
One-Pot Synthesis of Novel Multisubstituted 1-Alkoxyindoles


College of Pharmacy and Innovative Drug Center, Duksung Women’s University, Seoul 01369, Korea
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(5), 1466; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051466
Submission received: 17 January 2021
/
Revised: 3 March 2021
/
Accepted: 4 March 2021
/
Published: 8 March 2021
(This article belongs to the Special Issue Indole and Its Bioisosteric Replacements in Medicinal Chemistry)
Abstract
:Studies on a one-pot synthesis of novel multisubstituted 1-alkoxyindoles 1 and their mechanistic investigations are presented. The synthesis of 1 was successfully achieved through consecutive four step reactions from substrates 2. The substrates 2, prepared through a two-step synthetic sequence, underwent three consecutive reactions of nitro reduction, intramolecular condensation, and nucleophilic 1,5-addition to provide the intermediates, 1-hydroxyindoles 8, which then were alkylated in situ with alkyl halide to afford the novel target products 1. We optimized the reaction conditions for 1 focusing on the alkylation step, along with the consideration of formation of intermediates 8. The optimized condition was SnCl2·2H2O (3.3 eq) and alcohols (R1OH, 2.0 eq) for 1–2 h at 40 °C and then, base (10 eq) and alkyl halides (R2Y, 2.0 eq) for 1–4 h at 25–50 °C. Notably, all four step reactions were performed in one-pot to give 1 in good to modest yields. Furthermore, the mechanistic aspects were also discussed regarding the reaction pathways and the formation of side products. The significance lies in development of efficient one-pot reactions and in generation of new 1-alkoxyindoles.
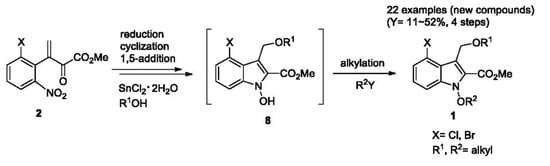
1. Introduction
1-Alkoxyindoles are the compounds that are similar to the indole structure, but have an alkoxy group (-OR) instead of H at the N(1) site, as shown in Figure 1. Due to the presence of alkoxy group, 1-alkoxyindole compounds are supposed to have different physical and chemical properties compared to indole compounds. While an indole structure is commonly found in natural products, the 1-alkoxyindole structure rarely appears. Recently, 1-alkoxyindole derivatives have been emerging as alternative compounds to indole compounds. Due to the 1-hydroxy group, 1-hydroxyindoles are known to be more polar and acidic (pKa 8.1–9.8) than indoles, leading to higher exposure of the polar 1-OH group and easier deprotonation followed by further functionalization [1]. Natural products and synthetic derivatives including the 1-hydroxyindole and 1-alkoxyindole structure have attracted much attention due to their various biological activities. For example, stephacidin B [2], tetrahydro-β-carboline derivatives [3], and (R)-paniculidine B [4] have cytotoxic activities and, in particular, nocathiacin is known as a promising natural antibiotic [5]. Synthetic 1-hydroxyindole compounds have also shown inhibitory activities for lactate dehydrogenase-A (LDH-A) [6]. N-alkoxyindole-3-carbinol (I3C) compounds are proved to regulate cell cycle-related gene transcription and as a result exhibit inhibitory activities in human breast cancer cell lines [7]. Despite these biological activities of natural and synthetic products containing 1-hydroxyindole and 1-alkoxyindole structures, their derivatizations have not been extensively studied due to the lack of tolerable synthetic methods and the instability of those compounds.
Since the hydroxy group in 1-hydroxyindole compounds is a kind of nucleophile, alkylations on that site could occur smoothly in the presence of bases. Thus, 1-methoxyindoles, a typical class of 1-alkoxyindoles, could be synthesized through the methylation of 1-hydroxyindole using methyl iodide, diazomethane, or dimethyl sulfate in the presence of an appropriate base [1,8]. As precursors of 1-alkoxyindoles, 1-hydroxyindoles have also drawn our attention. Previously, the 1-hydroxyindole derivative was successfully applied for the total synthesis of nocathiacin [9]. We also reported methods to synthesize 2,3-disubstituted 1-hydroxyindole compounds [10,11,12,13]. Other methods by reduction of the indole system to 2,3-dihydroindole, followed by an oxidation (Na2WO4/H2O2) were applied to give 1-hydroxyindoles [14]. The other methods include the direct synthesis of 1-alkoxyindoles through intramolecular cyclization of the alkoxyimine structure, without using 1-hydroxyindole as an intermediate [15]. However, these methods have suffered from a narrow range of derivative diversity, low yields, and poor reproducibility due to their chemical instabilities.
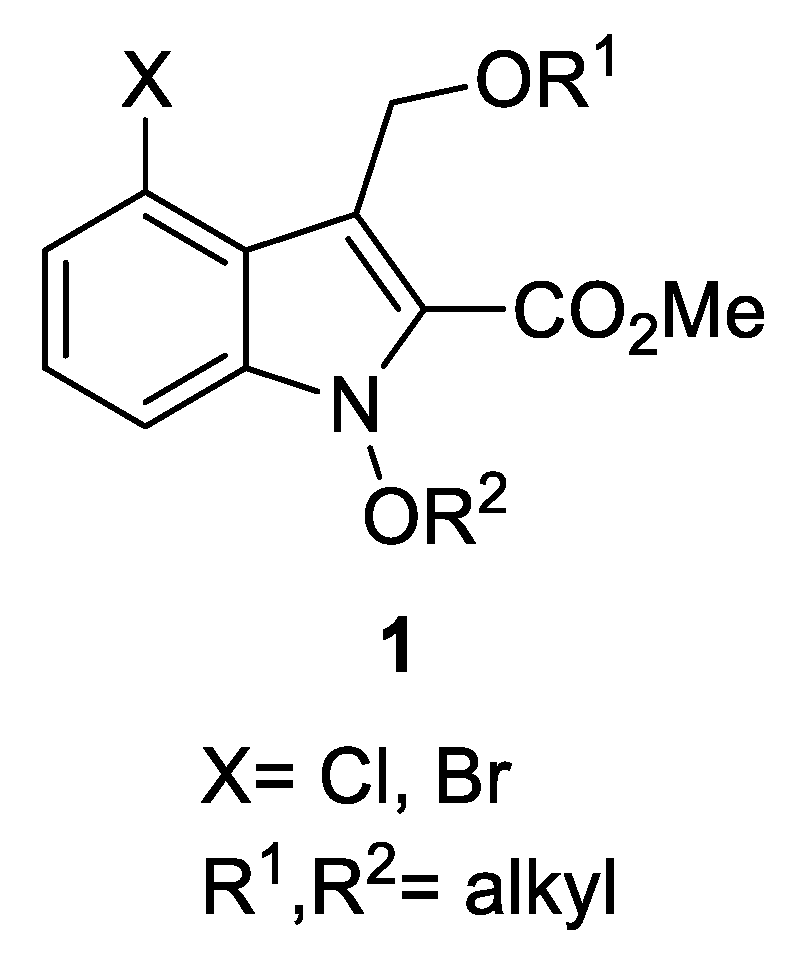
For decades, chemical instabilities have hampered the extensive studies on these compounds. So, we aimed to design and synthesize the compounds of improved chemical stability compared to 1-hydroxyindoles, with developing tolerable synthetic methods. It is believed that an alkyl or acyl group directly connected to the indole skeleton could stabilize the 1-hydroxyindole structure. For an example, 1-alkylation of the 1-hydroxy group is believed to improve the stability of those compounds [16]. Thus, we focus on creating new derivatives of multisubstituted alkoxyindole by introducing various groups at C(2), C(3), and C(4), and alkyl groups at N(1) in 1-hydroxyindoles, expecting to improve their chemical stabilities, as shown in Figure 2. It is also meaningful in medicinal chemistry to further expect the improved absorption in the body by lowering the polarity of the compounds with the alkylation. The significance of our study lies in the development of efficient one-pot reactions of a four-step sequence, and in creation of new 1-alkoxyindole compounds with improved stabilities, otherwise difficult to synthesize.
2. Results and Discussion
2.1. Synthesis of Conjugate Nitro Ketoesters
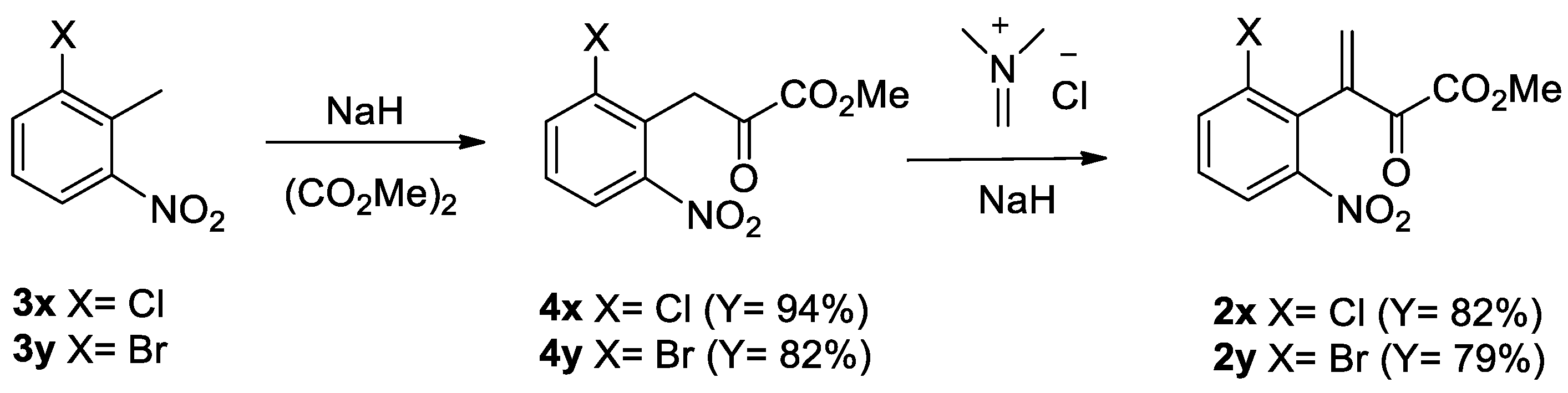
First, we needed to prepare the required substrates to synthesize the target compounds 1. For the synthesis of 2 we applied our previous two-step synthetic sequence [17,18,19] with minor modifications, as shown in Scheme 1. Nitrotoluenes 3 were reacted with sodium hydride and dimethyl oxalate in the N,N-dimethylformamide (DMF) solvent to afford ketoesters 4 in excellent yields. In this process, excess sodium hydride and efficient degassing processes were applied to produce 4 in improved yields. Then, ketoesters 4 were treated with sodium hydride and dimethylmethyleneiminium chloride in tetrahydrofuran (THF) to afford conjugate nitro ketoesters 2 in good yields. As a result, the synthesis of substrates 2 were achieved in improved yields compared with previous results [19].
