
A Novel Dual Drug Approach That Combines Ivermectin and Dihydromyricetin (DHM) to Reduce Alcohol Drinking and Preference in Mice

 , and
, and 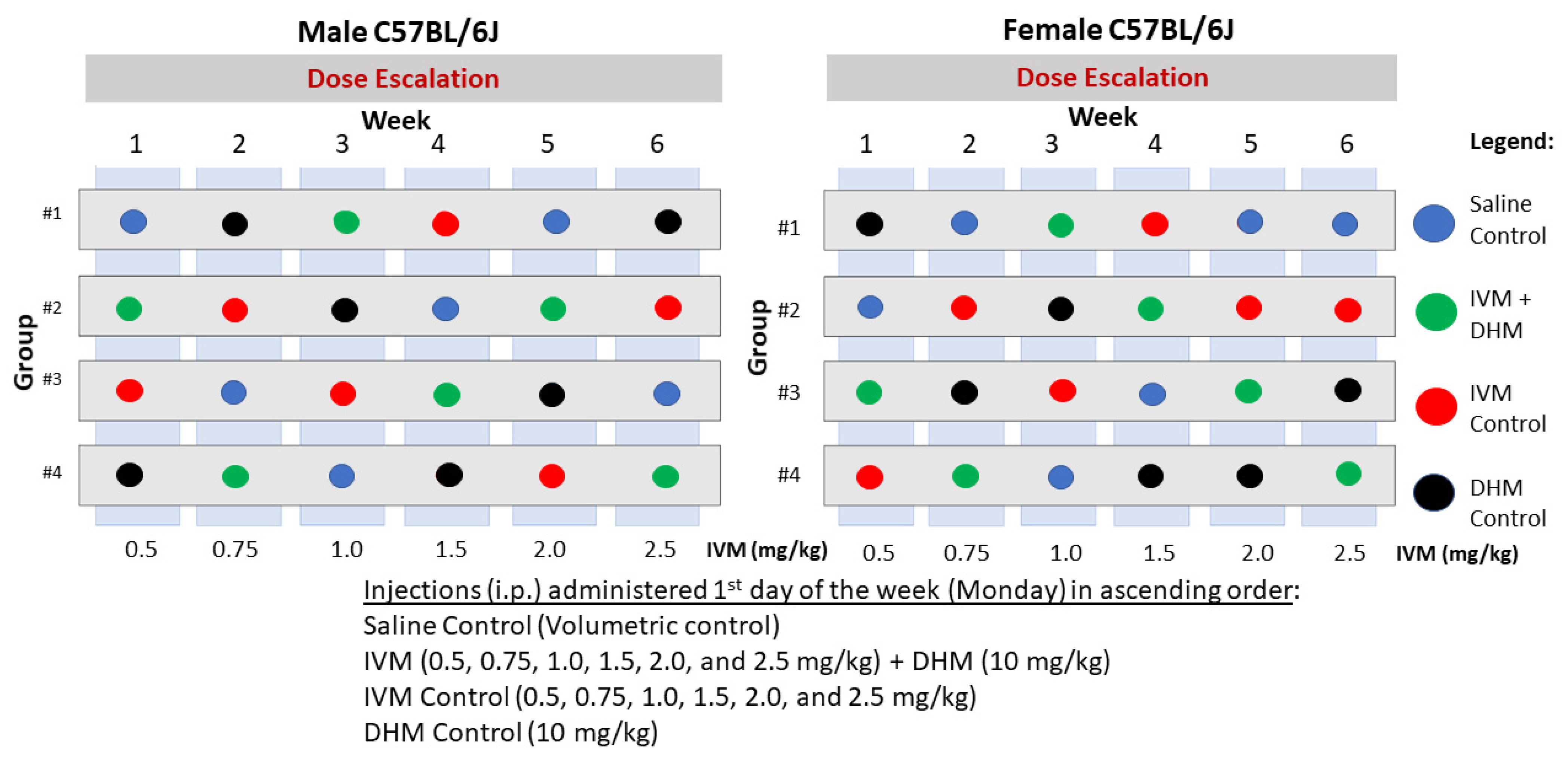{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
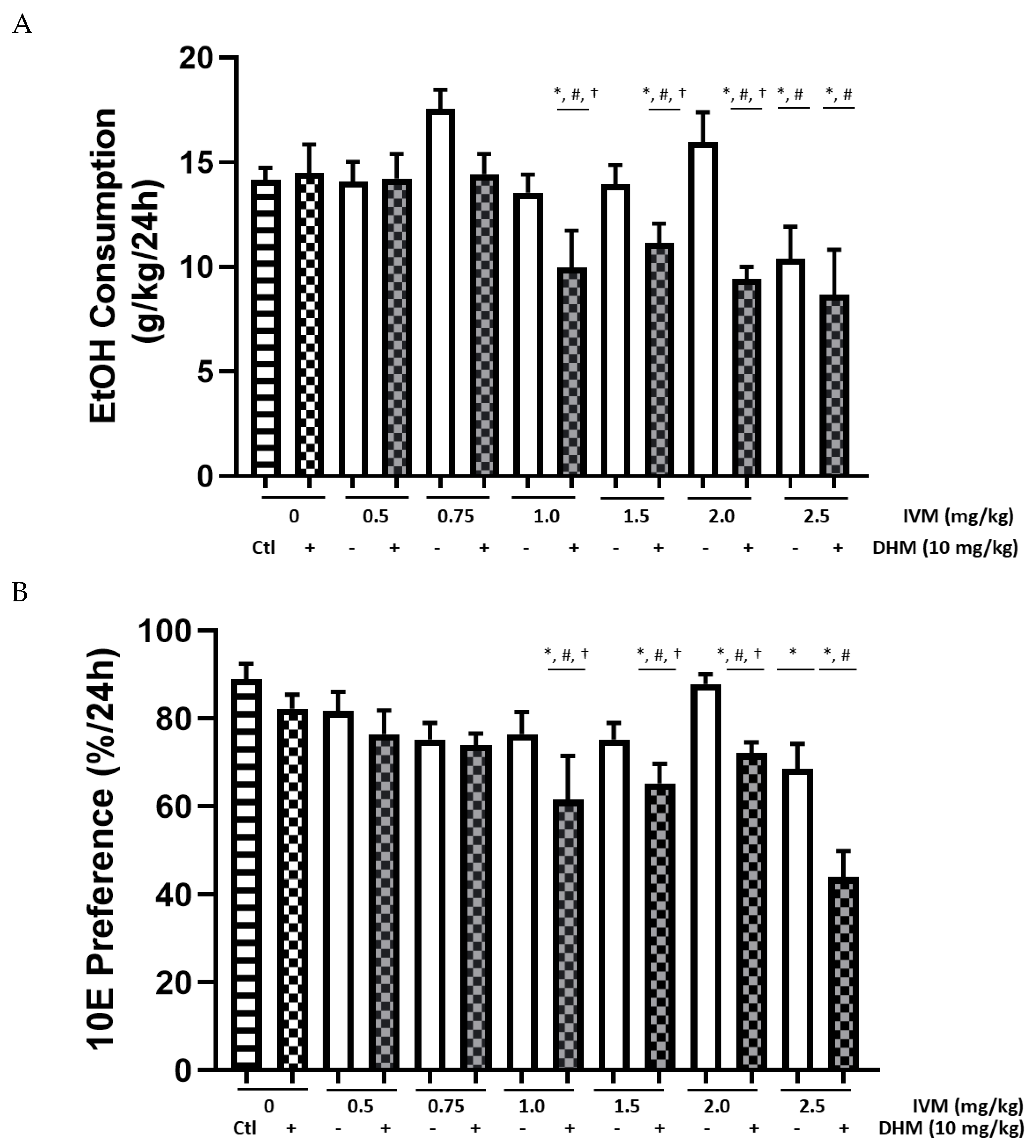
2.1. DHM Combined with IVM Significantly Increases IVM Potency on EtOH Intake and EtOH Preference
2.1.1. Male Group Baseline Values
2.1.2. EtOH Intake (g/kg) Averages Compared between Male Treatment Groups
2.1.3. 10E Preference Averages Compared between Male Treatment Groups
2.1.4. Female Group Baseline Values
2.1.5. EtOH Intake (g/kg) Averages Compared between Female Treatment Groups
2.1.6. 10E Preference Averages Compared between Female Treatment Groups
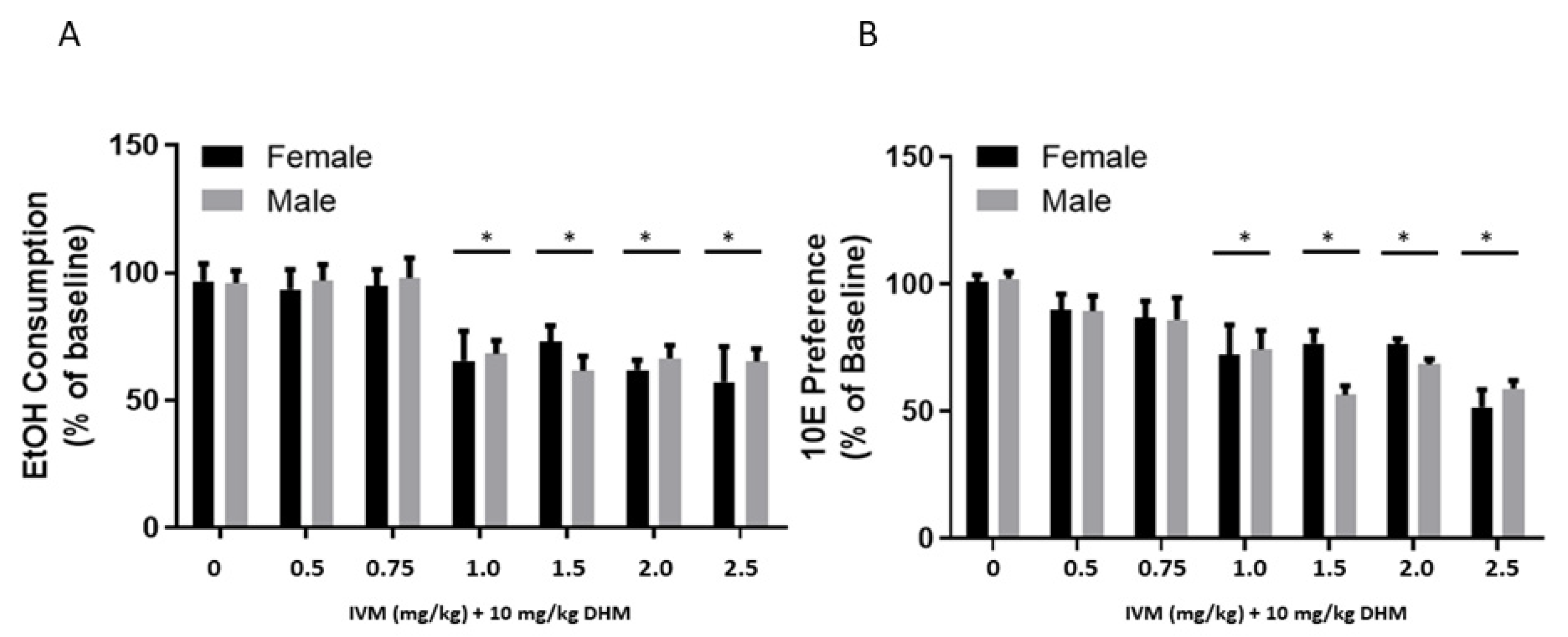
2.2. EtOH Consumption and Preference Averages Compared between Treatment Groups and Sex
2.2.1. EtOH Consumption Averages (24 h Post-Injection) Compared between Treatment Groups and Sex
2.2.2. 10E Preference Averages (24 h Post-Injection) Compared between Treatment Groups and Sex
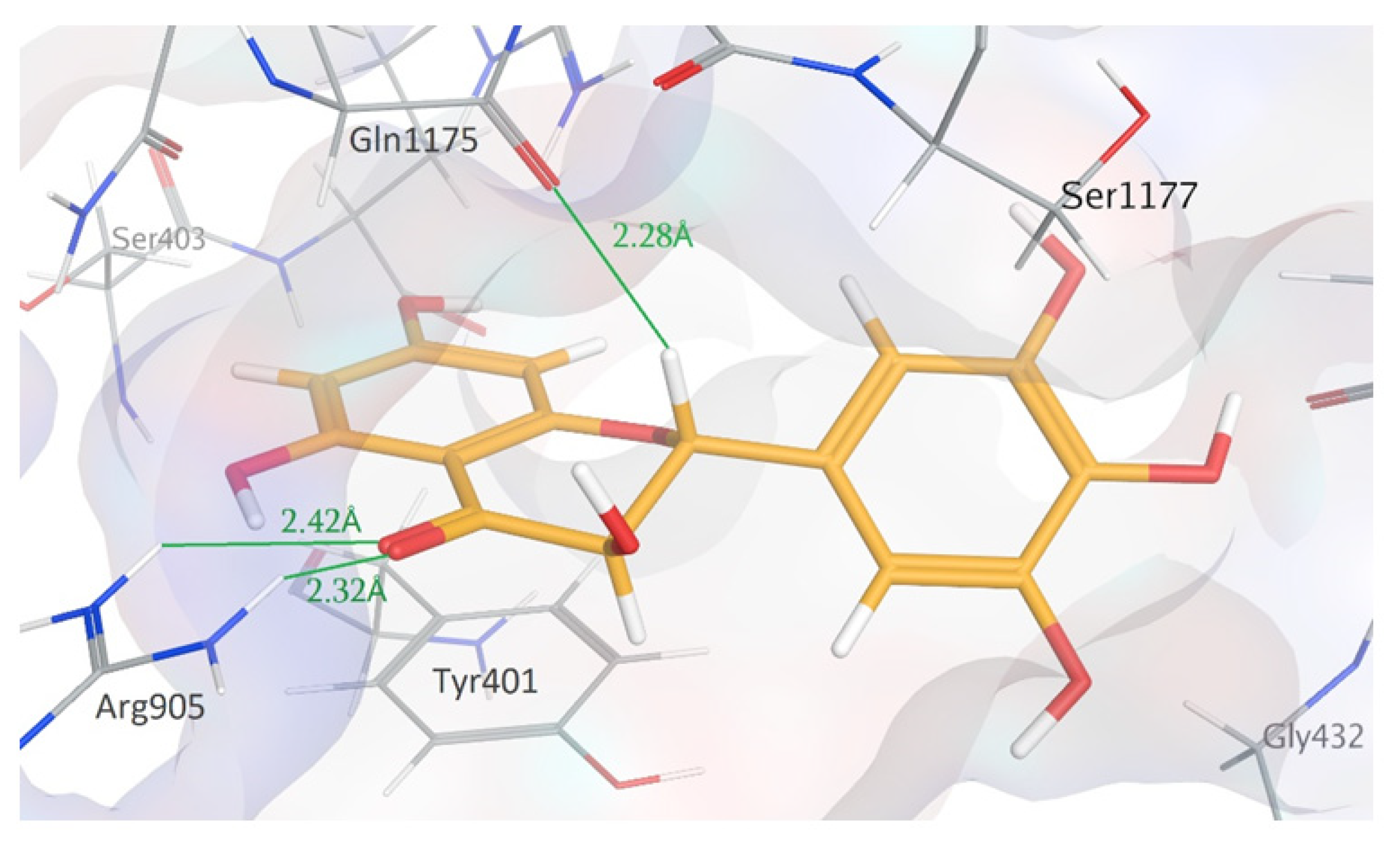
2.3. In Silico Modeling Studies
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Design
4.2. Two-Bottle Choice EtOH Drinking Behavior
4.3. Statistical Analyses
4.4. In Silico Modeling Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sacks, J.J.; Gonzales, K.R.; Bouchery, E.E.; Tomedi, L.E.; Brewer, R.D. 2010 National and State Costs of Excessive Alcohol Consumption. Am. J. Prev. Med. 2015, 49, e73–e79. [Google Scholar] [CrossRef] [PubMed]
- Stahre, M.; Roeber, J.; Kanny, D.; Brewer, R.D.; Zhang, X. Contribution of excessive alcohol consumption to deaths and years of potential life lost in the United States. Prev. Chronic. Dis. 2014. Epub ahead of print. [Google Scholar] [CrossRef] [Green Version]
- Esser, M.B.; Sherk, A.; Liu, Y.; Naimi, T.S.; Stockwell, T.; Stahre, M.; Kanny, D.; Landen, M.; Saitz, R.; Brewer, R.D. Deaths and Years of Potential Life Lost From Excessive Alcohol Use—United States, 2011–2015. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 981–987. [Google Scholar] [CrossRef]
- Kranzler, H.R.; Soyka, M. Diagnosis and pharmacotherapy of alcohol use disorder a review. JAMA—J. Am. Med. Assoc. 2018, 320, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Mark, T.L.; Kassed, C.A.; Vandivort-Warren, R.; Levit, K.R.; Kranzler, H.R. Alcohol and opioid dependence medications: Prescription trends, overall and by physician specialty. Drug Alcohol Depend. 