanthone derivatives; the main focus was in the area of antitumor and antimicrobial agents, especially taking into account MDR, which we believe should be pursuit in Academia; other areas of intervention include cardiovascular, antimalarial, anti-inflammatory, anti-obesity, hepatoprotection, etc. More recently, the area related to the discovery of environmental-benign AF agents was also explored;
- (6)
- explored the mechanisms of action, SAR and structure-activity-properties-relationship (SAPR);
- (7)
- formulated some compounds in nanoparticles, liposomes and proliposomes (drug delivery systems), especially with xanthones with potential antitumor activity;
- (8)
- determined the drug-likeness of some hit compounds (ADMET assays), as this should be carried out as soon as possible in the pipeline of drug discovery and development;
- (9)
- obtained several chiral derivatives of xanthones and studied enantioseparation and enantioselectivity with regard to various biological activities, as well as analytical applications as chiral selectors for liquid chromatography.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Biswas, S.C.; Sen, R.K. X-ray crystallographic studies of xanthones. Indian J. Pure Appl. Phys. 1982, 20, 414. [Google Scholar]
- Onuma, S.; Iijima, K.; Oonishi, I. Structure of xanthone. Acta Crystallogr. Sect. C 1990, 46, 1725–1727. [Google Scholar] [CrossRef] [Green Version]
- Gales, L.; Damas, A.M. Xanthones-A Structural Perspective. Curr. Med. Chem. 2005, 12, 2499–2515. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, H. Chemical Synthesis: Studies in the Investigation of Natural Organic Products; Blackie and Son: London, UK, 1924; Volume 44, pp. 120–121. [Google Scholar]
- Henry, L.; Cavenfou, J.-B. J. Pharm. Chim. 1821, 7, 178.
- Kamal, A.; Husain, S.A.; Noorani, R.; Murtaza, N.; Qureshi, I.H.; Qureshi, A.A. Studies in the biochemistry of microorganisms. XI. Isolation of tajixanthone, shamixanthone, ajamxanthone, shahenxanthone, najamxanthone, radixanthone and mannitol from mycelium of Aspergillus stellatus, Curzi. Pak. J. Sci. Indus. Res. 1970, 251–255, in press. [Google Scholar]
- Michael, A. On the action of aromatic oxy-acids on phenols. Am. Chem. J. 1883, 5, 81–97. [Google Scholar]
- Kostanecki, S.V. Über das Gentisin. Mon. Chem. Verwandte Teile And. Wiss. 1891, 12, 205–210. [Google Scholar] [CrossRef]
- Organic Syntheses. Available online: http://online.fliphtml5.com/jocd/yqzh/#p=1 (accessed on 2 December 2020).
- Goldberg, I. Ueber phenylirungen bei gegenwart von kupfer als katalysator. Ber. Dtsch. Chem. Ges. 1906, 39, 1691–1692. [Google Scholar] [CrossRef] [Green Version]
- Schmid, W. Ueber das Mangostin. Justus Liebigs Ann. Chem. 1855, 93, 83–88. [Google Scholar] [CrossRef]
- Rai, M.; Chikindas, M.L. Natural Antimicrobials in Food Safety and Quality; CAB International: Wallingford, UK, 2011. [Google Scholar]
- Negi, J.S.; Bisht, V.K.; Singh, P.; Rawat, M.S.M.; Joshi, G.P. Naturally Occurring Xanthones: Chemistry and Biology. J. Appl. Chem. 2013, 2013, 621459. [Google Scholar] [CrossRef] [Green Version]
- Madalena, M.M.P. Editorial [Hot Topic: Xanthone (Dibenzo-γ-Pyrone): An Interesting Framework In Medicinal Chemistry (Guest Editor: Madalena M.M. Pinto)]. Curr. Med. Chem. 2005, 12, 2411. [Google Scholar] [CrossRef]
- Vieira, L.M.M.; Kijjoa, A. Naturally-Occurring Xanthones: Recent Developments. Curr. Med. Chem. 2005, 12, 2413–2446. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.E.; Pinto, M.M.M. Synthesis of Xanthones: An Overview. Curr. Med. Chem. 2005, 12, 2447–2479. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.S.; Pinto, D.C.G.A. Structure Elucidation of Xanthone Derivatives: Studies of Nuclear Magnetic Resonance Spectroscopy. Curr. Med. Chem. 2005, 12, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.M.M.; Sousa, M.E.; Nascimento, M.S.J. Xanthone Derivatives: New Insights in Biological Activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar] [CrossRef] [PubMed]
- Riscoe, M.; Kelly, J.X.; Winter, R. Xanthones as Antimalarial Agents: Discovery, Mode of Action, and Optimization. Curr. Med. Chem. 2005, 12, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Lallemand, M.-C.; Boutefnouchet, S.; Michel, S.; Tillequin, F. Antitumor Psoropermum Xanthones and Sarcomelicope Acridones: Privileged Structures Implied in DNA Alkylation. J. Nat. Prod. 2009, 72, 527–539. [Google Scholar] [CrossRef]
- Lesch, B.; Bräse, S. A Short, Atom-Economical Entry to Tetrahydroxanthenones. Angew. Chem. Int. Ed. 2004, 43, 115–118. [Google Scholar] [CrossRef]
- Hesham, R.E.-S.; Dina, M.H.E.-G.; Mai, A.E.-B.; Mervat, F.Z.; Ulf, G.; Sonny, L.; Rob, V. Naturally Occurring Xanthones; Latest Investigations: Isolation, Structure Elucidation and Chemosystematic Significance. Curr. Med. Chem. 2009, 16, 2581–2626. [Google Scholar] [CrossRef]
- Demirkiran, O. Xanthones in Hypericum: Synthesis and BiologicalActivities. In Bioactive Heterocycles III; Khan, M.T.H., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 139–178. [Google Scholar] [CrossRef]
- Wezeman, T.; Masters, K.-S. Chapter 12 Xanthones are Privileged Scaffolds in Medicinal Chemistry—But are they Over-privileged? In Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; The Royal Society of Chemistry: London, UK, 2016; pp. 312–347. [Google Scholar] [CrossRef]
- Peres, V.; Nagem, T.J.; De Oliveira, F.F. Tetraoxygenated naturally occurring xanthones. Phytochemistry 2000, 55, 683–710. [Google Scholar] [CrossRef] [Green Version]