2.2. Optimization for Formation of 1-Alkoxyindoles 1
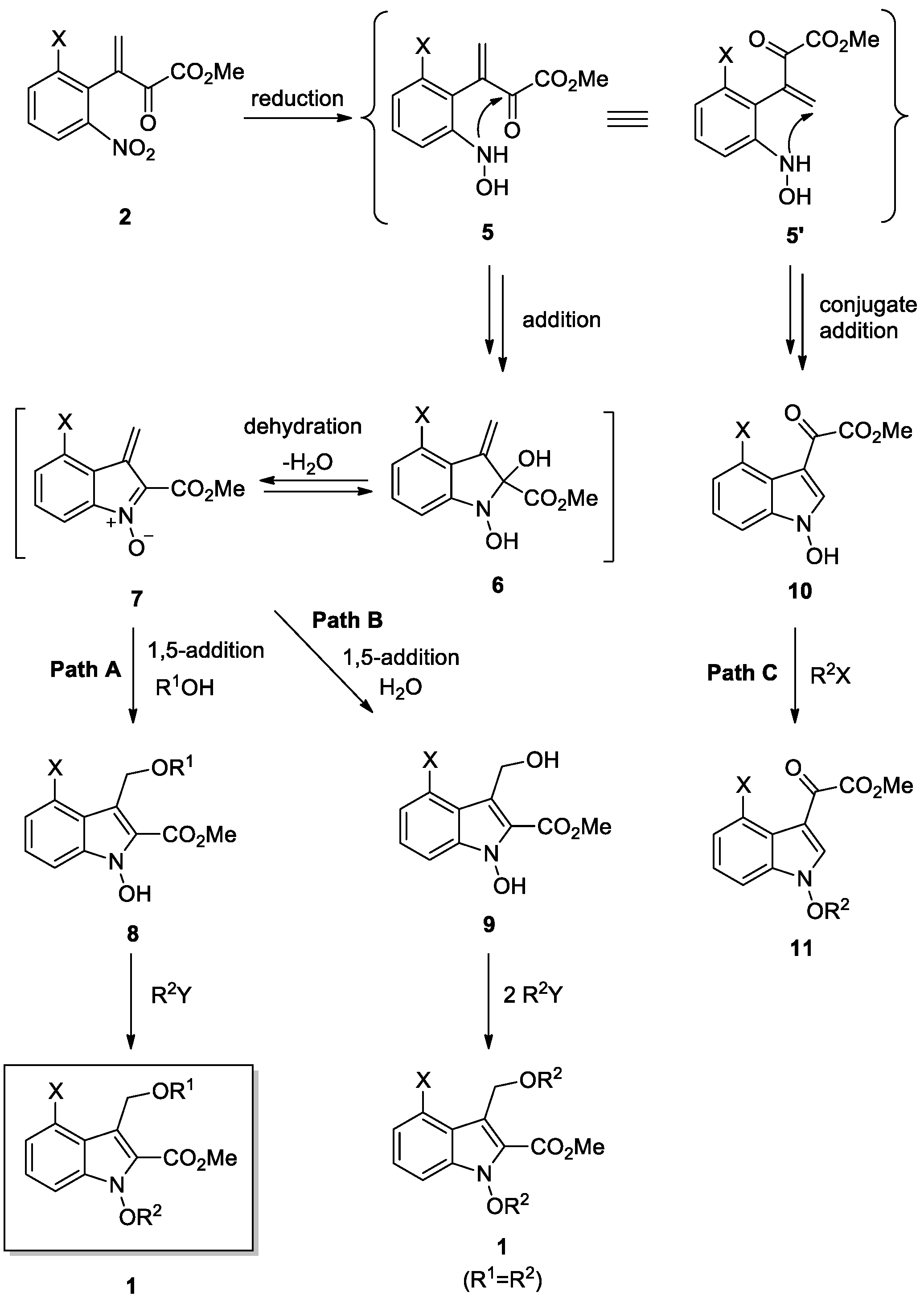
We adopted SnCl2·2H2O as a reducing agent for our reactions according to our previous procedures for intermediates 8 [9,17]. As shown in Scheme 2, substrates 2 undergo a reduction by SnCl2·2H2O to give hydroxyamines 5, intramolecular cyclization (addition) to give hydroxyindolines 6, dehydration to give conjugate nitrones 7, and 1,5-addition of nucleophile (R1OH) to afford the intermediate 1-hydroxyindoles 8 and then, 8 undergo alkylation with alkyl halides (R2Y) to provide the target compounds, 1-alkoxyindoles 1, which were all run in one-pot.
We then tried to optimize the reaction conditions for target compounds 1 including the intermediates 8. Although the procedures for intermediates 8 were already known [9,17], we had to reoptimize the whole processes since the following alkylation reaction to form 1 could be inevitably affected by the condition for intermediates 8. For examples, the remained nucleophile (R1OH) could interfere with the following alkylation step by reacting with alkyl halide (R2Y), and the excess amount of SnCl2·2H2O could also affect the alkylation step. Furthermore, the selection of the appropriate base for the alkylation step would be important. Considering these, we first tried to conduct the reactions with several bases in the preliminary-optimized condition (2 h, 40 °C for 8y; 2 h, 25 °C for 1y). So, we tested triethylamine (TEA), N,N-diisopropylethylamine (DIEA), 4-dimethylaminopyridine (DMAP), and 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU) as organic bases, and K2CO3 as an inorganic base. With 2y and SnCl2·2H2O (3.3 eq), we chose benzyl alcohol (BnOH, 2 eq) as a template nucleophile and benzyl bromide (BnBr, 2 eq) as a template alkyl halide with base (10 eq) affording 1-benzyloxyindole 1yh, and the results were shown in Table 1. Among organic bases, DBU provided the best yield (41%), followed by TEA (23%), DIEA (17%), and DMAP (13%). The inorganic base K2CO3 gave poor yield. The order of basicity of organic bases (DMAP < TEA < DIEA < DBU) [20,21] roughly corresponded with the order of yields, except TEA and DIEA cases. The nucleophilicity of 1-OH could be activated by base and so a stronger base could afford better yield. Accordingly, the DBU (a guanidine-type base) gave the best result. When we varied the amount of bases, the results were not improved much. Thus, we chose DBU for our further reactions.
With the selected base DBU and substrate 2x, we first attempted to perform the systematic studies on the reaction conditions suitable for formation of 1-benzyloxyindole 1xh. As the whole sequence of reactions could be triggered by reduction of the aromatic nitro group, the amount of the reducing agent would be considered one of the most important factors. Thus, we optimized the reaction conditions by varying the amount of SnCl2·2H2O (2.5–3.7 eq). We also varied the amount of nucleophile (BnOH), base (DBU) and alkyl halide (R2Y), and the results were shown in Table 2. Notably, since the reducing agent SnCl2·2H2O could be hydrolyzed to give HCl that makes the reaction media acidic, we used a large amount of base (DBU) in proportion to the amount of SnCl2·2H2O. We first tested the amount of SnCl2·2H2O (2.5–3.7 eq) and, correspondingly, DBU (7.6–11.2 eq). In general, lower levels and higher level of those reagents than 3.3 eq for SnCl2·2H2O and 10 eq for DBU, respectively, provided poor yields (entries 1, 2, and 9, Table 2). So, under this condition we further tested the amount of nucleophile (BnOH) and alkyl halide (BnBr). In general, higher amounts of nucleophile and alkyl halide than 2.0 eq provided higher yields (32 → 33 → 42 → 45%) (entries 5–8, Table 2). However, one issue was involved in those reactions with higher amounts of them. Excess alcohols in 1,5-addition reaction could react with alkyl halide in the alkylation step, producing dialkyl ethers. As expected, we observed the formation of large amount of dibenzyl ether as a byproduct, which is difficult to remove. In addition to the isolation problem, the economical reaction efficiencies were poor due to the excess amount of reagents and the large amount of byproducts. Considering these points, we adopted the condition of 2 eq of nucleophile and alkyl halide (entry 5, Table 2) despite the slightly lower yield. Taken together, we chose the optimized conditions for 1; 1.0 eq of 2x, 3.3 eq of SnCl2·2H2O, 2.0 eq of BnOH (2 h, 40 °C), and then 10.0 eq of DBU, 2 eq of BnBr (2 h, 25 °C), which was applied to all other reactions, unless otherwise noted. Notably, in this method, 1-alkoxyindoles 1 were synthesized in one-pot without separation of the intermediates 8. We compared the result of reactions with isolation and without isolation of the intermediate 8xh, and found that the yield (32%) of 1xh in the one-pot reaction was higher than that (28%) of 1xh in two separate reactions (45% for 8xh and 62% for 1xh).
2.3. Synthesis of New Derivatives of 1-Alkoxyindoles 1
Using the optimized condition, various new derivatives of 1 were synthesized, as shown in Table 3. The substrates 2 were treated with SnCl2·2H2O in dimethoxyethane (DME) for 1–2 h at 40 °C in the presence of a nucleophile and 4Å molecular sieves to give the intermediates 8, which were then alkylated in situ with alkyl halide in the presence of DBU for 1–4 h at 25–50 °C in one-pot, finally affording to the target compounds 1. In the alkylation step, vigorous stirring was required to make the reaction mixture a good suspension condition. Here, we used various alcohols as nucleophiles to synthesize 1xa-1ym (22 new derivatives). When primary alcohols and primary alkyl halides were used (entries 1–9, 12–19, and 22, Table 3), the reactions provided 1 in fairly good yields for four-step sequence (18–52%). Interestingly, when we compared the results of 1x series by the size of alkyl groups in both alcohols and alkyl halides, we found that the reaction using methanol with methyl iodide (methyl–methyl case, 1xa) gave the best yield (43%). The reaction using methanol with octyl bromide (methyl–octyl case, 1xm) gave 33% yield, and the reaction using octanol with methyl iodide (octyl–methyl case, 1xn) gave a similar yield (36%). The reactions for ethyl–ethyl (1xb), propyl–propyl (1xc), and butyl–butyl (1xd) cases gave modest yields (30–32%). The reactions for pentyl–pentyl (1xe), hexyl–hexyl (1xf), and octyl–octyl (1xg) cases gave relatively poor yields (20–22%). These observations implicated that the size of alkyl groups seems to affect the results, providing better yields with smaller alkyl groups, and that both nucleophilic addition of alcohol and alkylation of 1-OH group seem to influence the final results. Reactions with secondary alcohols and secondary alkyl halides (entries 10, 11, and 20, Table 3) gave 1 in relatively low yields (11–16%). The reaction with cyclohexyl bromide did not successfully proceed at 25 °C, so we elevated reaction temperature to 50 °C for an extended time (4 h), obtaining acceptable yield (11%, entry 11, Table 3). This implied that the steric effect of alkyl halides might have an influence on alkylation of 1-hydroxyindoles. Notably, the reactions with the methyl group (entries 1 and 15) and benzyl group (entries 8 and 19) afforded higher yields than the other cases. In addition, comparing the reactions for 1x (Cl group) and 1y (Br group), we found no consistent trends despite slightly higher yields for 1y in some cases.