2009, 99, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Leggio, L.; Lee, M.R. Treatment of Alcohol Use Disorder in Patients with Alcoholic Liver Disease. Am. J. Med. 2017, 130, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thursz, M.R.; Richardson, P.; Allison, M.; Austin, A.; Bowers, M.; Day, C.P.; Downs, N.; Gleeson, D.; MacGilchrist, A.; Grant, A.; et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N. Engl. J. Med. 2015, 372, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Fleury, M.J.; Djouini, A.; Huỳnh, C.; Tremblay, J.; Ferland, F.; Menard, J.M.; Belleville, G. Remission from substance use disorders: A systematic review and meta-analysis. Drug Alcohol Depend. 2016, 168, 293–306. [Google Scholar] [CrossRef]
- Kirouac, M.; Kruger, E.; Wilson, A.D.; Hallgren, K.A.; Witkiewitz, K. Consumption outcomes in clinical trials of alcohol use disorder treatment: Consideration of standard drink misestimation. Am. J. Drug Alcohol Abuse 2019, 45, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Popova, M.; Rodriguez, L.; Trudell, J.R.; Nguyen, S.; Bloomfield, M.; Davies, D.L.; Asatryan, L. Residues in transmembrane segments of the P2X4 receptor contribute to channel function and ethanol sensitivity. Int. J. Mol. Sci. 2020, 21, 2471. [Google Scholar] [CrossRef] [Green Version]
- Huynh, N.; Khoja, S.; Asatryan, L.; Jakowec, M.W.; Davies, D.L. The avermectin family as potential therapeutic compounds for alcohol use disorder: Implications for using P2X4 receptor as a drug-screening platform. In Neuroscience of Alcohol; Elsevier Inc.: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Wyatt, L.R.; Finn, D.A.; Khoja, S.; Yardley, M.M.; Asatryan, L.; Alkana, R.L.; Davies, D.L. Contribution of P2X4 receptors to ethanol intake in male C57BL/6 mice. Neurochem. Res. 2014, 39, 1127–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardley, M.M.; Wyatt, L.; Khoja, S.; Asatryan, L.; Ramaker, M.J.; Finn, D.A.; Alkana, R.L.; Huynh, N.; Louie, S.G.; Petasis, N.A.; et al. Ivermectin reduces alcohol intake and preference in mice. Neuropharmacology 2012, 63, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoja, S.; Huynh, N.; Warnecke, A.M.; Asatryan, L.; Jakowec, M.W.; Davies, D.L. Preclinical evaluation of avermectins as novel therapeutic agents for alcohol use disorders. Psychopharmacology 2018, 235, 1697–1709. [Google Scholar] [CrossRef]