- Loureiro, D.R.P.; Soares, J.X.; Costa, J.C.; Magalhães, Á.F.; Azevedo, C.M.G.; Pinto, M.M.M.; Afonso, C.M.M. Structures, activities and drug-likeness of anti-infective xanthone derivatives isolated from the marine environment: A review. Molecules 2019, 24, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumla, D.; Dethoup, T.; Gales, L.; Pereira, J.A.; Freitas-Silva, J.; Costa, P.M.; Silva, A.M.S.; Pinto, M.M.M.; Kijjoa, A. Erubescensoic Acid, a new polyketide and a xanthonopyrone SPF-3059-26 From the culture of the marine sponge-associated fungus penicillium erubescens KUFA 0220 and Antibacterial activity evaluation of some of its constituents. Molecules 2019, 24, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resende, D.; Pereira-Terra, P.; Inácio, Â.; Costa, P.; Pinto, E.; Sousa, E.; Pinto, M. Lichen Xanthones as Models for New Antifungal Agents. Molecules 2018, 23, 2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.M.M.; Castanheiro, R.A.P.; Kijjoa, A. Xanthones from Marine-Derived Microorganisms: Isolation, Structure Elucidation and Biological Activities. In Encyclopedia of Analytical Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Masters, K.-S.; Bräse, S. Xanthones from Fungi, Lichens, and Bacteria: The Natural Products and Their Synthesis. Chem. Rev. 2012, 112, 3717–3776. [Google Scholar] [CrossRef] [PubMed]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef]
- Roberts, J.C. Naturally Occurring Xanthones. Chem. Rev. 1961, 61, 591–605. [Google Scholar] [CrossRef]
- Narosa, G.B. (Ed.) Natural Prenylated xanthones: Chemistry and biological activities. In Natural Products: Chemistry, Biochemistry and Pharmacology; Publishing House PVT: New Dehli, India, 2009. [Google Scholar]
- Mesquita, A.A.L.; Gottlieb, O.R.; Madalena, M.D.M. Xanthonolignoids from Kielmeyera coriacea. Phytochemistry 1987, 26, 2045–2048. [Google Scholar] [CrossRef]
- Pinto, M.; Nascimento, M.S.J.; Gonzalez, M.J.; Mondranondra, I.-O. Anticomplementary activity and constituents of Cratoxylum maingayi DYER. Pharm. Pharmacol. Lett. 1997, 7, 128–130. [Google Scholar]
- Kijjoa, A.; José, M.; Gonzalez, T.G.; Pinto, M.M.M.; Damas, A.M.; Mondranondra, I.O.; Silva, A.M.S.; Herz, W. Xanthones from Cratoxylum maingayi. Phytochemistry 1998, 49, 2159–2162. [Google Scholar] [CrossRef]
- Gonzalez, M.J.; Nascimento, M.S.J.; Cidade, H.M.; Pinto, M.M.M.; Kijjoa, A.; Anantachoke, C.; Silva, A.M.S.; Herz, W. Immunomodulatory activity of xanthones from Calophyllum teysmannii var. inuphylloide. Planta Med. 1999, 65, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Kijjoa, A.; Gonzalez, M.J.; Pinto, M.M.M.; Silva, A.M.S.; Anantachoke, C.; Herz, W. Xanthones from Calophyllum teysmannii var. inophylloide. Phytochemistry 2000, 55, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Kijjoa, A.; Gonzalez, M.J.; Pinto, M.M.; Nascimento, M.S.J.; Campos, N.; Mondranondra, I.O.; Silva, A.M.S.; Eaton, G.; Herz, W. Cytotoxicity of prenylated xanthones and other constituents from the wood of Garcinia merguensis. Planta Med. 2008, 74, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Freitas, V.L.S.; Ribeiro da Silva, M.D.M.C. Influence of Hydroxyl Functional Group on the Structure and Stability of Xanthone: A Computational Approach. Molecules 2018, 23, 2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.; Nascimento, M.S.J. Anticomplementary activity of hydroxy- and methoxyxanthones. Pharm. Pharmacol. Lett. 1997, 7, 125–127. [Google Scholar]
- Fernandes, E.G.R.; Silva, A.M.S.; Cavaleiro, J.A.S.; Silva, F.M.; Fernanda, M.; Borges, M.; Pinto, M.M.M. 1H and 13C NMR Spectroscopy of mono-, di-, tri- and tetrasubstituted xanthones. Magn. Reson. Chem. 1998, 36, 305–309. [Google Scholar] [CrossRef]
- Gales, L.; De Sousa, M.E.; Pinto, M.M.M.; Kijjoa, A.; Damas, A.M. Naturally occurring 1,2,8-trimethoxy-xanthone and biphenyl ether intermediates leading to 1,2-dimethoxy-xanthone. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2001, 57, 1319–1323. [Google Scholar] [CrossRef]
- Gales, L.; Sousa, M.E.; Pinto, M.M.M.; Damas, A.M. 3,4-Dihydroxy-9H-xanthen-9-one trihydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, o2213–o2215. [Google Scholar] [CrossRef]
- Resende, D.I.S.P.; Durães, F.; Maia, M.; Sousa, E.; Pinto, M.M.M. Recent advances in the synthesis of xanthones and azaxanthones. Org. Chem. Front. 2020. [Google Scholar] [CrossRef]
- Teixeira, M.; Afonso, C.M.; Pinto, M.M.; Barbosa, C.M. A validated HPLC method for the assay of xanthone and 3-methoxyxanthone in PLGA nanocapsules. J. Chromatogr. Sci. 2003, 41, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Pedro, M.; Cerqueira, F.; Sousa, M.E.l.; Nascimento, M.S.J.; Pinto, M. Xanthones as inhibitors of growth of human cancer cell lines and Their effects on the proliferation of human lymphocytes In Vitro. Bioorg. Med. Chem. 2002, 10, 3725–3730. [Google Scholar] [CrossRef]
- Cidade, H.; Rocha, V.; Palmeira, A.; Marques, C.; Tiritan, M.E.; Ferreira, H.; Lobo, J.S.; Almeida, I.F.; Sousa, M.E.; Pinto, M. In silico and in vitro antioxidant and cytotoxicity evaluation of oxygenated xanthone derivatives. Arab. J. Chem. 2017. [Google Scholar] [CrossRef]
- Fernandes, E.R.; Carvalho, F.D.; Remião, F.G.; Bastos, M.L.; Pinto, M.M.; Gottlieb, O.R. Hepatoprotective Activity of Xanthones and Xanthonolignoids Against tert-Butylhydroperoxide-Induced Toxicity in Isolated Rat Hepatocytes—Comparison with Silybin. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1995, 12, 1756–1760. [Google Scholar] [CrossRef]
- Pinto, E.; Afonso, C.; Duarte, S.; Vale-Silva, L.; Costa, E.; Sousa, E.; Pinto, M. Antifungal Activity of Xanthones: Evaluation of their Effect on Ergosterol Biosynthesis by High-performance Liquid Chromatography. Chem. Biol. Drug Des. 2011, 77, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Palmeira, A.; Gomes, A.S.; Vasconcelos, V.; Sousa, E.; Pinto, M.; Da Costa, P.M. Synergistic effects between thioxanthones and oxacillin against methicillin-resistant staphylococcus aureus. Microb. Drug Resist. 2015, 21, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Resende, D.I.S.P.; Pereira-Terra, P.; Moreira, J.; Freitas-Silva, J.; Lemos, A.; Gales, L.; Pinto, E.; De Sousa, M.E.; Da Costa, P.M.; Pinto, M.M.M. Synthesis of a Small Library of Nature-Inspired Xanthones and Study of Their Antimicrobial Activity. Molecules 2020, 25, 2405. [Google Scholar] [CrossRef]