2.4. Mechanistic Investigations on Reaction Pathways
We investigated the reaction mechanisms and pathways based on the generated products, as shown in Scheme 3. Reduction of the substrates 2 could give two conformers of hydroxyamine, 5 and 5’. Here are three following pathways involved, pathways A, B, and C. Pathways A and B proceed through conformer 5, and pathway C through conformer 5’. We first investigated the reactions through conformer 5, which then could undergo intramolecular condensation (addition and dehydration) to give conjugate nitrone 7. Subsequent 1,5-addition of 7 with a nucleophile (R1OH) would give 1-hydroxyindoles 8, which then undergo alkylation with alkyl halide (R2Y) to give 1-alkoxyindoles 1 (Path A). Interestingly, in the process of 1,5-nucleophilic addition of 7, H2O could react as a nucleophile instead of alcohol, leading to the generation of dihydroxy species 9 (Path B). This species 9 could also react with 2.0 eq of alkyl halide (R2Y) to produce 1-alkoxindoles 1 (R1 = R2). Furthermore, the conformer 5’ could undergo intramolecular conjugate addition to give another type of 1-hydroxyindole compounds 10 with a different skeleton, which is consistent with the previous observation [9]. These compounds 10 would also undergo alkylation with alkyl halide to give 1-alkoxyindole-3-carboxylates 11 that are considered as rearranged products compared to 1. However, the formation of compounds 10 and 11 was variable and, sometimes, it was difficult to isolate and identify these compounds. When we conducted the reactions for 1xl and 1xm (entries 12 and 13), we observed the formation 11xl (R2 = Me, 32% yield) and 11xm (R2 = Oct, 3% yield) along with the main products 1xl and 1xm, respectively. In most of the reactions in Table 3, a substantial amount (10–35%) of rearranged products 11 were also formed, which might explain the low yields of the products 1. In addition, despite full conversion of the starting material in the reactions, significant tarring and side products might cause low yields.
3. Materials and Methods
3.1. General Methods
Reagents used in this study were purchased from Sigma-Aldrich (Darmstadt, Germany), Thermo Fisher (Waltham, MA, USA) and TCI (Tokyo, Japan). They were of commercial quality and used without further purification, unless otherwise stated. Reactions were periodically monitored by thin-layer chromatography (TLC) carried on 0.25 mm Merck silica gel plates (20 cm × 20 cm; Merck F254) (Darmstadt, Germany) and visualized under UV light. Purifications were performed by preparative TLC (PTLC) and column chromatography. PTLC separations were carried out on the same silica gel plates. Column chromatography was performed using Merck silica gels (230–400 mesh) (Zvornik, Bosnia and Herzegovina). Melting points were determined in Deckgläser Cover Glasses (Lauda-Königshofen, Germany) using a Thermo Scientific 00590Q apparatus (Dubuque, Iowa, USA). 1H (300 MHz) and 13C (75 MHz) NMR spectra were recorded on a Bruker DRX 300 spectrometer (Zürich, Switzerland) (See Supplementary Materials) and chemical shifts (δ) are expressed relative to tetramethylsilane (TMS). Mass spectra were obtained in EI or ESI ionization modes (Agilent, Santa Clara, CA, USA). High resolution mass spectra were obtained using JEOL apparatus (Tokyo, Japan) at the Korea Basic Science Institute, Republic of Korea. HPLC analyses were performed using the following Waters Associate Units: 515 A pump, 515 B pump, dual λ absorbance 2487 detector, 717 plus autosampler, and COSMOSIL 5C18-AR-Ⅱ Packed Column (4.6 mm × 250 mm) (Worcester, MA, USA). The product analyses were performed using a linear gradient condition: from 70% A (aqueous) and 30% B (acetonitrile) for 3 min (isocratic), then to 10% A and 90% B in 30 min (gradient). Finally, keep 10% A and 90% B for 5 min. The flow rate was 1 mL/min with eluent monitoring at 254 nm. HPLC solvents were filtered (aqueous solution with Millipore HVLP, 0.45 mm; MeCN with Millipore HV, 0.45 mm) and degassed before use.
3.2. Substrate Synthesis
Methyl 3-(2′-Chloro-6′-nitrophenyl)-2-oxopropanoate (4x) [19]
Dimethyl oxalate (4.02 g, 34.0 mmol, 5.0 eq) and 2-chloro-6-nitrotoluene (3x, 1.17 g, 6.8 mmol, 1.0 eq) was dissolved in anhydrous DMF (8.2 mL). To a stirred mixture of NaH (60% in mineral oil, 1.09 g, 27.2 mmol, 4.0 eq) in anhydrous DMF (4.1 mL) at 0 °C was added dropwise a solution of dimethyl oxalate and 2-chloro-6-nitrotoluene. Stirring was continued for 1 h at 0 °C during which it turned a reddish-brown. The reaction mixture was allowed to warm to 25 °C and stirred for an additional 4 h. The reaction mixture turned dark red. The reaction mixture was quenched with saturated NH4Cl (15 mL) at 0 °C, extracted with EtOAc (2 × 20 mL) and washed with H2O (2 × 20 mL). The organic layer was dried (MgSO4) and concentrated in vacuo. The residue was purified by column chromatography (1:4 → 1:2 EtOAc/hexanes) to get the compound 4x (1.65 g, 94%) as a pale-yellow solid. Mp 58–59 °C; Rf 0.24 (1:4 EtOAc/hexanes); HPLC tR21.2 min; 1H NMR (300 MHz, CDCl3): δ 8.00 (dd, J = 8.2, 1.1 Hz, 1H, Ar), 7.74 (dd, J = 8.2, 1.1 Hz, 1H, Ar), 7.46 (t, J = 8.2 Hz, 1H, Ar), 4.72 (s, 2H, C(2)CH2), 3.96 (s, 3H, CO2CH3); 13C NMR (75 MHz, CDCl3): δ 188.0 (COCO2CH3), 160.1 (CO2CH3), 150.5 (Ar), 137.7 (Ar), 134.7 (Ar), 129.0 (Ar), 127.6 (Ar), 124.0 (Ar), 53.8 (CO2CH3), 42.8 (C(2)CH2); MS m/z 257 [M]+; HRMS (+ESI) calcd for C10H8ClNNaO5 [M + Na]+ 279.9989, found 279.9983. Spectral data are in accordance with literature information [19].
Methyl 3-(2′-Bromo-6′-nitrophenyl)-2-oxopropanoate (4y) [19]
Dimethyl oxalate (2.01 g, 17.0 mmol, 5.0 eq) and 2-bromo-6-nitrotoluene (3y, 735 mg, 3.4 mmol, 1.0 eq) was dissolved in anhydrous DMF (4.1 mL). To a stirred mixture of NaH (60% in mineral oil, 544 mg, 13.6 mmol, 4.0 eq) in anhydrous DMF (2.04 mL) at 0 °C was added dropwise a solution of dimethyl oxalate and 2-bromo-6-nitrotoluene. Stirring was continued for 1 h at 0 °C during which it turned a reddish-brown. The reaction mixture was allowed to warm to 25 °C and stirred for additional 4 h. The reaction mixture turned dark red. The reaction mixture was quenched with saturated NH4Cl (10 mL) at 0 °C, extracted with EtOAc (2 × 10 mL), and washed with H2O (2 × 10 mL). The organic layer was dried (MgSO4) and concentrated in vacuo. The residue was purified by column chromatography (1:4 → 1:2 EtOAc/hexanes) to get the compound 4y (848 mg, 82%) as a pale-yellow solid. Mp 70 °C; Rf 0.22 (1:4 EtOAc/hexanes); HPLC tR 21.7 min; 1H NMR (300 MHz, CDCl3): δ 8.02 (dd, J = 8.2, 1.1 Hz, 1H, Ar), 7.92 (dd, J = 8.2, 1.1 Hz, 1H, Ar), 7.39 (t, J = 8.2 Hz, 1H, Ar), 4.75 (s, 2H, C(2)CH2), 3.97 (s, 3H, CO2CH3); 13C NMR (75 MHz, CDCl3): δ 187.8 (COCO2CH3), 160.7 (CO2CH3), 150.6 (Ar), 138.0 (Ar), 129.7 (Ar), 129.2 (Ar), 128.4 (Ar), 124.6 (Ar), 53.7 (CO2CH3), 43.9 (C(2)CH2); MS m/z 301 [M]+; HRMS (+ESI) calcd for C10H8BrNNaO5 [M + Na]+ 323.9484, found 323.9475. Spectral data are in accordance with literature information [19].
Methyl 3-(2′-Chloro-6′-nitrophenyl)-2-oxobut-3-enoate (2x) [19]
Ketoester (4x, 1.04 g, 4.03 mmol, 1.0 eq) was dissolved in anhydrous THF (34 mL). To a stirred mixture of NaH (60% in mineral oil, 178 mg, 4.43 mmol, 1.1 eq) in anhydrous THF (65 mL) at 0 °C was added dropwise a solution of ketoester. After stirring for 1 h at 0 °C, N,N-dimethylmethyleneiminium chloride (1.3 g, 12.08 mmol, 3.0 eq) was added and stirred for 1 h at 0 °C. The reaction mixture was allowed to warm to 25 °C and stirred for additional 3 h. The reaction mixture turned pale-yellow. The reaction mixture was quenched with saturated NH4Cl (10 mL) at 0 °C, extracted with EtOAc (2 × 50 mL), and washed with H2O (2 × 50 mL). The organic layer was dried (MgSO4) and concentrated in vacuo. The residue was purified by column chromatography (1:4 → 1:1 EtOAc/hexanes) to get the compound 2x (890 mg, 82%) as a pale-yellow solid. Mp 67–68 °C; Rf 0.44 (1:2 EtOAc/hexanes); HPLC tR 21.7 min; 1H NMR (300 MHz, CDCl3): δ 7.99 (dd, J = 8.2, 1.1 Hz, 1H, Ar), 7.75 (dd, J = 8.2, 1.1 Hz, 1H, Ar), 7.50 (t, J = 8.2 Hz, 1H, Ar), 6.80 (s, 1H, CHH), 6.19 (s, 1H, CHH), 3.93 (s, 1H, OCH3); 13C NMR (75 MHz, CDCl3): δ 183.0 (COCO2CH3), 162.5 (CO2CH3), 149.7 (C(2)CCH2), 139.8 (Ar), 136.3(C(2)CCH2), 134.7 (Ar), 134.5 (Ar), 130.6 (Ar), 130.1 (Ar), 123.2 (Ar), 53.3 (OCH3); MS m/z 269 [M]+; HRMS (+ESI) calcd for C11H8ClNNaO5 [M + Na]+ 291.9989, found 291.9983. Spectral data are in accordance with literature information [19].
Methyl 3-(2′-Bromo-6′-nitrophenyl)-2-oxobut-3-enoate (2y) [19]
Ketoester (4y, 829 mg, 2.74 mmol, 1.0 eq) was dissolved in anhydrous THF (23 mL). To a stirred mixture of NaH (60% in mineral oil, 121 mg, 3.02 mmol, 1.1 eq) in anhydrous THF (46 mL) at 0 °C was added dropwise a solution of ketoester. After stirring for 1 h at 0 °C, N,N-dimethylmethyleneiminium chloride (770 mg, 8.23 mmol, 3.0 eq) was added and stirred for 1 h at 0 °C. The reaction mixture was allowed to warm to 25 °C and stirred for additional 3 h. The reaction mixture turned pale yellow. The reaction mixture was quenched with saturated NH4Cl (10 mL) at 0 °C, extracted with EtOAc (2 × 50 mL) and washed with H2O (2 × 50 mL). The organic layer was dried (MgSO4) and concentrated in vacuo. The residue was purified by column chromatography (1:4 → 1:1 EtOAc/hexanes) to get the compound 2y (680 mg, 79%) as a pale-yellow solid. Mp 80 °C; Rf 0.44 (1:2 EtOAc/hexanes); HPLC tR 21.9 min; 1H NMR (300 MHz, CDCl3): δ 8.02 (dd, J = 8.2, 1.1 Hz, 1H, Ar), 7.94 (dd, J = 8.2, 1.1 Hz, 1H, Ar), 7.44 (t, J = 8.2 Hz, 1H, Ar), 6.79 (s, 1H, CHH), 6.17 (s, 1H, CHH), 3.94 (s, 3H, OCH3); 13C NMR (75 MHz, CDCl3): δ 183.0 (COCO2CH3), 162.5 (CO2CH3), 149.7 (C(2)CCH2), 141.7 (Ar), 137.8 (C(2)CCH2), 134.4 (Ar), 132.4 (Ar), 130.4 (Ar), 126.2 (Ar), 123.8 (Ar), 53.3 (OCH3); MS m/z 313 [M]+; HRMS (+ESI) calcd for C11H8BrNNaO5 [M + Na]+ 335.9484, found 335.9474. Spectral data are in accordance with literature information [19].