- Yardley, M.M.; Neely, M.; Huynh, N.; Asatryan, L.; Louie, S.G.; Alkana, R.L.; Davies, D.L. Multiday administration of ivermectin is effective in reducing alcohol intake in mice at doses shown to be safe in humans. Neuroreport 2014, 25, 1018–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asatryan, L.; Popova, M.; Perkins, D.; Trudell, J.R.; Alkana, R.L.; Davies, D.L. Ivermectin antagonizes ethanol inhibition in purinergic P2X4 receptors. J. Pharmacol. Exp. Ther. 2010, 334, 720–728. [Google Scholar] [CrossRef] [Green Version]
- Asatryan, L.; Popova, M.; Woodward, J.J.; King, B.F.; Alkana, R.L.; Davies, D.L. Roles of ectodomain and transmembrane regions in ethanol and agonist action in purinergic P2X2 and P2X3 receptors. Neuropharmacology 2008, 55, 835–843. [Google Scholar] [CrossRef] [Green Version]
- Franklin, K.M.; Asatryan, L.; Jakowec, M.W.; Trudell, J.R.; Bell, R.L.; Davies, D.L. P2X4 receptors (P2X4Rs) represent a novel target for the development of drugs to prevent and/or treat alcohol use disorders. Front. Neurosci. 2014, 8, 176. [Google Scholar] [CrossRef] [Green Version]
- Kidd, E.J.; Grahames, C.B.; Simon, J.; Michel, A.D.; Barnard, E.A.; Humphrey, P.P. Localization of P(2X) purinoceptor transcripts in the rat nervous system. Mol. Pharmacol. 1995, 48, 569–573. [Google Scholar]
- Li, C.; Aguayo, L.; Peoples, R.W.; Weight, F.F. Ethanol inhibits a neuronal ATP-gated ion channel. Mol. Pharmacol. 1993, 44, 871–875. [Google Scholar]
- Huynh, N.; Arabian, N.; Naito, A.; Louie, S.; Jakowec, M.W.; Asatryan, L.; Davies, D.L. Preclinical development of moxidectin as a novel therapeutic for alcohol use disorder. Neuropharmacology 2017, 113, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Menez, C.; Sutra, J.F.; Prichard, R.; Lespine, A. Relative Neurotoxicity of Ivermectin and Moxidectin in Mdr1ab (−/−) Mice and Effects on Mammalian GABA(A) Channel Activity. PLoS Negl. Trop. Dis 2012. Epub ahead of print November. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.; Blazquez, A.G.; Real, R.; Mendoza, G.; Prieto, J.G.; Merino, G.; Alvarez, A.I. In vitro and in vivo interaction of moxidectin with BCRP/ABCG2. Chem. Biol. Interact. 2009, 180, 106–112. [Google Scholar] [CrossRef]
- Amin, M.L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target. Insights 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Kemper, E.M.; Verheij, M.; Boogerd, W.; Beijnen, J.H.; Van Tellingen, O. Improved penetration of docetaxel into the brain by co-administration of inhibitors of P-glycoprotein. Eur. J. Cancer 2004, 40, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Donohue, T.M. Implication of altered proteasome function in alcoholic liver injury. World J. Gastroenterol. 2007, 13, 4931–4937. [Google Scholar] [CrossRef] [Green Version]