- Urbatzka, R.; Freitas, S.; Palmeira, A.; Almeida, T.; Moreira, J.; Azevedo, C.; Afonso, C.; Correia-da-Silva, M.; Sousa, E.; Pinto, M.; et al. Lipid reducing activity and toxicity profiles of a library of polyphenol derivatives. Eur. J. Med. Chem. 2018, 151, 272–284. [Google Scholar] [CrossRef]
- Almeida, J.R.; Palmeira, A.; Campos, A.; Cunha, I.; Freitas, M.; Felpeto, A.B.; Turkina, M.V.; Vasconcelos, V.; Pinto, M.; Correia-Da-silva, M.; et al. Structure-antifouling activity relationship and molecular targets of bio-inspired(Thio)xanthones. Biomolecules 2020, 10, 1126. [Google Scholar] [CrossRef]
- Gnerre, C.; Thull, U.; Gaillard, P.; Carrupt, P.-A.; Testa, B.; Fernandes, E.; Silva, F.; Pinto, M.; Pinto, M.M.M.; Wolfender, J.-L.; et al. Natural and Synthetic Xanthones as Monoamine Oxidase Inhibitors: Biological Assay and 3D-QSAR. Helv. Chim. Acta 2001, 84, 552–570. [Google Scholar] [CrossRef]
- Silva, R.; Sousa, E.; Carmo, H.; Palmeira, A.; Barbosa, D.J.; Gameiro, M.; Pinto, M.; De Lourdes Bastos, M.; Remião, F. Induction and activation of P-glycoprotein by dihydroxylated xanthones protect against the cytotoxicity of the P-glycoprotein substrate paraquat. Arch. Toxicol. 2014, 88, 937–951. [Google Scholar] [CrossRef]
- Martins, E.; Silva, V.; Lemos, A.; Palmeira, A.; Puthongking, P.; Sousa, E.; Rocha-Pereira, C.; Ghanem, C.I.; Carmo, H.; Remião, F.; et al. Newly Synthesized Oxygenated Xanthones as Potential P-Glycoprotein Activators: In Vitro, Ex Vivo, and In Silico Studies. Molecules 2019, 24, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, L.; Fresco, P.; Pinto, E.; Sousa, E.; Pinto, M.; Gonçalves, J. Synthesis and in vivo modulatory activity of protein kinase C of xanthone derivatives. Bioorg. Med. Chem. 2002, 10, 3219–3227. [Google Scholar] [CrossRef]
- Saraiva, L.; Fresco, P.; Pinto, E.; Sousa, E.; Pinto, M.; Gonçalves, J. Inhibition of protein kinase C by synthetic xanthone derivatives. Bioorg. Med. Chem. 2003, 11, 1215–1225. [Google Scholar] [CrossRef]
- Rosa, G.P.; Palmeira, A.; Resende, D.I.S.P.; Almeida, I.F.; Kane-Pagès, A.; Barreto, M.C.; Sousa, E.; Pinto, M.M.M. Xanthones for melanogenesis inhibition: Molecular docking and QSAR studies to understand their anti-tyrosinase activity. Bioorg. Med. Chem. 2021, 29, 115873. [Google Scholar] [CrossRef] [PubMed]
- Sousa, E.P.; Silva, A.M.S.; Pinto, M.M.M.; Pedro, M.M.; Cerqueira, F.A.M.; Nascimento, M.S.J. Isomeric Kielcorins and Dihydroxyxanthones: Synthesis, Structure Elucidation, and Inhibitory Activities of Growth of Human Cancer Cell Lines and on the Proliferation of Human Lymphocytes In Vitro. Helv. Chim. Acta 2002, 85, 2862–2876. [Google Scholar] [CrossRef]
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leão, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New inhibitor of the TAp73 interaction with MDM2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019, 446, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.; Cerqueira, F.; Nazareth, N.; Medeiros, R.; Sarmento, A.; Sousa, E.; Pinto, M. 1,2-Dihydroxyxanthone: Effect on A375-C5 Melanoma Cell Growth Associated with Interference with THP-1 Human Macrophage Activity. Pharmaceuticals 2019, 12, 85. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.S.; Brandão, P.; Fernandes, C.S.G.; Da Silva, M.; De Sousa, M.; Pinto, M.M.M. Drug-like Properties and ADME of Xanthone Derivatives: The Antechamber of Clinical Trials. Curr. Med. Chem. 2016, 23, 3654–3686. [Google Scholar] [CrossRef]
- Sousa, E.; Palmeira, A.; Cordeiro, A.S.; Sarmento, B.; Ferreira, D.; Lima, R.T.; Helena Vasconcelos, M.; Pinto, M. Bioactive xanthones with effect on P-glycoprotein and prediction of intestinal absorption. Med. Chem. Res. 2013, 22, 2115–2123. [Google Scholar] [CrossRef]
- Resende, D.I.S.P.; Almeida, M.C.; Maciel, B.; Carmo, H.; Sousa Lobo, J.; Dal Pozzo, C.; Cravo, S.M.; Rosa, G.P.; Kane-Pagès, A.; Do Carmo Barreto, M.; et al. Efficacy, Stability, and Safety Evaluation of New Polyphenolic Xanthones Towards Identification of Bioactive Compounds to Fight Skin Photoaging. Molecules 2020, 25, 2782. [Google Scholar] [CrossRef]
- Phyo, Y.Z.; Teixeira, J.; Tiritan, M.E.; Cravo, S.; Palmeira, A.; Gales, L.; Silva, A.M.S.; Pinto, M.M.M.; Kijjoa, A.; Fernandes, C. New chiral stationary phases for liquid chromatography based on small molecules: Development, enantioresolution evaluation and chiral recognition mechanisms. Chirality 2020, 32, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Tiritan, M.E.; Cravo, S.; Phyo, Y.Z.; Kijjoa, A.; Silva, A.M.S.; Cass, Q.B.; Pinto, M.M.M. New chiral stationary phases based on xanthone derivatives for liquid chromatography. Chirality 2017, 29, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.; Pedro, M.; Nascimento, M.S.J.; Pinto, M.M.M.; Barbosa, C.M. Development and characterization of PLGA nanoparticles containing 1,3-dihydroxy-2-methylxanthone with improved antitumor activity on a human breast cancer cell line. Pharm. Dev. Technol. 2019, 24, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.; Alonso, M.J.; Pinto, M.M.M.; Barbosa, C.M. Development and characterization of PLGA nanospheres and nanocapsules containing xanthone and 3-methoxyxanthone. Eur. J. Pharm. Biopharm. 2005, 59, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malta, R.; Loureiro, J.B.; Costa, P.; Sousa, E.; Pinto, M.; Saraiva, L.; Amaral, M.H. Development of lipid nanoparticles containing the xanthone LEM2 for topical treatment of melanoma. J. Drug Deliv. Sci. Technol. 2020. [Google Scholar] [CrossRef]