3.3. General Procedure for the Synthesis of 1-Alkoxyindoles 1
SnCl2·2H2O and 4Å molecular sieves stirred in DME for 30 min at 25 °C. To a stirred mixture was added alcohol and conjugate ketoester 2. The resulting mixture was stirred for 1–2 h at 40 °C. After checking that the starting material was disappeared by using TLC, DBU was added and stirred strongly for 30 min at 25 °C. The alkyl halide was then added and stirring was continued for 1–4 h at 25–50 °C until reaction completed. The reaction mixture was diluted with CH2Cl2 and washed with brine. The organic layer was dried (MgSO4) and concentrated in vacuo to afford a crude residue. The residue was purified by preparative TLC (PTLC) and column chromatography to give 1-alkoxyindoles 1. Spectral data of all compounds were in good accordance with the literature information.
Methyl 4-Chloro-1-methoxy-3-(methoxymethyl)-1H-indole-2-carboxylate (1xa)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2x (30 mg, 0.11 mmol, 1.0 eq) for 1 h at 40 °C then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and methyl iodide (14 μL, 0.22 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xa (13.7 mg, 43%) as a pale-yellow solid. Mp 59–60 °C; Rf 0.40 (1:4 EtOAc/hexanes); HPLC tR 20.8 min; UV vis (CH3CN-H2O) λmax 236, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.38 (d, J = 8.2 Hz, 1H, Ar), 7.26 (t, J = 8.4 Hz, 1H, Ar), 7.18 (d, J = 5.9 Hz, 1H, Ar), 5.07 (s, 2H, C(3)CH2O), 4.18 (s, 3H, N(1)OCH3), 4.01 (s, 3H, CO2CH3), 3.45 (s, 3H, CH2OCH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 135.9 (Ar), 128.6 (Ar), 126.4 (Ar), 125.3 (Ar), 123.1 (Ar), 119.7 (Ar), 116.4 (Ar), 108.2 (Ar), 66.4 (N(1)OCH3), 63.7(CH2OCH3), 58.1 (C(3)CH2O), 52.4 (CO2CH3); MS m/z 283 [M]+; HRMS (+ESI) calcd for C13H14ClNNaO4 [M + Na]+ 306.0509, found 306.0509.
Methyl 4-Chloro-1-ethoxy-3-(ethoxymethyl)-1H-indole-2-carboxylate (1xb)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), ethanol (22 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and bromoethane (28 μL, 0.37 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xb (17.8 mg, 31%) as a pale-yellow solid. Mp 42 °C; Rf 0.49 (1:4 EtOAc/hexanes); HPLC tR 25.8 min; UV vis (CH3CN-H2O) λmax 235, 298nm; 1H NMR (300 MHz, CDCl3): δ 7.34 (d, J = 8.1 Hz, 1H, Ar), 7.23 (t, J = 7.8 Hz, 1H, Ar), 7.15 (d, J = 7.0 Hz, 1H, Ar), 5.09 (s, 2H, C(3)CH2O), 4.41 (q, J = 7.1 Hz, 2H, N(1)OCH2), 3.99 (s, 3H, CO2CH3), 3.65 (q, J = 7.0 Hz, 2H, OCH2CH3), 1.44 (t, J = 7.1 Hz, 3H, N(1)OCH2CH3), 1.25 (t, J = 7.0 Hz, 3H, OCH2CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 136.2 (Ar), 128.6 (Ar), 126.1 (Ar), 125.3 (Ar), 122.8 (Ar), 119.5 (Ar), 116.4 (Ar), 108.4 (Ar), 74.9 (N(1)OCH2), 65.8 (OCH2CH3), 61.9 (C(3)CH2O), 52.3 (CO2CH3), 15.5 (N(1)OCH2CH3), 13.7 (OCH2CH3); MS m/z 311 [M]+; HRMS (+ESI) calcd for C15H18ClNNaO4 [M + Na]+ 334.0822, found 334.0820.
Methyl 4-Chloro-1-n-propyloxy-3-[(n-propyloxy)methyl]-1H-indole-2-carboxylate (1xc)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), n-propanol (28 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and 1-bromopropane (34 μL, 0.37 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xc (18.9 mg, 30%) as a white oil. Bp 170 °C (decomp.); Rf 0.54 (1:4 EtOAc/hexanes); HPLC tR 30.8 min; UV vis (CH3CN-H2O) λmax 230, 298nm; 1H NMR (300 MHz, CDCl3): δ 7.35 (d, J = 7.9 Hz, 1H, Ar), 7.23 (t, J = 8.0 Hz, 1H, Ar), 7.15 (d, J = 7.1 Hz, 1H, Ar), 5.09 (s, 2H, C(3)CH2O), 4.29 (t, J = 6.4 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.54 (t, J = 6.5 Hz, 2H, OCH2), 1.86 (sextet, J = 6.9 Hz, 2H, N(1)OCH2CH2), 1.64 (sextet, J = 7.0 Hz, 2H, OCH2CH2), 1.11 (t, J = 7.2 Hz, 3H, N(1)OCH2CH2CH3), 0.93 (t, J = 7.3 Hz, 3H, OCH2CH2CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.1 (Ar), 128.6 (Ar), 126.0 (Ar), 125.3(Ar), 122.8 (Ar), 119.6 (Ar), 116.4 (Ar), 108.3 (Ar), 80.6 (N(1)OCH2), 72.4 (OCH2), 62.1 (C(3)CH2O), 52.2 (CO2CH3), 23.1 (N(1)OCH2CH2), 21.8 (OCH2CH2), 10.9 (N(1)O(CH2)2CH3), 10.6 (O(CH2)2CH3); MS m/z 339 [M]+; HRMS (+ESI) calcd for C17H22ClNO4 [M + Na]+ 362.1135, found 362.1134.
Methyl 4-Chloro-1-n-butyloxy-3-[(n-butyloxy)methyl]-1H-indole-2-carboxylate (1xd)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-butanol (21 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and 1-bromobutane (24 μL, 0.22 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xd (12.9 mg, 32%) as a white oil. Bp 198 °C (decomp.); Rf 0.54 (1:4 EtOAc/hexanes); HPLC tR 34.3 min; UV vis (CH3CN-H2O) λmax 235, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.34 (d, J = 8.2 Hz, 1H, Ar), 7.23 (t, J = 8.2 Hz, 1H, Ar), 7.15 (d, J = 7.4 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.5 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.58 (t, J = 6.5 Hz, 2H, OCH2), 1.82 (quintet, J = 7.0 Hz, 2H, N(1)OCH2CH2), 1.65–1.52 (m, 4H, OCH2CH2, N(1)OCH2CH2CH2), 1.39 (sextet, J = 7.5 Hz, 2H, O(CH2)2CH2), 1.00 (t, J = 7.4 Hz, 3H, N(1)O(CH2)3CH3), 0.90 (t, J = 7.4 Hz, 3H, O(CH2)3CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.1 (Ar), 128.6 (Ar), 126.0 (Ar), 125.3 (Ar), 122.8 (Ar), 119.6 (Ar), 116.4 (Ar), 108.3 (Ar), 79.0 (N(1)OCH2), 70.4 (OCH2), 62.1 (C(3)CH2O), 52.3 (CO2CH3), 32.1 (N(1)OCH2CH2), 30.5 (OCH2CH2) 19.6 (N(1)O(CH2)2CH2), 19.4 (O(CH2)2CH2) 14.2 (N(1)O(CH2)3CH3), 14.1 (O(CH2)3CH3); MS m/z 367 [M]+; HRMS (+ESI) calcd for C19H26ClNO4 [M + Na]+ 390.1448, found 390.1447.
Methyl 4-Chloro-1-n-pentyloxy-3-[(n-pentyloxy)methyl]-1H-indole-2-carboxylate (1xe)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-pentanol (24 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and 1-bromopentane (28 μL, 0.22 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xe (8.7 mg, 20%) as a white oil. Bp 184 °C (decomp.); Rf 0.58 (1:4 EtOAc/hexanes); HPLC tR 32.9 min; UV vis (CH3CN-H2O) λmax 235, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.34 (d, J = 8.1 Hz, 1H, Ar), 7.23 (t, J = 8.1 Hz, 1H, Ar), 7.15 (d, J = 7.5 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.59 (t, J = 6.7 Hz, 2H, OCH2), 1.84 (quintet, J = 7.4 Hz, 2H, N(1)OCH2CH2), 1.64–1.25 (m, 10H, OCH2(CH2)3CH3, N(1)OCH2CH2(CH2)2CH3), 0.95 (t, J = 7.2 Hz, 3H, N(1)O(CH2)4CH3), 0.87 (t, J = 6.9 Hz, 3H, O(CH2)4CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 136.0 (Ar), 128.6 (Ar), 126.0 (Ar), 125.3 (Ar), 122.8 (Ar), 119.6 (Ar), 116.4 (Ar), 108.2 (Ar), 79.3 (N(1)OCH2), 70.7 (OCH2), 62.1 (C(3)CH2O), 52.2 (CO2CH3), 29.7, 28.6, 28.3, 28.1, 22.7, 22.6, (N(1)OCH2(CH2)3, OCH2(CH2)3), 14.2 (N(1)O(CH2)4CH3), 14.1 (O(CH2)4CH3); MS m/z 395 [M]+; HRMS (+ESI) calcd for C21H30ClNO4 [M + Na]+ 418.1761, found 418.1760.
Methyl 4-Chloro-1-n-hexyloxy-3-[(n-hexyloxy)methyl]-1H-indole-2-carboxylate (1xf)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), n-hexanol (74 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and 1-bromohexane (52 μL, 0.37 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xf (16.5 mg, 21%) as a pale-yellow oil. Bp 152 °C (decomp.); Rf 0.63 (1:4 EtOAc/hexanes); HPLC tR 29.7 min; UV vis (CH3CN-H2O) λmax 236, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.33 (d, J = 8.2 Hz, 1H, Ar), 7.23 (t, J = 8.0 Hz, 1H, Ar), 7.14 (d, J = 8.0 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, OCH3), 3.57 (t, J = 6.6 Hz, 2H, OCH2), 1.83 (quintet, J = 6.7 Hz, 2H, N(1)OCH2CH2), 1.63–1.50 (m, 4H, OCH2CH2, N(1)O(CH2)2CH2), 1.37–1.25 (m, 10H, OCH2CH2(CH2)3, N(1)OCH2CH2CH2(CH2)2), 0.92–0.84 (m, 6H, N(1)O(CH2)5CH3, O(CH2)5CH3); 13C NMR (75 MHz, CDCl3): δ 160.7 (C=O), 135.9 (Ar), 128.5 (Ar), 125.8 (Ar), 125.1(Ar), 122.6 (Ar), 119.4 (Ar), 116.2 (Ar), 108.1 (Ar), 79.1 (N(1)OCH2), 70.5 (OCH2), 61.9 (C(3)CH2O), 52.0 (CO2CH3), 31.7, 31.6, 29.8, 28.2, 25.9, 25.6, 22.6, 22.5 (N(1)OCH2(CH2)4, OCH2(CH2)4), 14.1 (N(1)O(CH2)5CH3), 14.0 (O(CH2)5CH3); MS m/z 423 [M]+; HRMS (+ESI) calcd for C23H34ClNO4 [M + Na]+ 446.2074, found 446.2071.