- Potschka, H. Targeting the brain—Surmounting or bypassing the blood-brain barrier. Handb. Exp. Pharmacol. 2010, 197, 411–431. [Google Scholar]
- Silva, J.; Khoja, S.; Asatryan, L.; Pacifici, E.; Davies, D.L. A novel pharmacotherapy approach using P-glycoprotein (PGP/ABCB1) efflux inhibitor combined with ivermectin to reduce alcohol drinking and preference in mice. Alcohol 2020. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.L.; Lin, H.Y.; Chan, M.C.; Lin, W.L.; Lin, W.C. Treatment of chronic liver injuries in mice by oral administration of ethanolic extract of the fruit of Hovenia dulcis. Am. J. Chin. Med. 2007, 35, 693–703. [Google Scholar] [CrossRef]
- Qiu, P.; Dong, Y.; Zhu, T.; Luo, Y.Y.; Kang, X.J.; Pang, M.X.; Li, H.Z.; Xu, H.; Gu, C.; Pan, S.H.; et al. Semen hoveniae extract ameliorates alcohol-induced chronic liver damage in rats via modulation of the abnormalities of gut-liver axis. Phytomedicine 2019, 52, 40–50. [Google Scholar] [CrossRef]
- Silva, J.; Yu, X.; Moradian, R.; Folk, C.; Spatz, M.H.; Kim, P.; Bhatti, A.A.; Davies, D.L.; Liang, J. Dihydromyricetin Protects the Liver via Changes in Lipid Metabolism and Enhanced Ethanol Metabolism. Alcohol Clin. Exp. Res. 2020. Epub ahead of print. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, C.; Meng, Q.; Liu, Z.; Huo, X.; Sun, P.; Sun, H.; Ma, X.; Peng, J.; Liu, K. Targeting P-glycoprotein and SORCIN: Dihydromyricetin strengthens anti-proliferative efficiency of adriamycin via MAPK/ERK and Ca2+-mediated apoptosis pathways in MCF-7/ADR and K562/ADR. J. Cell Physiol. 2018, 233, 3066–3079. [Google Scholar] [CrossRef]
- Wong, I.L.; Wang, B.C.; Yuan, J.; Duan, L.X.; Liu, Z.; Liu, T.; Li, X.M.; Hu, X.; Zhang, X.Y.; Jiang, T.; et al. Potent and Nontoxic Chemosensitizer of P-Glycoprotein-Mediated Multidrug Resistance in Cancer: Synthesis and Evaluation of Methylated Epigallocatechin, Gallocatechin, and Dihydromyricetin Derivatives. J. Med. Chem. 2015, 58, 4529–4549. [Google Scholar] [CrossRef]
- Deng, Y.; Guo, L.; Cai, H.; Chen, L.; Tan, S.; Zhang, B.; Fang, P.; Xiang, D.; Li, H.; He, G.; et al. Dihydromyricetin affect the pharmacokinetics of triptolide in rats. Xenobiotica 2020, 50, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Fang, Y.; Cao, W.; Liang, F.; Pan, S.; Xu, X. Quantitative structure-activity relationships for the flavonoid-mediated inhibition of P-Glycoprotein in KB/MDR1 Cells. Molecules 2019, 24, 1661. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Liu, X.; Chow, L.M.C. Flavonoids as P-gp Inhibitors: A Systematic Review of SARs. Curr. Med. Chem. 2018, 26, 4799–4831. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, A.; Conseil, G.; Perez-Victoria, J.M.; Dayan, G.; Baubichon-Cortay, H.; Trompier, D.; Steinfels, E.; Jault, J.M.; De Wet, H.; Maitrejean, M.; et al. Modulation by flavonoids of cell multidrug resistance mediated by P-glycoprotein and related ABC transporters. Cell Mol. Life Sci. 2002, 59, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Wongrattanakamon, P.; Lee, V.S.; Nimmanpipug, P.; Jiranusornkul, S. Nucleotide-binding domain 1 modelling: A novel molecular docking approach for screening of P-glycoprotein inhibitory activity of bioflavonoids. Chem. Data Collect. 2016, 2, 10–16. [Google Scholar] [CrossRef]
- Kadioglu, O.; Efferth, T. A Machine Learning-Based Prediction Platform for P-Glycoprotein Modulators and Its Validation by Molecular Docking. Cells 2019, 8, 1286. [Google Scholar] [CrossRef] [Green Version]
- Wongrattanakamon, P.; Nimmanpipug, P.; Sirithunyalug, B.; Chansakaow, S.; Jiranusornkul, S. A significant mechanism of molecular recognition between bioflavonoids and P-glycoprotein leading to herb-drug interactions. Toxicol. Mech. Methods 2018, 28, 11. [Google Scholar] [CrossRef]
- Silva, J.; Spatz, M.H.; Folk, C.; Chang, A.; Cadenas, E.; Liang, J.; Davies, D.L. Dihydromyricetin improves mitochondrial outcomes in the liver of alcohol-fed mice via the AMPK/Sirt-1/PGC-1α signaling axis. Alcohol 2020, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Chen, Z.; Zhang, C.; Xu, X.; Jin, J.; Zhan, W.; Han, T.; Wang, J. Dihydromyricetin ameliorates oleic acid-induced lipid accumulation in L02 and HepG2 cells by inhibiting lipogenesis and oxidative stress. Life Sci. 2016, 157, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhao, X.; Wan, J.; Ran, L.; Qin, Y.; Wang, X.; Gao, Y.; Shu, F.; Zhang, Y.; Liu, P.; et al. Dihydromyricetin improves glucose and lipid metabolism and exerts anti-inflammatory effects in nonalcoholic fatty liver disease: A randomized controlled trial. Pharmacol. Res. 2015, 99, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yang, J.; Hu, O.; Huang, J.; Ran, L.; Chen, M.; Zhang, Y.; Zhou, X.; Zhu, J.; Zhang, Q.; et al. Dihydromyricetin Ameliorates Nonalcoholic Fatty Liver Disease by Improving Mitochondrial Respiratory Capacity and Redox Homeostasis Through Modulation of SIRT3 Signaling. Antioxid. Redox Signal. 2019, 30, 163–183. [Google Scholar] [CrossRef]