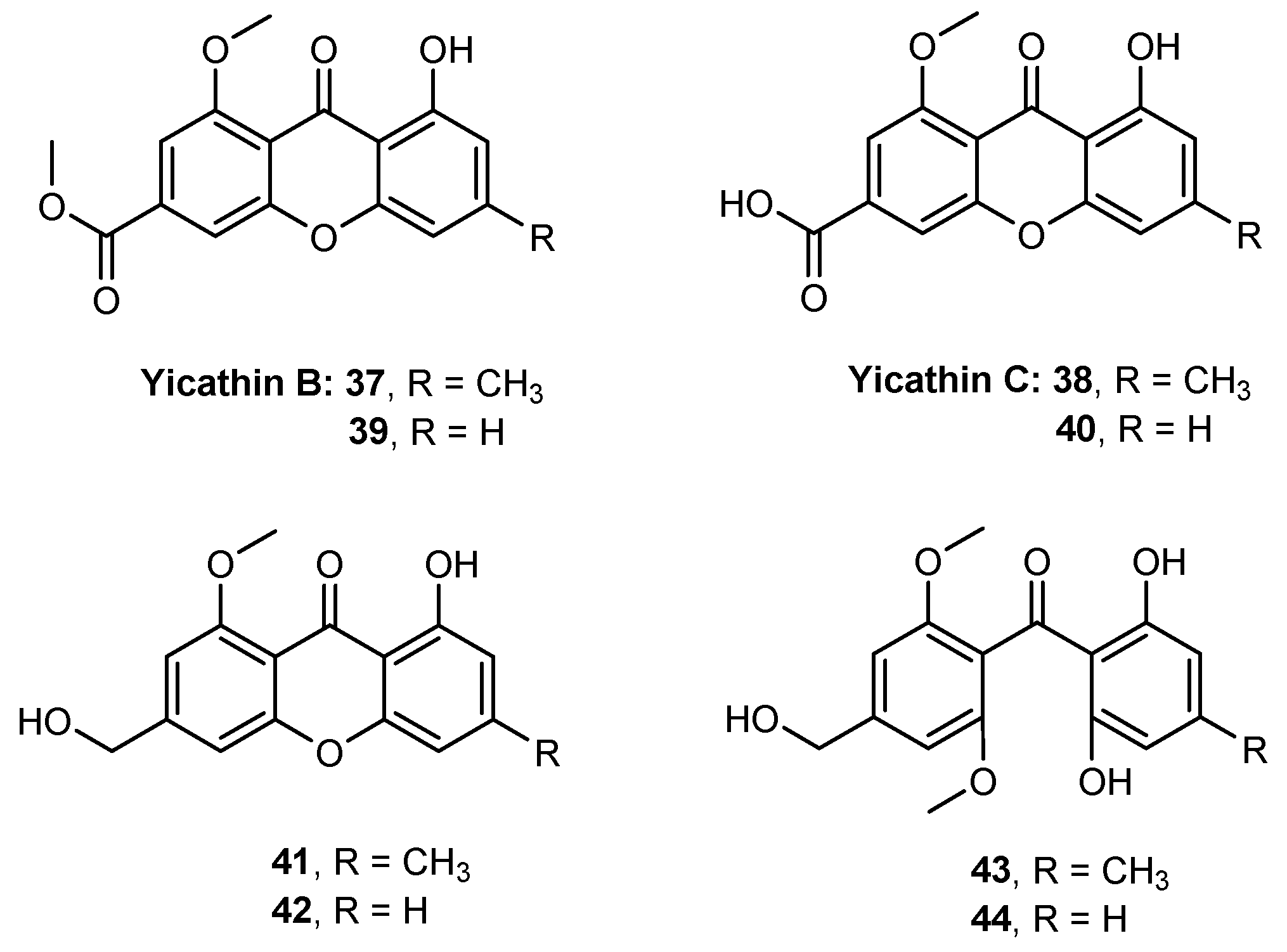
- Loureiro, D.R.P.; Magalhães, Á.F.; Soares, J.X.; Pinto, J.; Azevedo, C.M.G.; Vieira, S.; Henriques, A.; Ferreira, H.; Neves, N.; Bousbaa, H.; et al. Yicathins B and C and Analogues: Total Synthesis, Lipophilicity and Biological Activities. ChemMedChem 2020, 15, 749–755. [Google Scholar] [CrossRef]
- Silva, V.; Gil-Martins, E.; Rocha-Pereira, C.; Lemos, A.; Palmeira, A.; Puthongking, P.; Sousa, E.; De Lourdes Bastos, M.; Remião, F.; Silva, R. Oxygenated xanthones as P-glycoprotein modulators at the intestinal barrier: In vitro and docking studies. Med. Chem. Res. 2020, 29, 1041–1057. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.M.M.; Castanheiro, R.A.P. Synthesis of prenylated xanthones: An overview. Curr. Org. Chem. 2009, 13, 1215–1240. [Google Scholar] [CrossRef]
- Azevedo, C.M.G.; Afonso, C.M.M.; Soares, J.X.; Reis, S.; Sousa, D.; Lima, R.T.; Vasconcelos, M.H.; Pedro, M.; Barbosa, J.; Gales, L.; et al. Pyranoxanthones: Synthesis, growth inhibitory activity on human tumor cell lines and determination of their lipophilicity in two membrane models. Eur. J. Med. Chem. 2013, 69, 798–816. [Google Scholar] [CrossRef] [PubMed]
- Castanheiro, R.A.P.; Pinto, M.M.M.; Cravo, S.M.M.; Pinto, D.C.G.A.; Silva, A.M.S.; Kijjoa, A. Improved methodologies for synthesis of prenylated xanthones by microwave irradiation and combination of heterogeneous catalysis (K10 clay) with microwave irradiation. Tetrahedron 2009, 65, 3848–3857. [Google Scholar] [CrossRef]
- Azevedo, C.M.G.; Afonso, C.M.M.; Sousa, D.; Lima, R.T.; Helena Vasconcelos, M.; Pedro, M.; Barbosa, J.; Corrêa, A.G.; Reis, S.; Pinto, M.M.M. Multidimensional optimization of promising antitumor xanthone derivatives. Bioorg. Med. Chem. 2013, 21, 2941–2959. [Google Scholar] [CrossRef] [PubMed]
- França, F.; Silva, P.M.A.; Soares, J.X.; Henriques, A.C.; Loureiro, D.R.P.; Azevedo, C.M.G.; Afonso, C.M.M.; Bousbaa, H. A Pyranoxanthone as a Potent Antimitotic and Sensitizer of Cancer Cells to Low Doses of Paclitaxel. Molecules 2020, 25, 5845. [Google Scholar] [CrossRef] [PubMed]
- Epifano, F.; Genovese, S.; Menghini, L.; Curini, M. Chemistry and pharmacology of oxyprenylated secondary plant metabolites. Phytochemistry 2007, 68, 939–953. [Google Scholar] [CrossRef]
- Nakatani, K.; Nakahata, N.; Arakawa, T.; Yasuda, H.; Ohizumi, Y. Inhibition of cyclooxygenase and prostaglandin E2 synthesis by γ-mangostin, a xanthone derivative in mangosteen, in C6 rat glioma cells11Abbreviations: AA, arachidonic acid; BSA, bovine serum albumin; COX, cyclooxygenase; COX-1, constitutive COX.; COX-2, inducible COX; cPLA2, cytosolic PLA2; EMEM, Eagle’s minimum essential medium; ERK, extracellular signal regulated kinase; HEPES, 2-[4-(2-Hydroxyethyl)-1-piperazinyl]ethanesulfonic acid; MAPK, mitogen-activated protein kinase;PGE2, prostaglandin E2; sPLA2, secretory phospholipase A2; TBST, Tris-buffered saline containing 0.05% Tween 20. Biochem. Pharmacol. 2002, 63, 73–79. [Google Scholar] [CrossRef]
- Castanheiro, R.A.P.; Pinto, M.M.M.; Silva, A.M.S.; Cravo, S.M.M.; Gales, L.; Damas, A.M.; Nazareth, N.; Nascimento, M.S.J.; Eaton, G. Dihydroxyxanthones prenylated derivatives: Synthesis, structure elucidation, and growth inhibitory activity on human tumor cell lines with improvement of selectivity for MCF-7. Bioorg. Med. Chem. 2007, 15, 6080–6088. [Google Scholar] [CrossRef]
- Castanheiro, R.A.P.; Silva, A.M.S.; Campos, N.A.N.; Nascimento, M.S.J.; Pinto, M.M.M. Antitumor activity of some prenylated xanthones. Pharmaceuticals 2009, 2, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, A.; Paiva, A.; Sousa, E.; Seca, H.; Almeida, G.M.; Lima, R.T.; Fernandes, M.X.; Pinto, M.; Vasconcelos, M.H. Insights into the in vitro antitumor mechanism of action of a new pyranoxanthone. Chem. Biol. Drug Des. 2010, 76, 43–58. [Google Scholar] [CrossRef]
- Paiva, A.M.; Sousa, M.E.; Camões, A.; Nascimento, M.S.J.; Pinto, M.M.M. Prenylated xanthones: Antiproliferative effects and enhancement of the growth inhibitory action of 4-hydroxytamoxifen in estrogen receptor-positive breast cancer cell line. Med. Chem. Res. 2012, 21, 552–558. [Google Scholar] [CrossRef]
- Leão, M.; Gomes, S.; Pedraza-Chaverri, J.; Machado, N.; Sousa, E.; Pinto, M.; Inga, A.; Pereira, C.; Saraiva, L. α-Mangostin and Gambogic Acid as Potential Inhibitors of the p53–MDM2 Interaction Revealed by a Yeast Approach. J. Nat. Prod. 2013, 76, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Lemos, A.; Leão, M.; Soares, J.; Palmeira, A.; Pinto, M.; Saraiva, L.; Sousa, M.E. Medicinal Chemistry Strategies to Disrupt the p53–MDM2/MDMX Interaction. Med. Chem. Res. 2016, 36, 789–844. [Google Scholar] [CrossRef] [PubMed]
- Inga, A.; Pinto, M.M.d.M.; Saraiva, L.H.A.; Sousa, M.E.d.S.P.d.; Pereira, C.I.F.; Paiva, A.M.G.; Leão, M.V.C.F. Inhibitors of p53-mdm2 Interaction. WO/2013/105037, 7 February 2013. [Google Scholar]
- Liu, J.; Zhou, F.; Zhang, L.; Wang, H.; Zhang, J.; Zhang, C.; Jiang, Z.; Li, Y.; Liu, Z.; Chen, H. DMXAA-pyranoxanthone hybrids enhance inhibition activities against human cancer cells with multi-target functions. Eur. J. Med. Chem. 2018, 143, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Paiva, A.M.; Pinto, R.A.; Teixeira, M.; Barbosa, C.M.; Lima, R.T.; Vasconcelos, M.H.; Sousa, E.; Pinto, M. Development of noncytotoxic PLGA nanoparticles to improve the effect of a new inhibitor of p53-MDM2 interaction. Int. J. Pharm. 2013, 454, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Paiva, A.M.; Teixeira, M.; Pereira, R.; Barbosa, C.M.; Sousa, E.; Pinto, M.M.M. Development and validation of an HPLC method for the quantification of a cytotoxic dihydropyranoxanthone in biodegradable nanoparticles. Int. J. Drug Deliv. 2013, 5, 224–232. [Google Scholar]