Methyl 4-Chloro-1-n-octyloxy-3-[(n-octyloxy)methyl]-1H-indole-2-carboxylate (1xg)
Use of SnCl2·2H2O (166 mg, 0.74 mmol, 3.3 eq), n-octanol (70 μL, 0.45 mmol, 2.0 eq) and 2x (60 mg, 0.22 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (350 μL, 2.20 mmol, 10.0 eq) and 1-bromooctane (80 μL, 0.45 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xg (23.2 mg, 22%) as a white solid. Mp 19–20 °C; Rf 0.70 (1:4 EtOAc/hexanes); HPLC tR 35.5 min; UV vis (CH3CN-H2O) λmax 237, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.33 (d, J = 8.2 Hz, 1H, Ar), 7.23 (t, J = 8.1 Hz, 1H, Ar), 7.14 (d, J = 7.4 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.56 (t, J = 6.7 Hz, 2H, OCH2), 1.83 (quintet, J = 7.0 Hz, 2H, N(1)OCH2CH2), 1.66–1.25 (m, 22H, OCH2(CH2)6CH3), N(1)OCH2CH2(CH2)5CH3), 0.89–0.85 (m, 6H, N(1)O(CH2)7CH3, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 136.1 (Ar), 128.7 (Ar), 126.0 (Ar), 125.3(Ar), 122.8 (Ar), 120.0 (Ar), 116.4 (Ar), 108.3(Ar), 79.3 (N(1)OCH2), 70.7 (OCH2), 62.1 (C(3)CH2O), 52.2 (CO2CH3), 32.0, 31.9, 30.0, 29.9, 29.6, 29.5, 29.4, 28.5, 26.4, 26.2, 22.8, 22.6 5 (N(1)CH2(CH2)6, OCH2(CH2)6), 14.3 (N(1)O(CH2)7CH3), 13.5 (O(CH2)7CH3); MS m/z 479 [M]+; HRMS (+ESI) calcd for C27H42ClNNaO4 [M + Na]+ 502.2700, found 502.2926.
Methyl 4-Chloro-1-benzyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1xh)
Use of SnCl2·2H2O (38.4 mg, 0.17 mmol, 3.3 eq), benzyl alcohol (12 μL, 0.11 mmol, 2.0 eq) and 2x (14.5 mg, 0.05 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (165 μL, 0.55 mmol, 10.0 eq) and benzyl bromide (14 μL, 0.11 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xh (7.4 mg, 32%) as a white solid. Mp 74 °C; Rf 0.54 (1:2 EtOAc/hexanes); HPLC tR 31.8 min; UV vis (CH3CN-H2O) λmax 212, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.50–7.10 (m, 13H, Ar), 5.32 (s, 2H, C(3)CH2O), 5.17 (s, 2H, N(1)OCH2), 4.66 (s, 2H, C(3)CH2OCH2), 3.87 (s, 3H, CO2CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 138.8 (Ar), 136.4 (Ar), 134.3 (Ar), 130.0 (Ar), 129.5 (Ar), 128.9 (Ar), 128.6 (Ar), 128.5 (Ar), 128.2 (Ar), 127.7 (Ar), 126.2 (Ar), 125.9 (Ar), 123.0 (Ar), 119.7 (Ar), 116.2 (Ar), 108.6 (Ar), 81.0 (N(1)OCH2), 72.6 (OCH2Ph), 61.8 (C(3)CH2O), 52.2 (CO2CH3); MS m/z 435 [M]+; HRMS (+ESI) calcd for C25H22ClNNaO4 [M + Na]+ 458.1135, found 458.1133.
Methyl 4-Chloro-1-phenylethyloxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1xi)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), 2-phenylethyl alcohol (46 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and 2-(bromoethyl)benzene (51 μL, 0.37 mmol, 2.0 eq) for 1 h at 25 °C in general procedure afforded the title compound 1xi (15.2 mg, 18%) as a pale yellow solid. Mp 50 °C; Rf 0.60 (1:2 EtOAc/hexanes); HPLC tR 34.5 min; UV vis (CH3CN-H2O) λmax 212, 236, 297 nm; 1H NMR (300 MHz, CDCl3): δ 7.38–6.90 (m, 13H, Ar), 5.14 (s, 2H, C(3)CH2O), 4.55 (t, J = 6.7 Hz, 2H, N(1)OCH2), 3.86 (s, 3H, CO2CH3), 3.79 (t, J = 7.4 Hz, 2H, C(3)CH2OCH2), 3.14 (t, J = 6.7 Hz, 2H, N(1)OCH2CH2), 2.94 (t, J = 7.5 Hz, 2H, C(3)CH2OCH2CH2); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 139.2 (Ar), 137.8 (Ar), 136.2 (Ar), 129.3 (Ar), 129.1 (Ar), 128.8 (Ar), 128.5 (Ar), 128.4 (Ar), 126.9 (Ar), 126.2 (Ar), 125.9 (Ar), 125.4 (Ar), 122.9 (Ar), 119.6 (Ar), 116.4 (Ar), 108.3 (Ar), 79.6 (N(1)OCH2), 71.4 (OCH2), 62.2 (C(3)CH2O), 52.2 (CO2CH3), 36.5 (N(1)OCH2CH2), 35.0 (OCH2CH2); MS m/z 463 [M]+; HRMS (+ESI) calcd for C27H26ClNNaO4 [M + Na]+ 486.1448, found 486.1445.
Methyl 4-Chloro-1-isopropyloxy-3-[(isopropyloxy)methyl]-1H-indole-2-carboxylate (1xj)
Use of SnCl2·2H2O (166 mg, 0.74 mmol, 3.3 eq), isopropanol (35 μL, 0.45 mmol, 2.0 eq) and 2x (60 mg, 0.22 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (350 μL, 2.20 mmol, 10.0 eq) and 2-bromopropane (43 μL, 0.45 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1xj (9.2 mg, 12%) as a white oil. Bp 164 °C (decomp.); Rf 0.50 (1:4 EtOAc/hexanes); HPLC tR 29.1 min; UV vis (CH3CN-H2O) λmax 233, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.35 (d, J = 8.2 Hz, 1H, Ar), 7.20 (t, J = 7.6 Hz, 1H, Ar), 7.13 (d, J = 7.4 Hz, 1H, Ar), 5.09 (s, 2H, C(3)CH2O), 4.72 (septet, J = 6.2 Hz, 1H, N(1)OCH(CH3)2), 3.80 (septet, 1H, C(3)CH2OCH(CH3)2), 1.33 (d, J = 6.2 Hz, 6H, N(1)OCH(CH3)2), 1.24 (d, J = 6.1 Hz, 6H, C(3)CH2OCH(CH3)2); 13C NMR (75 MHz, CDCl3): δ 161.1 (C=O), 137.5 (Ar), 128.4 (Ar), 126.1 (Ar), 125.8 (Ar), 122.6 (Ar), 119.5 (Ar), 116.8 (Ar), 109.4 (Ar), 82.1 (N(1)OCH), 71.5 (OCH), 60.0 (C(3)CH2O), 52.2 (CO2CH3), 22.4 (N(1)OCH(CH3)2), 21.3 (OCH(CH3)2); MS m/z 339 [M]+; HRMS (+ESI) calcd for C17H22ClNNaO4 [M + Na]+ 362.1135, found 362.1131.
Methyl 4-Chloro-1-cyclohexyloxy-3-[(cyclohexyloxy)methyl]-1H-indole-2-carboxylate (1xk)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), cyclohexanol (23 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and bromocyclohexane (27 μL, 0.22 mmol, 2.0 eq) for 4 h at 50 °C in general procedure afforded the title compound 1xk (4.7 mg, 11%) as a white solid. Mp 60–64 °C; Rf 0.57 (1:4 EtOAc/hexanes); HPLC tR 31.9 min; UV vis (CH3CN-H2O) λmax 237, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.37 (d, J = 8.2 Hz, 1H, Ar), 7.20 (t, J = 7.6 Hz, 1H, Ar), 7.12 (d, J = 7.5 Hz, 1H, Ar), 5.11 (s, 2H, C(3)CH2O), 4.35–4.28 (m, 1H, N(1)OCH), 3.97 (s, 3H, CO2CH3), 3.45–3.38 (m, 1H, C(3)CH2OCH), 2.38–0.86 (m, 20H, N(1)OCH(CH2)5, C(3)CH2OCH(CH2)5); 13C NMR (75 MHz, CDCl3): δ 161.1 (C=O), 137.3 (Ar) 128.4 (Ar), 126.0 (Ar), 125.7 (Ar), 122.5 (Ar), 119.2 (Ar), 116.7 (Ar), 109.4 (Ar), 87.9 (N(1)OCH), 78.3 (C(3)CH2OCH), 59.7 (C(3)CH2O), 52.2 (CO2CH3), 31.7, 29.9. 26.1, 25.6, 24.7, 24.6 (N(1)OCH(CH2)5, OCH(CH2)5); MS m/z 419 [M]+; HRMS (+ESI) calcd for C23H30ClNNaO4 [M + Na]+ 442.1760, found 442.1760.
Methyl 4-Chloro-1-methoxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1xl)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), 2-phenylethyl alcohol (46 μL, 0.37 mmol, 2.0 eq) and 2x (50 mg, 0.185 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and methyl iodide (23 μL, 0.37 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1xl (15.2 mg, 22%) as a white solid. Mp 68 °C; Rf 0.30 (1:4 EtOAc/hexanes); HPLC tR 29.0 min; UV vis (CH3CN-H2O) λmax 215, 234, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.38 (d, J = 8.2 Hz, 2H, Ar), 7.29–7.16 (m, 6H, Ar), 5.15 (s, 2H, C(3)CH2O,), 4.19 (s, 3H, N(1)OCH3), 3.96 (s, 3H, CO2CH3), 3.81 (t, J = 7.4 Hz, 2H, OCH2), 2.95 (t, J = 7.4 Hz, 2H, OCH2CH2); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 139.3 (Ar), 135.9 (Ar), 129.1 (Ar), 128.7 (Ar), 128.5 (Ar), 126.3 (Ar), 126.2 (Ar), 125.3 (Ar), 123.0 (Ar), 119.7 (Ar), 116.5 (Ar), 108.2 (Ar), 71.5 (N(1)OCH3), 66.4 (OCH2), 62.2 (C(3)CH2O), 52.4 (CO2CH3), 36.5 (OCH2CH2); MS m/z 373 [M]+; HRMS (+ESI) calcd for C20H20ClNNaO4 [M + Na]+ 396.0979, found 396.0977.