- Krause, R.M.; Buisson, B.; Bertrand, S.; Corringer, P.J.; Galzi, J.L.; Changeux, J.P.; Bertrand, D. Ivermectin: A positive allosteric effector of the α7 neuronal nicotinic acetylcholine receptor. Mol. Pharmacol. 1998, 53, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Krůšek, J.; Zemková, H. Effect of ivermectin on γ-aminobutyric acid-induced chloride currents in mouse hippocampal embryonic neurones. Eur. J. Pharmacol. 1994, 259, 121–128. [Google Scholar] [CrossRef]
- Shan, Q.; Haddrill, J.L.; Lynch, J.W. Ivermectin, an Unconventional Agonist of the Glycine Receptor Chloride Channel. J. Biol. Chem. 2001, 276, 12556–12564. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Clarke, D.M. Tariquidar inhibits P-glycoprotein drug efflux but activates ATPase activity by blocking transition to an open conformation. Biochem. Pharmacol. 2014, 92, 558–566. [Google Scholar] [CrossRef]
- Shen, Y.; Lindemeyer, A.K.; Gonzalez, C.; Shao, X.M.; Spigelman, I.; Olsen, R.W.; Liang, J. Dihydromyricetin as a novel anti-alcohol intoxication medication. J. Neurosci. 2012, 32, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef]
- Silva, J.; Yu, X.; Qi, L.; Davies, D.L.; Liang, J. Antialcohol Effects of Dihydromyricetin in Combination with Other Flavonoids. Nat. Prod. Commun. 2020, 15, 2–6. [Google Scholar]
- Hou, X.L.; Tong, Q.; Wang, W.Q.; Shi, C.Y.; Xiong, W.; Chen, J.; Liu, X.; Fang, J.G. Suppression of Inflammatory Responses by Dihydromyricetin, a Flavonoid from Ampelopsis grossedentata, via Inhibiting the Activation of NF-κB and MAPK Signaling Pathways. J. Nat. Prod. 2015, 78, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zhang, T.; Shi, L.; Kang, C.; Wan, J.; Zhou, Y.; Zhu, J.; Mi, M. Ampelopsin protects endothelial cells from hyperglycemia-induced oxidative damage by inducing autophagy via the AMPK signaling pathway. BioFactors 2015, 41, 463–475. [Google Scholar] [CrossRef]
- Chu, J.; Wang, X.; Bi, H.; Li, L.; Ren, M.; Wang, J. Dihydromyricetin relieves rheumatoid arthritis symptoms and suppresses expression of pro-inflammatory cytokines via the activation of Nrf2 pathway in rheumatoid arthritis model. Int. Immunopharmacol. 2018, 59, 174–180. [Google Scholar] [CrossRef]