- Pedro Gonçalves, A.; Silva, N.; Oliveira, C.; Kowbel, D.J.; Glass, N.L.; Kijjoa, A.; Palmeira, A.; Sousa, E.; Pinto, M.; Videira, A. Transcription profiling of the Neurospora crassa response to a group of synthetic (thio)xanthones and a natural acetophenone. Genom. Data 2015, 4, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Pogam, P.; Boustie, J. Xanthones of Lichen Source: A 2016 Update. Molecules 2016, 21, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayan, F.E.; Romagni, J.G. Lichens as a potential source of pesticides. Pestic. Outlook 2001, 12, 229–232. [Google Scholar] [CrossRef]
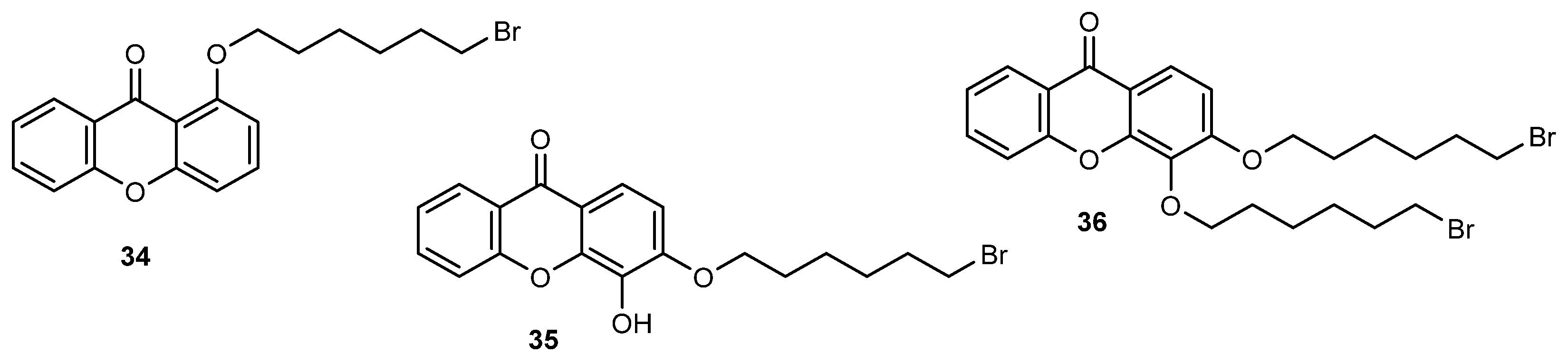
- Sousa, E.; Paiva, A.; Nazareth, N.; Gales, L.; Damas, A.M.; Nascimento, M.S.J.; Pinto, M. Bromoalkoxyxanthones as promising antitumor agents: Synthesis, crystal structure and effect on human tumor cell lines. Eur. J. Med. Chem. 2009, 44, 3830–3835. [Google Scholar] [CrossRef]
- Sun, R.-R.; Miao, F.-P.; Zhang, J.; Wang, G.; Yin, X.-L.; Ji, N.-Y. Three new xanthone derivatives from an algicolous isolate of Aspergillus wentii. Magn. Reson. Chem. 2013, 51, 65–68. [Google Scholar] [CrossRef]
- Cruz, M.I.; Cidade, H.; Pinto, M. Dual/multitargeted xanthone derivatives for Alzheimer’s disease: Where do we stand? Future Med. Chem. 2017, 9, 1611–1630. [Google Scholar] [CrossRef] [PubMed]
- Cruz, I.; Puthongking, P.; Cravo, S.; Palmeira, A.; Cidade, H.; Pinto, M.; Sousa, E. Xanthone and Flavone Derivatives as Dual Agents with Acetylcholinesterase Inhibition and Antioxidant Activity as Potential Anti-Alzheimer Agents. J. Chem. 2017, 2017, 16. [Google Scholar] [CrossRef]
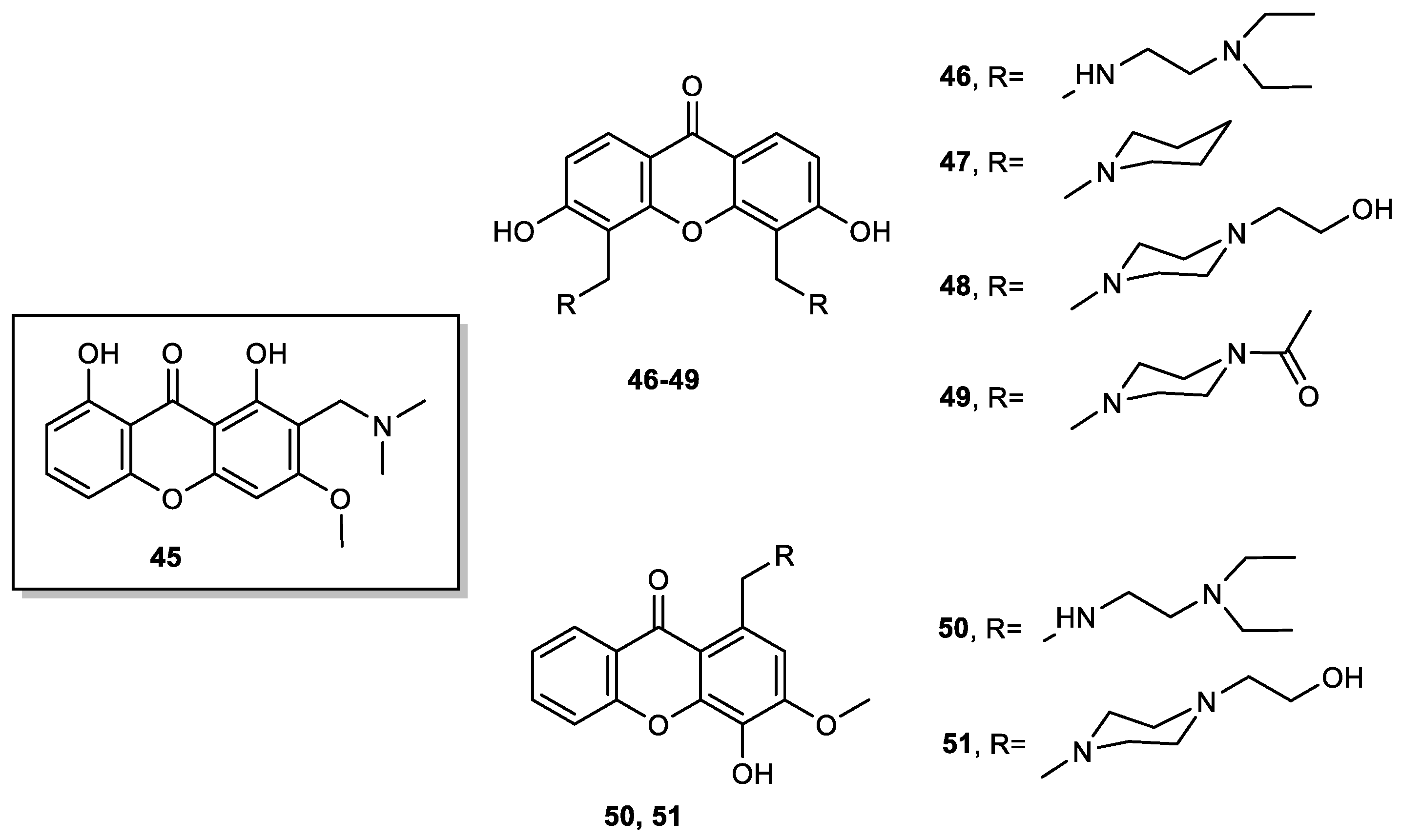
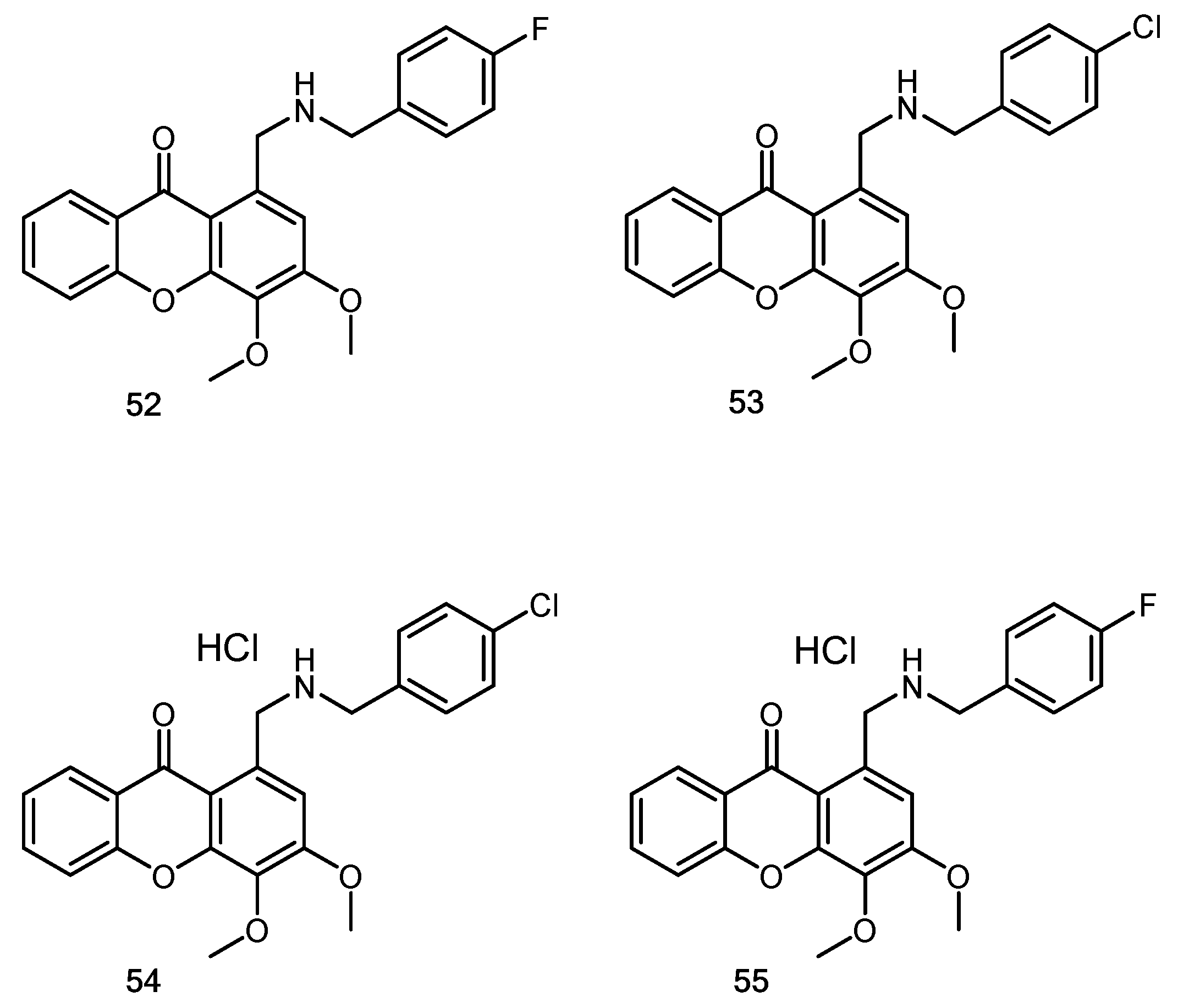
- Lemos, A.; Gomes, A.S.; Loureiro, J.B.; Brandão, P.; Palmeira, A.; Pinto, M.M.M.; Saraiva, L.; Sousa, M.E. Synthesis, Biological Evaluation, and In Silico Studies of Novel Aminated Xanthones as Potential p53-Activating Agents. Molecules 2019, 24, 1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portela, C.; Afonso, C.M.M.; Pinto, M.M.M.; João Ramos, M. Definition of an electronic profile of compounds with inhibitory activity against hematin aggregation in malaria parasite. Bioorg. Med. Chem. 2004, 12, 3313–3321. [Google Scholar] [CrossRef] [PubMed]
- Portela, C.; Afonso, C.M.M.; Pinto, M.M.M.; Ramos, M.J. Receptor–drug association studies in the inhibition of the hematin aggregation process of malaria. FEBS Lett. 2003, 547, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Portela, C.; Afonso, C.M.M.; Pinto, M.M.M.; João Ramos, M. Computational studies of new potential antimalarial compounds—Stereoelectronic complementarity with the receptor. J. Comput.-Aided Mol. Des. 2003, 17, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Portela, C.; Afonso, C.M.M.; Pinto, M.M.M.; Lopes, D.; Nogueira, F.; Do Rosário, V. Synthesis and Antimalarial Properties of New Chloro-9H-xanthones with an Aminoalkyl Side Chain. Chem. Biodivers. 2007, 4, 1508–1519. [Google Scholar] [CrossRef]
- Neves, A.R.; Correia-da-Silva, M.; Sousa, E.; Pinto, M. Strategies to overcome heparins’ low oral bioavailability. Pharmaceuticals 2016, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Desai, U.R. The promise of sulfated synthetic small molecules as modulators of glycosaminoglycan function. Future Med. Chem. 2013, 5, 1363–1366. [Google Scholar] [CrossRef]
- Correia-da-Silva, M.; Sousa, E.; Duarte, B.; Marques, F.; Carvalho, F.; Cunha-Ribeiro, L.M.; Pinto, M.M.M. Polysulfated Xanthones: Multipathway Development of a New Generation of Dual Anticoagulant/Antiplatelet Agents. J. Med. Chem. 2011, 54, 5373–5384. [Google Scholar] [CrossRef]
- Imran, M.; Arshad, M.S.; Butt, M.S.; Kwon, J.-H.; Arshad, M.U.; Sultan, M.T. Mangiferin: A natural miracle bioactive compound against lifestyle related disorders. Lipids Health Dis. 2017, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Rashid, Q.; Abid, M.; Jairajpuri, M. Elucidating the specificity of non-heparin-based conformational activators of antithrombin for factor Xa inhibition. J. Nat. Sci. Biol. Med. 2014, 5, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.; Sousa, M.E.; Da Silva, M.C.; Marques, F.; Carvalho, F. Patente Portuguesa Nº 104739: Xantonas Sulfatadas e Análogos Xantónicos Glicosilados Sulfatados com Actividade Anticoagulante e Processos Para a Sua Preparação. Portuguese Patent nº 104739, Boletim da Propriedade Industrial nº 47/2011, 9 March 2011. [Google Scholar]
- Neves, A.R.; Correia-Da-Silva, M.; Sousa, E.; Pinto, M. Structure-activity relationship studies for multitarget antithrombotic drugs. Future Med. Chem. 2016, 8, 2305–2355. [Google Scholar] [CrossRef] [PubMed]
- Rashid, Q.; Singh, P.; Abid, M.; Jairajpuri, M.A. Limitations of conventional anticoagulant therapy and the promises of non-heparin based conformational activators of antithrombin. J. Thromb. Thrombolysis 2012, 34, 251–259. [Google Scholar] [CrossRef]
- Kaltenbach, D.D.; Jaishankar, D.; Hao, M.; Beer, J.C.; Volin, M.V.; Desai, U.R.; Tiwari, V. Sulfotransferase and Heparanase: Remodeling Engines in Promoting Virus Infection and Disease Development. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Daniel, K.A.; Rami, A.A.-H. Sulfated Non-Saccharide Glycosaminoglycan Mimetics as Novel Drug Discovery Platform for Various Pathologies. Curr. Med. Chem. 2020, 27, 3412–3447. [Google Scholar] [CrossRef]
- Lima, R.T.; Seca, H.; Palmeira, A.; Fernandes, M.X.; Castro, F.; Correia-da-Silva, M.; Nascimento, M.S.J.; Sousa, E.; Pinto, M.; Vasconcelos, M.H. Sulfated small molecules targeting EBV in Burkitt lymphoma: From in silico screening to the evidence of in vitro effect on viral episomal DNA. Chem. Biol. Drug Des. 2013, 81, 631–644. [Google Scholar] [CrossRef]