Methyl 4-Chloro-1-n-octyloxy -3-[(methoxymethyl]-1H-indole-2-carboxylate (1xm)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and 1-bromooctane (38 μL, 0.22 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1xm (13.7 mg, 33%) as a yellow oil. Bp 178 °C (decomp.); Rf 0.67 (1:2 EtOAc/hexanes); HPLC tR 26.3 min; UV vis (CH3CN-H2O) λmax 236, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.36 (dd, J = 8.2, 1.0 Hz, 1H, Ar), 7.25 (t, J = 7.8 Hz, 1H, Ar), 7.16 (dd, J = 7.5, 1.0 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O,), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 4.00 (s, 3H, CO2CH3), 3.46 (s, 3H, OCH3), 1.83 (quintet, J = 7.1 Hz, 2H, OCH2CH2), 1.54–1.47 (m, 2H, O(CH2)2CH2), 1.34–1.25 (m, 8H, O(CH2)3(CH2)4, 0.90 (t, J = 7.0 Hz, 3H, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 136.0 (Ar), 128.5 (Ar), 126.1 (Ar), 125.2 (Ar), 122.9 (Ar), 119.5 (Ar), 116.1 (Ar), 108.4 (Ar), 79.4 (N(1)OCH2), 63.7 (OCH3), 58.1 (C(3)CH2O), 52.3 (CO2CH3), 32.0, 29.6, 29.4, 28.4, 26.2, 22.8 (N(1)OCH2(CH2)6), 14.3 N(1)O(CH2)7CH3; MS m/z 381 [M]+; HRMS (+ESI) calcd for C20H28ClNO4 [M]+ 381.1707, found 381.1707.
Methyl 4-Chloro-1-methoxy-3-[(n-octyloxy)methyl]-1H-indole-2-carboxylate (1xn)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-octanol (35 μL, 0.22 mmol, 2.0 eq) and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and methyl iodide (14 μL, 0.22 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1xn (15.0 mg, 36%) as a white solid. Mp 36 °C; Rf 0.74 (1:2 EtOAc/hexanes); HPLC tR 27.0 min; UV vis (CH3CN-H2O) λmax 236, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.37 (d, J = 8.2 Hz, 1H, Ar), 7.26 (t, J = 7.8 Hz, 1H, Ar), 7.16 (d, J = 7.1 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O,), 4.19 (s, 3H, N(1)OCH3), 4.00 (s, 3H, CO2CH3), 3.57 (t, J = 6.7 Hz, 2H, OCH2), 1.66–1.59 (m, 2H, OCH2CH2), 1.43–1.25 (m, 10H, O(CH2)2(CH2)5), 0.87 (t, J = 6.6 Hz, 3H, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.0 (Ar), 128.8 (Ar), 126.3 (Ar), 125.3 (Ar), 122.9 (Ar), 119.7 (Ar), 116.7 (Ar), 108.1 (Ar), 70.8 (N(1)OCH3), 66.4 (OCH2), 62.1 (C(3)CH2O), 52.4 (CO2CH3), 32.0, 30.0, 29.6, 29.5, 26.4, 22.9 (OCH2(CH2)6), 14.3 (O(CH2)7CH3); MS m/z 381 [M]+; HRMS (+ESI) calcd for C20H28ClNO4 [M]+ 381.1707, found 381.1707.
Methyl 4-Bromo-1-methoxy-3-(methoxymethyl)-1H-indole-2-carboxylate (1ya)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), methanol (8 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and methyl iodide (13 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1ya (17.1 mg, 52%) as a white solid. Mp 50–52 °C; Rf 0.29 (1:4 EtOAc/hexanes); HPLC tR 34.3 min; UV vis (CH3CN-H2O) λmax 215, 298 nm; 1H NMR (300 MHz, CDCl3): δ 7.49–7.38 (m, 2H, Ar), 7.19 (t, J = 8.1 Hz, 1H, Ar), 5.08 (s, 2H, C(3)CH2O), 4.18 (s, 3H, N(1)OCH3), 4.01 (s, 3H, CO2CH3), 3.47 (s, 3H, CH2OCH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 135.8 (Ar), 131.2 (Ar), 126.7 (Ar), 125.6 (Ar), 123.8 (Ar), 120.9 (Ar), 116.7 (Ar), 108.8 (Ar), 66.4 (N(1)OCH3), 63.0 (CH2OCH3), 58.0 (C(3)CH2O), 52.4 (CO2CH3); MS m/z 327 [M]+; HRMS (+ESI) calcd for C13H14BrNNaO4 [M + Na]+ 350.0004, found 350.0002.
Methyl 4-Bromo-1-ethoxy-3-(ethoxymethyl)-1H-indole-2-carboxylate (1yb)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), ethanol (12 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and bromoethane (16 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yb (9.3 mg, 26%) as a white solid. Mp 42 °C; Rf 0.51 (1:2 EtOAc/hexanes); HPLC tR 26.2 min; UV vis (CH3CN-H2O) λmax 235, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.45–7.33 (m, 2H, Ar), 7.16 (t, J = 7.9 Hz, 1H, Ar), 5.10 (s, 2H, C(3)CH2O), 4.41 (q, J = 7.1 Hz, 2H, N(1)OCH2), 3.99 (s, 3H, CO2CH3), 3.66 (q, J = 7.0 Hz, 2H, OCH2), 1.44 (t, J = 7.1 Hz, 3H, N(1)OCH2CH3), 1.26 (t, J = 7.0 Hz 3H, OCH2CH3), 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.2 (Ar), 126.5 (Ar), 126.3 (Ar), 120.8 (Ar), 125.5(Ar), 116.7 (Ar), 116.4 (Ar), 109.0 (Ar), 75.0 (N(1)OCH2), 65.8 (OCH2), 61.4 (C(3)CH2O), 52.3 (CO2CH3), 15.6 (N(1)OCH2CH3), 13.8 (OCH2CH3); MS m/z 355 [M]+; HRMS (+ESI) calcd for C15H19BrNNaO4 [M + Na]+ 378.0317, found 378.0315.
Methyl 4-Bromo-1-n-propyloxy-3-[(n-propyloxy)methyl]-1H-indole-2-carboxylate (1yc)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), n-propanol (15 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and 1-bromopropane (18 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yc (10.9 mg, 28%) as a white oil. Bp 224 °C (decomp.); Rf 0.62 (1:2 EtOAc/hexanes); HPLC tR 31.3 min; UV vis (CH3CN-H2O) λmax 236, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.40 (d, J = 8.3 Hz, 1H, Ar), 7.39 (d, J = 8.3 Hz, 1H, Ar), 7.15 (t, J = 7.7 Hz, 1H, Ar), 5.10 (s, 2H, C(3)CH2O), 4.29 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.55 (t, J = 6.6 Hz, 2H, OCH2), 1.86 (sextet, J = 7.2 Hz, 2H, N(1)OCH2CH2), 1.66 (sextet, J = 7.2 Hz, 2H, OCH2CH2), 1.11 (t, J = 7.4 Hz, 3H, N(1)OCH2CH2CH3), 0.94 (t, J = 7.4 Hz, 3H, OCH2CH2CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.0 (Ar), 126.5 (Ar), 126.3 (Ar), 125.7(Ar), 120.9 (Ar), 116.8 (Ar), 116.4 (Ar), 108.9 (Ar), 80.7 (N(1)OCH2), 72.3 (OCH2), 61.5 (C(3)CH2O), 52.3 (CO2CH3), 23.2 (N(1)OCH2CH2), 21.8 (OCH2CH2), 11.0 (N(1)O(CH2)2CH3), 10.7 (O(CH2)2CH3); MS m/z 383 [M]+; HRMS (+ESI) calcd for C17H22BrNNaO4 [M + Na]+ 406.0630, found 408.0608.
Methyl 4-Bromo-1-n-octyloxy-3-[(n-octyloxy)methyl]-1H-indole-2-carboxylate (1yg)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), n-octanol (9 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 1 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and 1-bromooctane (35 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yg (11.0 mg, 21%) as a yellow oil. Bp 194 °C (decomp.); Rf 0.40 (1:10 EtOAc/hexanes); HPLC tR 36.1 min; UV vis (CH3CN-H2O) λmax 236, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.37 (t, J = 7.9 Hz, 2H, Ar), 7.16 (t, J = 7.9 Hz, 1H, Ar), 5.09 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.98 (s, 3H, CO2CH3), 3.57 (t, J = 6.6 Hz, 2H, OCH2), 1.83 (quintet, J = 7.5 Hz, 2H, N(1)OCH2CH2), 1.66–1.15 (m, 22H, N(1)OCH2CH2(CH2)5, OCH2(CH2)6), 1.00–0.77 (m, 6H, N(1)O(CH2)7CH3, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 160.9 (C=O), 136.0 (Ar), 126.4 (Ar), 126.3 (Ar), 125.6 (Ar), 120.9 (Ar), 116.8 (Ar), 116.4 (Ar), 108.9(Ar), 79.4 (N(1)OCH2), 70.6 (OCH2), 61.5 (C(3)CH2O), 53.3 (CO2CH3), 53.3, 52.3, 32.0, 30.1, 30.0, 29.7, 29.5, 29.4, 28.5, 26.5, 26.2, 22.9 (N(1)OCH2(CH2)6,OCH2(CH2)6), 14.3 (N(1)O(CH2)7CH3), 13.5 (O(CH2)7CH3); MS m/z 523 [M]+; HRMS (+ESI) calcd for C27H42BrNNaO4 [M + Na]+ 546.2195, found 546.2190.
Methyl 4-Bromo-1-benzyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1yh)
Use of SnCl2·2H2O (38 mg, 0.17 mmol, 3.3 eq), benzyl alcohol (11 μL, 0.10 mmol, 2.0 eq) and 2y (15.7 mg, 0.05 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (75 μL, 0.50 mmol, 10.0 eq) and benzyl bromide (12 μL, 0.10 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yh (9.8 mg, 41%) as a pale-yellow solid. Mp 84 °C; Rf 0.29 (1:2 EtOAc/hexanes); HPLC tR 33.3 min; UV vis (CH3CN-H2O) λmax 228, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.55–7.22 (m, 12H, Ar), 7.13 (t, J = 7.9 Hz, 1H, Ar), 5.32 (s, 2H, C(3)CH2O), 5.19 (s, 2H, N(1)OCH2), 4.68 (s, 2H, OCH2), 3.88 (s, 3H, CO2CH3); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 138.8 (Ar), 136.3 (Ar), 134.3 (Ar), 130.0 (Ar), 129.5 (Ar), 129.0 (Ar), 128.5 (Ar), 128.3 (Ar), 127.7 (Ar), 126.7 (Ar), 126.4 (Ar), 126.2 (Ar), 121.0 (Ar), 116.5 (Ar), 116.3 (Ar) 109.2 (Ar), 81.0 (N(1)OCH2), 72.6 (OCH2Ph), 61.2(C(3)CH2O), 52.3 (CO2CH3); MS m/z 479 [M]+; HRMS (+ESI) calcd for C25H22BrNNaO4 [M + Na]+ 502.0630, found 502.0626.