- Qiu, P.; Dong, Y.; Li, B.; Kang, X.J.; Gu, C.; Zhu, T.; Luo, Y.Y.; Pang, M.X.; Du, W.F.; Ge, W.H. Dihydromyricetin modulates p62 and autophagy crosstalk with the Keap-1/Nrf2 pathway to alleviate ethanol-induced hepatic injury. Toxicol. Lett. 2017, 274, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, D.J.; Yardley, M.M.; Lunny, K.F.; Louie, S.G.; Davies, D.L.; Miotto, K.; Ray, L.A. A Pilot Study of the Safety and Initial Efficacy of Ivermectin for the Treatment of Alcohol Use Disorder. Alcohol Clin. Exp. Res. 2016, 40, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Reus, V.I.; Fochtmann, L.J.; Bukstein, O.; Eyler, A.E.; Hilty, D.M.; Horvitz-Lennon, M.; Mahoney, J.; Pasic, J.; Weaver, M.; Wills, C.D.; et al. The American psychiatric association practice guideline for the pharmacological treatment of patients with alcohol use disorder. Am. J. Psychiatry 2018, 175, 86–90. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis Prim. 2018. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Mellinger, J.L.; Shedden, K.; Winder, G.S.; Tapper, E.; Adams, M.; Fontana, R.J.; Volk, M.L.; Blow, F.C.; Lok, A.S. The high burden of alcoholic cirrhosis in privately insured persons in the United States. Hepatology 2018, 68, 872–882. [Google Scholar] [CrossRef] [Green Version]
- Chacko, K.R.; Reinus, J. Spectrum of Alcoholic Liver Disease. Clin. Liver Dis. 2016, 20, 419–427. [Google Scholar] [CrossRef]
- Baraona, E.; Lieber, C.S. Effects of ethanol on lipid metabolism. J. Lipid Res. 1979, 20, 289–315. [Google Scholar] [CrossRef]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcoholic liver disease: New insights in pathogenesis lead to new treatments. J. Hepatol. 2000, 32, 113–128. [Google Scholar] [CrossRef]
- Lieber, C.S. Alcohol and the liver: 1994 update. Gastroenterology 1994, 106, 1085–1105. [Google Scholar] [CrossRef]
- Jin, L.; Wang, R.; Zhu, Y.; Zheng, W.; Han, Y.; Guo, F.; Ye, F.B.; Li, Y. Selective targeting of nuclear receptor FXR by avermectin analogues with therapeutic effects on nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 1–15. [Google Scholar]
- Yang, J.S.; Qi, W.; Farias-Pereira, R.; Choi, S.; Clark, J.M.; Kim, D.; Park, Y. Permethrin and ivermectin modulate lipid metabolism in steatosis-induced HepG2 hepatocyte. Food Chem. Toxicol. 2019, 125, 595–604. [Google Scholar] [CrossRef]
- Jin, L.; Feng, X.; Rong, H.; Pan, Z.; Inaba, Y.; Qiu, L.; Zheng, W.; Lin, S.; Wang, R.; Wang, Z.; et al. The antiparasitic drug ivermectin is a novel FXR ligand that regulates metabolism. Nat. Commun. 2013, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lv, L.; Pi, H.; Qin, W.; Chen, J.; Guo, D.; Lin, J.; Chi, X.; Jiang, Z.; Yang, H.; et al. Dihydromyricetin protects against liver ischemia/reperfusion induced apoptosis via activation of FOXO3a-mediated autophagy. Oncotarget 2016, 7, 76508–76522. [Google Scholar] [CrossRef]