- Vilas-Boas, C.; Sousa, E.; Pinto, M.; Correia-da-Silva, M. An antifouling model from the sea: A review of 25 years of zosteric acid studies. Biofouling 2017, 33, 927–942. [Google Scholar] [CrossRef]
- Almeida, J.R.; Correia-Da-Silva, M.; Sousa, E.; Antunes, J.; Pinto, M.; Vasconcelos, V.; Cunha, I. Antifouling potential of Nature-inspired sulfated compounds. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Alves, A.; Correia-da-Silva, M.; Nunes, C.; Campos, J.; Sousa, E.; Silva, P.M.A.; Bousbaa, H.; Rodrigues, F.; Ferreira, D.; Costa, P.C.; et al. Discovery of a new xanthone against glioma: Synthesis and Development of (Pro)liposome Formulations. Molecules 2019, 24, 409. [Google Scholar] [CrossRef] [Green Version]
- Neves, A.R.; Correia-da-Silva, M.; Silva, P.M.A.; Ribeiro, D.; Sousa, E.; Bousbaa, H.; Pinto, M. Synthesis of new glycosylated flavonoids with inhibitory activity on cell growth. Molecules 2018, 23, 1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiritan, M.E.; Ribeiro, A.R.; Fernandes, C.; Pinto, M.M.M. Chiral pharmaceuticals. In Encyclopedia of Chemical Technology; Kirk-Othmer, M., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Lanzotti, V. Drugs based on natural compounds: Recent achievements and future perspectives. Phytochem. Rev. 2014, 13, 725–726. [Google Scholar] [CrossRef] [Green Version]
- Carlos Miguel Goncalves, A.; Carlos Manuel Magalhaes, A.; Madalena Maria Magalhaes, P. Routes to Xanthones: An Update on the Synthetic Approaches. Curr. Org. Chem. 2012, 16, 2818–2867. [Google Scholar] [CrossRef]
- Blaser, H.-U. Chirality and its implications for the pharmaceutical industry. Rend. Lincei 2013, 24, 213–216. [Google Scholar] [CrossRef]
- Fernandes, C.; Carraro, M.L.; Ribeiro, J.; Araujo, J.; Tiritan, M.E.; Pinto, M.M.M. Synthetic chiral derivatives of xanthones: Biological activities and enantioselectivity studies. Molecules 2019, 24, 791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, J.; Fernandes, C.; Pinto, M.; Elizabeth Tiritan, M. Chiral derivatives of xanthones with antimicrobial activity. Molecules 2019, 24, 314. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.M.M.; Sousa, E.P. Natural and synthetic xanthonolognoids: Chemistry and biological activities. Curr. Med. Chem. 2003, 10, 1–12. [Google Scholar] [CrossRef]
- Fernandes, E.G.R.; Pinto, M.M.M.; Silva, A.M.S.; Cavaleiro, J.A.S.; Gottlieb, O.R. Synthesis and structural elucidation of xanthonolignoids: Trans-(±)- kielcorin B and trans-(±)-isokielcorin B. Heterocycles 1999, 51, 821–828. [Google Scholar] [CrossRef]
- Emília Sousa, M.; Afonso, C.M.M.; Pinto, M.M.M. Quantitative Analysis of Kielcorins in Biomimetic Synthesis by Liquid Chromatography/UV Detection. J. Liq. Chromatogr. Relat. Technol. 2003, 26, 29–41. [Google Scholar] [CrossRef]
- Saraiva, L.; Fresco, P.; Pinto, E.; Sousa, E.; Pinto, M.; Gonçalves, J. Inhibition of α, βI, δ, η and ζ protein kinase C isoforms by xanthonolignoids. J. Enzym. Inhib. Med. Chem. 2003, 18, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Sousa, E.P.; Tiritan, M.E.; Oliveira, R.V.; Afonso, C.M.M.; Cass, Q.B.; Pinto, M.M.M. Enantiomeric resolution of kielcorin derivatives by HPLC on polysaccharide stationary phases using multimodal elution. Chirality 2004, 16, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Pereira, J.A.; Cravo, S.; Araújo, A.M.; Fernandes, C.; Pinto, M.M.M.; De Pinho, P.G.; Remião, F. Multi-milligram resolution and determination of absolute configuration of pentedrone and methylone enantiomers. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1100–1101, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.M.M.; Fernandes, C.; Tiritan, M.E. Chiral Separations in Preparative Scale: A Medicinal Chemistry Point of View. Molecules 2020, 25, 1931. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.E.; Tiritan, M.E.; Belaz, K.R.A.; Pedro, M.; Nascimento, M.S.J.; Cass, Q.B.; Pinto, M.M.M. Multimilligram enantioresolution of low-solubility xanthonolignoids on polysaccharide chiral stationary phases using a solid-phase injection system. J. Chromatogr. A 2006, 1120, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Masawang, K.; Tiritan, M.E.; Sousa, E.; De Lima, V.; Afonso, C.; Bousbaa, H.; Sudprasert, W.; Pedro, M.; Pinto, M.M. New chiral derivatives of xanthones: Synthesis and investigation of enantioselectivity as inhibitors of growth of human tumor cell lines. Bioorg. Med. Chem. 2014, 22, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.L.; Marques, S.; Silva, A.S.; Freitas, B.; Silva, P.M.A.; Pedrosa, J.; De Marco, P.; Bousbaa, H.; Fernandes, C.; Tiritan, M.E.; et al. Synthesis of New Chiral Derivatives of Xanthones with Enantioselective Effect on Tumor Cell Growth and DNA Crosslinking. ChemistrySelect 2020, 5, 10285–10291. [Google Scholar] [CrossRef]
- Fernandes, C.; Oliveira, L.; Tiritan, M.E.; Leitao, L.; Pozzi, A.; Noronha-Matos, J.B.; Correia-de-Sa, P.; Pinto, M.M. Synthesis of new chiral xanthone derivatives acting as nerve conduction blockers in the rat sciatic nerve. Eur. J. Med. Chem. 2012, 55, 1–11. [Google Scholar] [CrossRef]
- Ribeiro, J.; Veloso, C.; Fernandes, C.; Tiritan, M.E.; Pinto, M.M.M. Carboxyxanthones: Bioactive agents and molecular scaffold for synthesis of analogues and derivatives. Molecules 2019, 24, 180. [Google Scholar] [CrossRef] [Green Version]
- Twibanire, J.-d.K.; Grindley, T.B. Efficient and Controllably Selective Preparation of Esters Using Uronium-Based Coupling Agents. Org. Lett. 2011, 13, 2988–2991. [Google Scholar] [CrossRef]