Methyl 4-Bromo-1-isopropyloxy-3-[(isopropyloxy)methyl]-1H-indole-2-carboxylate (1yj)
Use of SnCl2·2H2O (75 mg, 0.33 mmol, 3.3 eq), isopropanol (15 μL, 0.20 mmol, 2.0 eq) and 2y (31.4 mg, 0.10 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (150 μL, 1.00 mmol, 10.0 eq) and 2-bromopropane (19 μL, 0.20 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yj (6.2 mg, 16%) as a white oil. Bp 178 °C (decomp.); Rf 0.69 (1:2 EtOAc/hexanes); HPLC tR 30.5 min; UV vis (CH3CN-H2O) λmax 235, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.40 (d, J = 8.3 Hz, 1H, Ar), 7.35 (d, J = 7.5 Hz, 1H, Ar), 7.13 (t, J = 7.9 Hz, 1H, Ar), 5.11 (s, 2H, C(3)CH2O), 4.72 (septet, J = 6.2 Hz, 1H, N(1)OCH(CH3)2), 3.97 (s, 3H, CO2CH3), 3.82 (septet, J = 6.0 Hz, 1H, C(3)CH2OCH(CH3)2), 1.34 (d, J = 6.2 Hz, 6H, N(1)OCH(CH3)2), 1.26 (d, J = 6.1 Hz, 6H, OCH(CH3)2); 13C NMR (75 MHz, CDCl3): δ 161.1 (C=O), 137.3 (Ar) 126.3 (Ar), 126.1 (Ar), 120.7 (Ar), 117.1 (Ar), 116.1 (Ar), 109.9 (Ar), (one Ar peak was not detected and believed to overlap with the observed peak), 82.2 (N(1)OCH(CH3)2), 71.5 (OCH(CH3)2), 59.4 (C(3)OCH2), 52.2 (CO2CH3), 22.4 (N(1)OCH(CH3)2), 21.3 (OCH(CH3)2); MS m/z 383 [M]+; HRMS (+ESI) calcd for C17H22BrNNaO4 [M + Na]+ 406.0630, found 406.0632.
Methyl 4-Bromo-1-methoxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1yl)
Use of SnCl2·2H2O (37 mg, 0.17 mmol, 3.3 eq), 2-phenylethyl alcohol (12 μL, 0.10 mmol, 2.0 eq) and 2y (15.7 mg, 0.05 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (75 μL, 0.50 mmol, 10.0 eq) and methyl iodide (4 μL, 0.10 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1yl (5.2 mg, 25%) as a white solid. Mp 59 °C; Rf 0.57 (1:2 EtOAc/hexanes); HPLC tR 29.7 min; UV vis (CH3CN-H2O) λmax 235, 299 nm; 1H NMR (300 MHz, CDCl3): δ 7.43 (d, J = 8.3 Hz, 1H, Ar), 7.38 (d, J = 7.4 Hz, 1H, Ar), 7.29–7.16 (m, 6H, Ar), 5.15 (s, 2H, C(3)CH2O), 4.19 (s, 3H, N(1)OCH3), 3.96 (s, 3H, CO2CH3), 3.82 (t, J = 7.4 Hz, 2H, OCH2), 2.96 (t, J = 7.4 Hz, 2H, OCH2CH2); 13C NMR (75 MHz, CDCl3): δ 160.8 (C=O), 139.3 (Ar), 135.9 (Ar), 129.1 (Ar), 128.5 (Ar), 126.7 (Ar), 126.5 (Ar), 126.3 (Ar), 125.6 (Ar), 120.9 (Ar), 116.8 (Ar), 116.4 (Ar), 108.8 (Ar), 71.5 (N(1)OCH3), 66.4 (OCH2), 61.6 (C(3)CH2O), 52.4 (CO2CH3), 36.5 (OCH2CH2); MS m/z 419 [M]+; HRMS (+ESI) calcd for C20H20BrNNaO4 [M + Na]+ 440.0473, found 440.0471.
Methyl 4-Bromo-1-pentyloxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1ym)
Use of SnCl2·2H2O (37 mg, 0.17 mmol, 3.3 eq), 2-phenylethyl alcohol (12 μL, 0.10 mmol, 2.0 eq), and 2y (15.7 mg, 0.05 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (75 μL, 0.50 mmol, 10.0 eq) and 1-bromopentane (13 μL, 0.10 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the title compound 1ym (5.0 mg, 21%) as a white oil. Bp 224 °C (decomp.); Rf 0.70 (1:2 EtOAc/hexanes); HPLC tR 36.7 min; UV vis (CH3CN-H2O) λmax 236, 299 nm; 1H NMR (300 MHz, CD3CN): δ 7.38 (t, J = 8.1 Hz, 2H, Ar), 7.30–7.10 (m, 6H, Ar), 5.16 (s, 2H, C(3)CH2O), 4.32 (t, J = 6.6 Hz, 2H, N(1)OCH2), 3.95 (s, 3H, CO2CH3), 3.81 (t, J = 7.5 Hz, 2H, OCH2CH2Ph), 2.96 (t, J = 7.4 Hz, 2H, OCH2CH2Ph), 1.95–1.73 (m, 2H, N(1)OCH2CH2), 1.58–1.30 (m, 4H, N(1)OCH2CH2(CH2)2), 0.96 (t, J = 7.1 Hz, 3H, N(1)O(CH2)4CH3); 13C NMR (75 MHz, CD3CN): δ 160.7 (C=O), 139.3 (Ar), 136.0 (Ar), 129.1 (Ar), 128.5 (Ar), 126.5 (Ar), 126.4 (Ar), 126.3 (Ar), 125.6 (Ar), 120.8 (Ar), 116.5 (Ar), 116.4 (Ar), 108.9 (Ar), 79.4 (N(1)OCH2), 71.4 (OCH2CH2Ph), 61.6 (C(3)CH2O), 52.3 (CO2CH3), 36.6 (OCH2CH2Ph), 28.3 (N(1)OCH2CH2), 28.2 (N(1)O(CH2)2CH2), 22.8 (N(1)O(CH2)3CH2), 14.2 (N(1)O(CH2)4CH3); MS m/z 473 [M]+; HRMS (+ESI) calcd for C24H28BrNNaO4 [M+Na]+ 496.1099, found 495.1097.
Methyl 2-(4′-Chloro-1′-hydroxy-1′H-indol-3′-yl)-2-oxoacetate (10x) [19]
Brown solid. Mp 84–86 °C; Rf 0.15 (2:1 EtOAc/hexanes); 1H NMR (300 MHz, CD3CN): δ 9.45 (br s, 1H, OH), 8.27 (s, 1H), 7.51 (dd, J = 5.0, 1.0 Hz, 1H), 7.40 (t, J = 4.7 Hz, 1H), 7.33 (dd, J = 4.2, 1.0 Hz, 1H), 3.90 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 179.5, 164.3, 137.5, 127.2, 126.5, 126.4, 126.1, 117.4, 110.0, 109.5, 53.8; MS m/z 276 [M + Na]+; HRMS (+ESI) Calcd for C11H8ClNNaO4 [M + Na]+ 276.0040, found 276.0034.
Methyl 2-(4′-Chloro-1′-methoxy-1′H-indol-3′-yl)-2-oxoacetate (11xl)
Use of SnCl2·2H2O (140 mg, 0.62 mmol, 3.3 eq), 2-phenylethyl alcohol (46 μL, 0.37 mmol, 2.0 eq), and 2x (50 mg, 0.185 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (272 μL, 1.85 mmol, 10.0 eq) and methyl iodide (23 μL, 0.37 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the compound 11xl (15.8 mg, 32%) as a brown solid. Mp 64 °C; Rf 0.50 (2:1 EtOAc/hexanes); HPLC tR 16.6 min; UV vis (CH3CN-H2O) λmax 218, 262, 321 nm; 1H NMR (300 MHz, CDCl3): δ 8.40 (s, 1H, C(2)H), 7.43–7.15 (m, 3H, Ar), 4.19 (s, 3H, N(1)OCH3), 3.95 (s, 3H, OCH3); 13C NMR (75 MHz, CDCl3): δ 177.7 (C(3)C=O), 164.1 (C=O), 134.3 (Ar), 133.5 (Ar), 128.1 (Ar), 125.4 (Ar), 125.3 (Ar), 120.7 (Ar), 109.6 (Ar), 107.8 (Ar), 67.6 (N(1)OCH3), 53.2 (CO2CH3); MS m/z 267 [M]+; HRMS (+ESI) calcd for C12H10ClNNaO4 [M + Na]+ 290.0196, found 290.0193.
Methyl 2-(4′-Chloro-1′-octyloxy-1′H-indol-3′-yl)-2-oxoacetate (11xm)
Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2x (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (165 μL, 1.10 mmol, 10.0 eq) and 1-bromooctane (38 μL, 0.22 mmol, 2.0 eq) for 2 h at 25 °C in general procedure afforded the compound 11xm (1.1 mg, 3%) as a brown solid. Mp 40 °C; Rf 0.57 (1:2 EtOAc/hexanes); HPLC tR 33.4 min; UV vis (CH3CN-H2O) λmax 219, 262, 320 nm; 1H NMR (300 MHz, CDCl3): δ 8.37 (s, 1H, C(2)H), 7.40–7.15 (m, 3H, Ar), 4.31 (t, J = 6.7 Hz, 2H, N(1)OCH2), 3.95 (s, 3H, OCH3), 1.82 (quintet, J = 7.1 Hz, 2H, N(1)OCH2CH2), 1.57–1.25 (m, 10H, O(CH2)2(CH2)5, 0.90 (t, J = 6.6 Hz, 3H, O(CH2)7CH3); 13C NMR (75 MHz, CDCl3): δ 177.6 (C(3)C=O), 164.2 (C=O), 134.9 (Ar), 134.2 (Ar), 128.1 (Ar), 125.4 (Ar), 125.2 (Ar), 109.4 (Ar), 108.0 (Ar), (one Ar peak was not detected and believed to overlap with the observed peak), 80.5 (N(1)OCH2), 53.1 (CO2CH3), 32.1, 29.9, 29.5, 28.3, 25.9, 22.8 (N(1)OCH2(CH2)6), 14.3 (N(1)O(CH2)7CH3); MS m/z 365 [M]+; HRMS (+ESI) calcd for C19H24ClNO4 [M]+ 365.1394, found 365.1394.
4. Conclusions
We reported the studies on one-pot synthesis of novel multisubstituted 1-alkoxyindoles 1 through four step reactions. With substrates 2 obtained by two-step synthetic sequences, we performed the reactions using SnCl2·2H2O as a reducing agent and alcohols (R1OH) as nucleophiles, through reduction, condensation, and 1,5-addition, affording the intermediates, 1-hydroxyindoles 8. Subsequent alkylation reactions of 8 using alkyl halides (R2Y) in basic condition gave target compounds, 1-alkoxyindoles 1. The optimized condition was established as follows: 1) conjugate ketoester (1.0 eq), SnCl2·2H2O (3.3 eq), and alcohols (2.0 eq) in DME for 1–2 h at 40 °C and 2) DBU (10.0 eq) and alkyl halides (2.0 eq) for 1–4 h at 25–50 °C. Considering the yields and reaction efficiency, we chose 2.0 eq of both alcohols and alkyl halides, focusing on the optimization of the final alkylation step. All four step reactions were performed in one-pot, providing 1-alkoxyindoles 1 in modest to good yields (22 examples, 11–52% yields for four steps). Mechanistic investigations on reaction pathways (Path A, B, and C) were presented along with the formation of side products 11.