- Huynh, N.; Arabian, N.M.; Asatryan, L.; Davies, D.L. Murine drinking models in the development of pharmacotherapies for alcoholism: Drinking in the dark and two-bottle choice. J. Vis. Exp. 2019, 2019, 2–8. [Google Scholar] [CrossRef]
- Tordoff, M.G.; Bachmanov, A.A. Influence of test duration on the sensitivity of the two-bottle choice test. Chem. Sens. 2002, 27, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Biddle, F.G.; Eales, B.A. The degree of lateralization of paw usage (handedness) in the mouse is defined by three major phenotypes. Behav. Genet. 1996, 26, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Bachmanov, A.A.; Reed, D.R.; Beauchamp, G.K.; Tordoff, M.G. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 2002, 32, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Belknap, J.K.; Crabbe, J.C.; Young, E.R. Voluntary consumption of ethanol in 15 inbred mouse strains. Psychopharmacology 1993, 112, 503–510. [Google Scholar] [CrossRef]
- Rodgers, D.A. Factors Underlying Differences in Alcohol Preference Among Inbred Strains of Mice. Psychosom Med. 1966, 28, 498–513. [Google Scholar] [CrossRef]
- Hwa, L.S.; Chu, A.; Levinson, S.A.; Kayyali, T.M.; DeBold, J.F.; Miczek, K.A. Persistent escalation of alcohol drinking in C57BL/6J mice with intermittent access to 20% ethanol. Alcohol Clin. Exp. Res. 2011, 35, 1938–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, C.E.; Griffin, W.C.; Luderman, L.N.; Kates, M.M.; McGinty, J.F.; Becker, H.C. Oxytocin Reduces Ethanol Self-Administration in Mice. Alcohol Clin. Exp. Res. 2017, 41, 955–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, J.; Carry, E.; Xue, C.; Zhang, J.; Liang, J.; Roberge, J.Y.; Davies, D.L. A Novel Dual Drug Approach That Combines Ivermectin and Dihydromyricetin (DHM) to Reduce Alcohol Drinking and Preference in Mice. Molecules 2021, 26, 1791. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061791
Silva J, Carry E, Xue C, Zhang J, Liang J, Roberge JY, Davies DL. A Novel Dual Drug Approach That Combines Ivermectin and Dihydromyricetin (DHM) to Reduce Alcohol Drinking and Preference in Mice. Molecules. 2021; 26(6):1791. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061791
Chicago/Turabian StyleSilva, Joshua, Eileen Carry, Chen Xue, Jifeng Zhang, Jing Liang, Jacques Y. Roberge, and Daryl L. Davies. 2021. "A Novel Dual Drug Approach That Combines Ivermectin and Dihydromyricetin (DHM) to Reduce Alcohol Drinking and Preference in Mice" Molecules 26, no. 6: 1791. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26061791