- Fernandes, C.; Palmeira, A.; Ramos, I.I.; Carneiro, C.; Afonso, C.; Tiritan, M.E.; Cidade, H.; Pinto, P.C.A.G.; Saraiva, M.L.M.F.S.; Reis, S.; et al. Chiral derivatives of xanthones: Investigation of the effect of enantioselectivity on inhibition of cyclooxygenases (COX-1 and COX-2) and binding interaction with human serum albumin. Pharmaceuticals 2017, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Santos, Á.; Soares, J.X.; Cravo, S.; Tiritan, M.E.; Reis, S.; Afonso, C.; Fernandes, C.; Pinto, M.M.M. Lipophilicity assessement in drug discovery: Experimental and theoretical methods applied to xanthone derivatives. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1072, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Palmeira, A.; Santos, A.; Tiritan, M.E.; Afonso, C.; Pinto, M.M. Enantioresolution of Chiral Derivatives of Xanthones on (S,S)-Whelk-O1 and l-Phenylglycine Stationary Phases and Chiral Recognition Mechanism by Docking Approach for (S,S)-Whelk-O1. Chirality 2013, 25, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.L.; Palmeira, A.; Tiritan, M.E.; Fernandes, C.; Pinto, M.M.M. Resolution, determination of enantiomeric purity and chiral recognition mechanism of new xanthone derivatives on (S,S)-whelk-O1 stationary phase. Chirality 2017, 29, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Tiritan, M.E.; Cass, Q.; Kairys, V.; Fernandes, M.X.; Pinto, M. Enantioseparation and chiral recognition mechanism of new chiral derivatives of xanthones on macrocyclic antibiotic stationary phases. J. Chromatogr. A 2012, 1241, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Phyo, Y.Z.; Cravo, S.; Palmeira, A.; Tiritan, M.E.; Kijjoa, A.; Pinto, M.M.M.; Fernandes, C. Enantiomeric Resolution and Docking Studies of Chiral Xanthonic Derivatives on Chirobiotic Columns. Molecules 2018, 23, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do Carmo, J.P.; Phyo, Y.Z.; Palmeira, A.; Tiritan, M.E.; Afonso, C.; Kijjoa, A.; M Pinto, M.M.; Fernandes, C. Enantioseparation, recognition mechanisms and binding of xanthones on human serum albumin by liquid chromatography. Bioanalysis 2019, 11, 1255–1274. [Google Scholar] [CrossRef]
- Pinto, M.; Tiritan, M.E.; Fernandes, C.; Cass, Q. Fases Estacionárias Quirais baseadas em Derivados Xantónicos. WO/2011/010284A2, 27 January 2011. [Google Scholar]
- Fernandes, C.; Tiritan, M.E.; Pinto, M. Small Molecules as Chromatographic Tools for HPLC Enantiomeric Resolution: Pirkle-Type Chiral Stationary Phases Evolution. Chromatographia 2013, 76, 871–897. [Google Scholar] [CrossRef]
- Corbett, T.H.; Panchapor, C.; Polin, L.; Lowichik, N.; Pugh, S.; White, K.; Kushner, J.; Meyer, J.; Czarnecki, J.; Chinnukroh, S.; et al. Preclinical efficacy of thioxanthone SR271425 against transplanted solid tumors of mouse and human origin. Investig. New Drugs 1999, 17, 17–27. [Google Scholar] [CrossRef]
- Stevenson, J.P.; DeMaria, D.; Reilly, D.; Purvis, J.D.; Graham, M.A.; Lockwood, G.; Drozd, M.; O’Dwyer, P.J. Phase I/pharmacokinetic trial of the novel thioxanthone SR233377 (WIN33377) on a 5-day schedule. Cancer Chemother. Pharmacol. 1999, 44, 228–234. [Google Scholar] [CrossRef]
- Pica-Mattoccia, L.; Cioli, D. Studies on the Mode of Action of Oxamniquine and Related Schistosomicidal Drugs. Am. J. Trop. Med. Hyg. 1985, 34, 112–118. [Google Scholar] [CrossRef]
- Rosi, D.; Peruzzotti, G.; Dennis, E.W.; Berberian, D.A.; Freele, H.; Tullar, B.F.; Archer, S. Hycanthone,1 a New Active Metabolite of Lucanthone2. J. Med. Chem. 1967, 10, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Naidu, M.D.; Agarwal, R.; Pena, L.A.; Cunha, L.; Mezei, M.; Shen, M.; Wilson, D.M., III; Liu, Y.; Sanchez, Z.; Chaudhary, P.; et al. Lucanthone and Its Derivative Hycanthone Inhibit Apurinic Endonuclease-1 (APE1) by Direct Protein Binding. PLoS ONE 2011, 6, e23679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioli, D.; Pica-Mattoccia, L.; Archer, S. Antischistosomal drugs: Past, present … and future? Pharmacol. Ther. 1995, 68, 35–85. [Google Scholar] [CrossRef]
- Ong, T.m.; De Serres, F.J. Mutagenic evaluation of antischistosomal drugs and their derivatives in Neurospora crassa. J. Toxicol. Environ. Health 1975, 1, 271–279. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Foster, B.J.; Wozniak, A.; Heilbrun, L.K.; McCormick, J.I.; Ruble, P.E.; Graham, M.A.; Purvis, J.; Rake, J.; Drozd, M.; et al. Phase I Pharmacokinetic Study of the Novel Antitumor Agent SR233377. Clin. Cancer Res. 2000, 6, 3088–3094. [Google Scholar] [PubMed]
- Goncalves, P.H.; High, F.; Juniewicz, P.; Shackleton, G.; Li, J.; Boerner, S.; LoRusso, P.M. Phase I dose-escalation study of the thioxanthone SR271425 administered intravenously once every 3 weeks in patients with advanced malignancies. Investig. New Drugs 2008, 26, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, A.C.; Calvo, E.; Tolcher, A.W.; Rowinsky, E.K.; Shackleton, G.; Morrison, J.G.; Rafi, R.; VerMeulen, W.; Rothenberg, M.L. A Phase I Dose-Escalation Study of SR271425, an Intravenously Dosed Thioxanthone Analog, Administered Weekly in Patients With Refractory Solid Tumors. Am. J. Clin. Oncol. 2009, 32, 9–14. [Google Scholar] [CrossRef]
- Soria, J.-C.; Dieras, V.; Girre, V.; Yovine, A.; Mialaret, K.; Armand, J.-P. QTc Monitoring During a Phase I Study: Experience With SR271425. Am. J. Clin. Oncol. 2007, 30, 106–112. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmeira, A.; Sousa, E.; Helena Vasconcelos, M.; Pinto, M.; Fernandes, M.X. Structure and Ligand-based design of P-glycoprotein inhibitors: A historical perspective. Curr. Pharm. Des. 2012, 18, 4197–4214. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, C.A.; Callaghan, R. How can we best use structural information on P-glycoprotein to design inhibitors? Pharmacol. Ther. 2007, 113, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.B.; Kuo, C.L.; Lien, L.L.; Lien, E.J. Structure–activity relationship: Analyses of p-glycoprotein substrates and inhibitors. J. Clin. Pharm. Ther. 2003, 28, 203–228. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp Inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Richard, M.; Zoran, R. Designing Multiple Ligands—Medicinal Chemistry Strategies and Challenges. Curr. Pharm. Des. 2009, 15, 587–600. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, A.; Vasconcelos, M.H.; Paiva, A.; Fernandes, M.X.; Pinto, M.; Sousa, E. Dual inhibitors of P-glycoprotein and tumor cell growth: (Re)discovering thioxanthones. Biochem. Pharmacol. 2012, 83, 57–68. [Google Scholar] [CrossRef]