Supplementary Materials
The charts for 1H- and 13C-NMR spectroscopies are available online.
Author Contributions
Conceptualization, S.H.L. and H.C.; methodology, Y.E.K., Y.J.L., C.K. and H.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) Grant (2018R1D1A1B07048631, 2016R1D1A1B03930981, and 2019M3E5D5066543), and by Priority Research Centers Program through NRF (2016R1A6A1A03007648) funded by the Ministry of Education, Science and Technology (MEST).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in insert article or supplementary material here.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 1xa–1ym are available from the authors.
References
- Somei, M. Recent advances in the chemistry of 1-hydroxytryptophans, and 1-hydroxytryptamines. Adv. Heterocycl. Chem. 2002, 82, 101–154. [Google Scholar]
- Escolano, C. Stephacidin B, the avrainvillamide dimer: A formidable synthetic challenge. Angew. Chem. Int. Ed. 2005, 44, 7670–7673. [Google Scholar] [CrossRef]
- Wang, J.; Pearce, A.N.; Chan, S.T.S.; Taylor, R.B.; Page, M.J.; Valentine, A.; Bourguet-Kondracki, M.-L.; Dalton, J.P.; Wiles, S.; Copp, B.R. Biologically active acetylenic amino alcohol and N-hydroxylated 1,2,3,4-tetrahydro-β-carboline constituents of the New Zealand ascidian pseudodistoma opacum. J. Nat. Prod. 2016, 79, 607–610. [Google Scholar] [CrossRef]
- Kinoshita, T.; Tatara, S.; Ho, F.-C.; Sankawa, U. 3-Prenylindoles from murraya paniculata and their biogenetic significance. Phytochemistry 1989, 28, 147–151. [Google Scholar] [CrossRef]
- Bagley, M.C.; Dale, J.W.; Merritt, E.A.; Xiong, X. Thiopeptide antibiotics. Chem. Rev. 2005, 105, 685–714. [Google Scholar] [CrossRef]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Jump, S.M.; Kung, J.; Staub, R.; Kinseth, M.A.; Cram, E.J.; Yudina, L.N.; Preobrazhenskaya, M.N.; Bjeldanes, L.F.; Firestone, G.L. N-Alkoxy derivatization of indole-3-carbinol increases the efficacy of the G1 cell cycle arrest and of I3C-specific regulation of cell cycle gene transcription and activity in human breast cancer cells. Biochem. Pharmacol. 2008, 75, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acheson, R.M. 1-Hydroxypyrroles, 1-hydroxyindoles and 9-hydroxycarbazoles. Adv. Heterocycl. Chem. 1990, 51, 105–175. [Google Scholar]
- Nicolaou, K.C.; Lee, S.H.; Estrada, A.A.; Zak, M. Construction of substituted N-hydroxyindoles: Synthesis of a Nocathiacin Ⅰ model system. Angew. Chem, Int. Ed. 2005, 44, 3736–3740. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Kim, H.; Kim, D.S.; Cho, H.; Moon, A.; Jeong, C.; Yoon, H.-R.; Lee, S.H. Synthesis of new 2,3-disubstituted 4-chloro-1-hydroxyindoles. Bull. Kor. Chem. Soc. 2015, 36, 2095–2100. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, H.; Park, Y.K.; Cho, H. Synthetic of new 3-[(alkylthio)methyl]-1-hydroxy-2-phenylindoles. Synlett 2015, 26, 1069–1072. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.H. Synthesis of new 3-substituted 1-hydroxy-2-phenylindoles using sulfur-containing nucleophiles. Heterocycles 2016, 92, 2004–2017. [Google Scholar]
- Cho, H.; Kim, H.; Lim, Y.J.; Lee, S.H. Synthesis of new 3-[(alkylthio)methyl]-1-hydroxy-2-(4′-substituted phenyl)indoles and their mechanistic studies on substituent effects. Arkivoc 2018, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Somei, M.; Kawasaki, T. A new and simple synthesis of 1-hydroxyindole derivatives. Heterocycles 1989, 29, 1251–1254. [Google Scholar] [CrossRef] [Green Version]
- Yun, Z.; Cheng, R.; Sun, J.; Zhang-Negrerie, D.; Du, Y. Iodobenzene dichloride/zinc chloride-mediated synthesis of N-alkoxyindole-3-carbonitriles from 3-alkoxyimino-2-arylalkylnitriles via intramolecular heterocyclization. Adv. Synth. Catal. 2018, 360, 250–254. [Google Scholar] [CrossRef]
- Somei, M. 1-Hydroxyindoles. Heterocycles 1999, 50, 1157–1211. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Estrada, A.A.; Lee, S.H.; Freestone, G.C. Synthesis of highly substituted N-hydroxyindoles through 1,5-addition of carbon nucleophiles to in situ generated unsaturated nitrones. Angew. Chem. Int. Ed. 2006, 45, 5364–5368. [Google Scholar] [CrossRef]
- Bellamy, F.D.; Ou, K. Selective reduction of aromatic nitro compounds with stannous chloride in non acidic and non aqueous medium. Tetrahedron Lett. 1984, 25, 839–842. [Google Scholar] [CrossRef]
- Park, Y.K.; Lee, S.H. Synthesis of new 1-hydroxyindole-2-carboxylates and mechanistic studies on reaction pathways. J. Heterocyclic Chem. 2017, 54, 1995–2002. [Google Scholar] [CrossRef]
- Kolthoff, I.M.; Chantooni, J.M.K.; Bhowmik, S. Dissociation constant of uncharged and monovalent cation acids in dimethyl sulfoxide. J. Am. Chem. Soc. 1986, 90, 23–28. [Google Scholar] [CrossRef]
- Streitwieser, A.; Kim, Y.J. Ion pair basicity of some amines in THF: Implications for ion pair acidity scales. J. Am. Chem. Soc. 2000, 122, 11783–11786. [Google Scholar] [CrossRef]
Figure 1.
Indole, 1-hydroxyindole, and 1-alkoxyindoles.

Figure 2.
Structures of multisubstituted 1-alkoxyindoles 1.

Scheme 1.
Synthesis of conjugate nitro ketoesters 2.

Scheme 2.
Synthetic pathway for multisubstituted 1-alkoxyindoles 1.

Scheme 3.
Proposed pathways for 1 and 11.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Synthesis of 1-alkoxyindole 1yh in different base conditions a.

| Entry | Base | Yield (%) |
|---|---|---|
| 1 | TEA | 23 |
| 2 | DIEA | 17 |
| 3 | DMAP | 13 |
| 4 | DBU | 41 |
| 5 | K2CO3 b | 5 |
a All reactions were run in the 0.05 mmol scale of conjugate ketoester 2y (1.0 eq, [c] = 0.12 M) and BnOH (2.0 eq) for formation of 8yh in DME for 2 h at 40 °C; base (10 eq) and BnBr (2.0 eq) for 2 h at 25 °C for formation of 1yh. bAqueous K2CO3 was used with acetone.
Table 2.
Optimization of the reaction conditions for 1xh a.

| Entry | SnCl2 (eq) | BnOH (eq) | DBU (eq) | BnBr (eq) | Yield (%) |
|---|---|---|---|---|---|
| 1 | 2.5 | 2.0 | 7.6 | 2.0 | 25 |
| 2 | 2.9 | 2.0 | 8.8 | 2.0 | 26 |
| 3 | 3.3 | 1.5 | 10.0 | 1.5 | 22 |
| 4 | 3.3 | 1.5 | 10.0 | 3.0 | 23 |
| 5 | 3.3 | 2.0 | 10.0 | 2.0 | 32 |
| 6 | 3.3 | 2.0 | 10.0 | 5.0 | 33 |
| 7 | 3.3 | 3.0 | 10.0 | 3.0 | 42 |
| 8 | 3.3 | 5.0 | 10.0 | 5.0 | 45 |
| 9 | 3.7 | 2.0 | 11.2 | 2.0 | 23 |
a All reactions were run in the 0.05 mmol scale of conjugate ketoester 2x (1.0 eq, [c] = 0.12 M) and BnOH (2.0 eq) for formation of 8xh in DME for 2 h at 40 °C; BnBr (2.0 eq) for 2 h at 25 °C for the formation of 1xh.
Table 3.
Synthesis of derivatives of 1-alkoxyindole 1 a.
| Entry | ROH | RX | Product | Yield (%) |
|---|---|---|---|---|
| 1 | MeOH | MeI |  1xa 1xa | 43 |
| 2 | EtOH | EtBr |  1xb 1xb | 31 |
| 3 | n-PrOH | n-PrBr |  1xc 1xc | 30 |
| 4 | n-BuOH | n-BuBr |  1xd 1xd | 32 |
| 5 | n-PenOH | n-PenBr |  1xe 1xe | 20 |
| 6 | n-HexOH | n-HexBr |  1xf 1xf | 21 |
| 7 | n-OctOH | n-OctBr |  1xg 1xg | 22 |
| 8 | BnOH | BnBr |  1xh 1xh | 32 |
| 9 | PhCH2CH2OH | PhCH2CH2Br |  1xi 1xi | 18 |
| 10 | i-PrOH | i-PrBr |  1xj 1xj | 12 |
| 11 | c-HexOH | c-HexBr |  1xk 1xk | 11 |
| 12 | PhCH2CH2OH | MeI |  1xl 1xl | 22 |
| 13 | MeOH | n-OctBr |  1xm 1xm | 33 |
| 14 | n-OctOH | MeI |  1xn 1xn | 36 |
| 15 | MeOH | MeI |  1ya 1ya | 52 |
| 16 | EtOH | EtBr |  1yb 1yb | 26 |
| 17 | n-PrOH | n-PrBr |  1yc 1yc | 28 |
| 18 | n-OctOH | n-OctBr |  1yg 1yg | 21 |
| 19 | BnOH | BnBr |  1yh 1yh | 41 |
| 20 | i-PrOH | i-PrBr |  1yj 1yj | 16 |
| 21 | PhCH2CH2OH | MeI |  1yl 1yl | 25 |
| 22 | PhCH2CH2OH | n-PenBr |  1ym 1ym | 21 |
a Reactions were run in 0.05–0.22 mmol scale of conjugate ketoester 2 (1.0 eq) and R1OH (2.0 eq) for formation of 8 in DME for 1–2 h at 40 °C; R2Y (2.0 eq) for 1–4 h at 25–50 °C for formation of 1.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, Y.E.; Cho, H.; Lim, Y.J.; Kim, C.; Lee, S.H. One-Pot Synthesis of Novel Multisubstituted 1-Alkoxyindoles. Molecules 2021, 26, 1466. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051466
AMA Style
Kim YE, Cho H, Lim YJ, Kim C, Lee SH. One-Pot Synthesis of Novel Multisubstituted 1-Alkoxyindoles. Molecules. 2021; 26(5):1466. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051466
Chicago/Turabian StyleKim, Ye Eun, Hyunsung Cho, Yoo Jin Lim, Chorong Kim, and Sang Hyup Lee. 2021. "One-Pot Synthesis of Novel Multisubstituted 1-Alkoxyindoles" Molecules 26, no. 5: 1466. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051466