- Palmeira, A.; Rodrigues, F.; Sousa, E.; Pinto, M.; Vasconcelos, M.H.; Fernandes, M.X. New Uses for Old Drugs: Pharmacophore-Based Screening for the Discovery of P-Glycoprotein Inhibitors. Chem. Biol. Drug Des. 2011, 78, 57–72. [Google Scholar] [CrossRef]
- Garrigos, M.; Mir, L.M.; Orlowski, S. Competitive and Non-Competitive Inhibition of the Multidrug-Resistance-Associated P-glycoprotein ATPase. Eur. J. Biochem. 1997, 244, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Zeuthen, T.; Skovsgaard, T.; Stein, W.D. Competitive, non-competitive and cooperative interactions between substrates of P-glycoprotein as measured by its ATPase activity. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1997, 1361, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; Barbosa, D.J.; Palmeira, A.; Sousa, E.; Carvalho, F.; Bastos, M.d.L.; Remião, F. Colchicine effect on P-glycoprotein expression and activity: In silico and in vitro studies. Chem. Biol. Interact. 2014, 218, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, J.; Lima, R.T.; Sousa, D.; Gomes, A.S.; Palmeira, A.; Seca, H.; Choosang, K.; Pakkong, P.; Bousbaa, H.; Pinto, M.M.; et al. Screening a small library of xanthones for antitumor activity and identification of a hit compound which induces apoptosis. Molecules 2016, 21, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safety and Efficacy Study of Lucanthone When Used in Combination With Temozolomide(TMZ) and Radiation to Treat Glioblastoma Multiforme(GBM). Available online: https://clinicaltrials.gov/ct2/show/NCT01587144 (accessed on 25 November 2020).
- Lima, R.T.; Sousa, D.; Paiva, A.M.; Palmeira, A.; Barbosa, J.; Pedro, M.; Pinto, M.M.; Sousa, E.; Vasconcelos, M.H. Modulation of autophagy by a thioxanthone decreases the viability of melanoma cells. Molecules 2016, 21, 1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, R.T.; Sousa, D.; Gomes, A.S.; Mendes, N.; Matthiesen, R.; Pedro, M.; Marques, F.; Pinto, M.M.; Sousa, E.; Helena Vasconcelos, M. The antitumor activity of a lead thioxanthone is associated with alterations in cholesterol localization. Molecules 2018, 23, 3301. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, A.; Sousa, E.; Fernandes, M.X.; Pinto, M.M.; Helena Vasconcelos, M. Multidrug resistance reversal effects of aminated thioxanthones and interaction with cytochrome P450 3A4. J. Pharm. Pharm. Sci. 2011, 15, 31–45. [Google Scholar] [CrossRef]
- Kannan, P.; Telu, S.; Shukla, S.; Ambudkar, S.V.; Pike, V.W.; Halldin, C.; Gottesman, M.M.; Innis, R.B.; Hall, M.D. The “Specific” P-Glycoprotein Inhibitor Tariquidar Is Also a Substrate and an Inhibitor for Breast Cancer Resistance Protein (BCRP/ABCG2). ACS Chem. Neurosci. 2011, 2, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Martins, E.; Silva, R.; Pinto, M.M.M.; Remião, F.; Sousa, E.; Fernandes, C. Chiral Thioxanthones as Modulators of P-glycoprotein: Synthesis and Enantioselectivity Studies. Molecules 2018, 23, 626. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; De Lourdes Bastos, M.; Remião, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef]
- Maglich, J.M.; Stoltz, C.M.; Goodwin, B.; Hawkins-Brown, D.; Moore, J.T.; Kliewer, S.A. Nuclear Pregnane X Receptor and Constitutive Androstane Receptor Regulate Overlapping but Distinct Sets of Genes Involved in Xenobiotic Detoxification. Mol. Pharmacol. 2002, 62, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, R.; Palmeira, A.; Carmo, H.; Barbosa, D.J.; Gameiro, M.; Gomes, A.; Paiva, A.M.; Sousa, E.; Pinto, M.; Bastos, M.L.; et al. P-glycoprotein induction in Caco-2 cells by newly synthetized thioxanthones prevents paraquat cytotoxicity. Arch. Toxicol. 2015, 89, 1783–1800. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.F.; Ponte, F.; Silva, R.; Rocha-Pereira, C.; Sousa, E.; Pinto, M.; Bastos, M.D.L.; Remião, F. Quantification of 1-(propan-2-ylamino)-4-propoxy-9H-thioxanthen-9-one (TX5), a newly synthetized P-glycoprotein inducer/activator, in biological samples: Method development and validation. Biomed. Chromatogr. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.; Gil-Martins, E.; Silva, B.; Rocha-Pereira, C.; Sousa, M.E.; Remião, F.; Silva, R. Xanthones as P-glycoprotein modulators and their impact on drug bioavailability. Expert Opin. Drug Metab. Toxicol. 2020. [Google Scholar] [CrossRef]



























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, M.M.M.; Palmeira, A.; Fernandes, C.; Resende, D.I.S.P.; Sousa, E.; Cidade, H.; Tiritan, M.E.; Correia-da-Silva, M.; Cravo, S. From Natural Products to New Synthetic Small Molecules: A Journey through the World of Xanthones. Molecules 2021, 26, 431. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020431
Pinto MMM, Palmeira A, Fernandes C, Resende DISP, Sousa E, Cidade H, Tiritan ME, Correia-da-Silva M, Cravo S. From Natural Products to New Synthetic Small Molecules: A Journey through the World of Xanthones. Molecules. 2021; 26(2):431. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020431
Chicago/Turabian StylePinto, Madalena M. M., Andreia Palmeira, Carla Fernandes, Diana I. S. P. Resende, Emília Sousa, Honorina Cidade, Maria Elizabeth Tiritan, Marta Correia-da-Silva, and Sara Cravo. 2021. "From Natural Products to New Synthetic Small Molecules: A Journey through the World of Xanthones" Molecules 26, no. 2: 431. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020431