
The Future of Cancer Diagnosis, Treatment and Surveillance: A Systemic Review on Immunotherapy and Immuno-PET Radiotracers

 ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Material and Methods
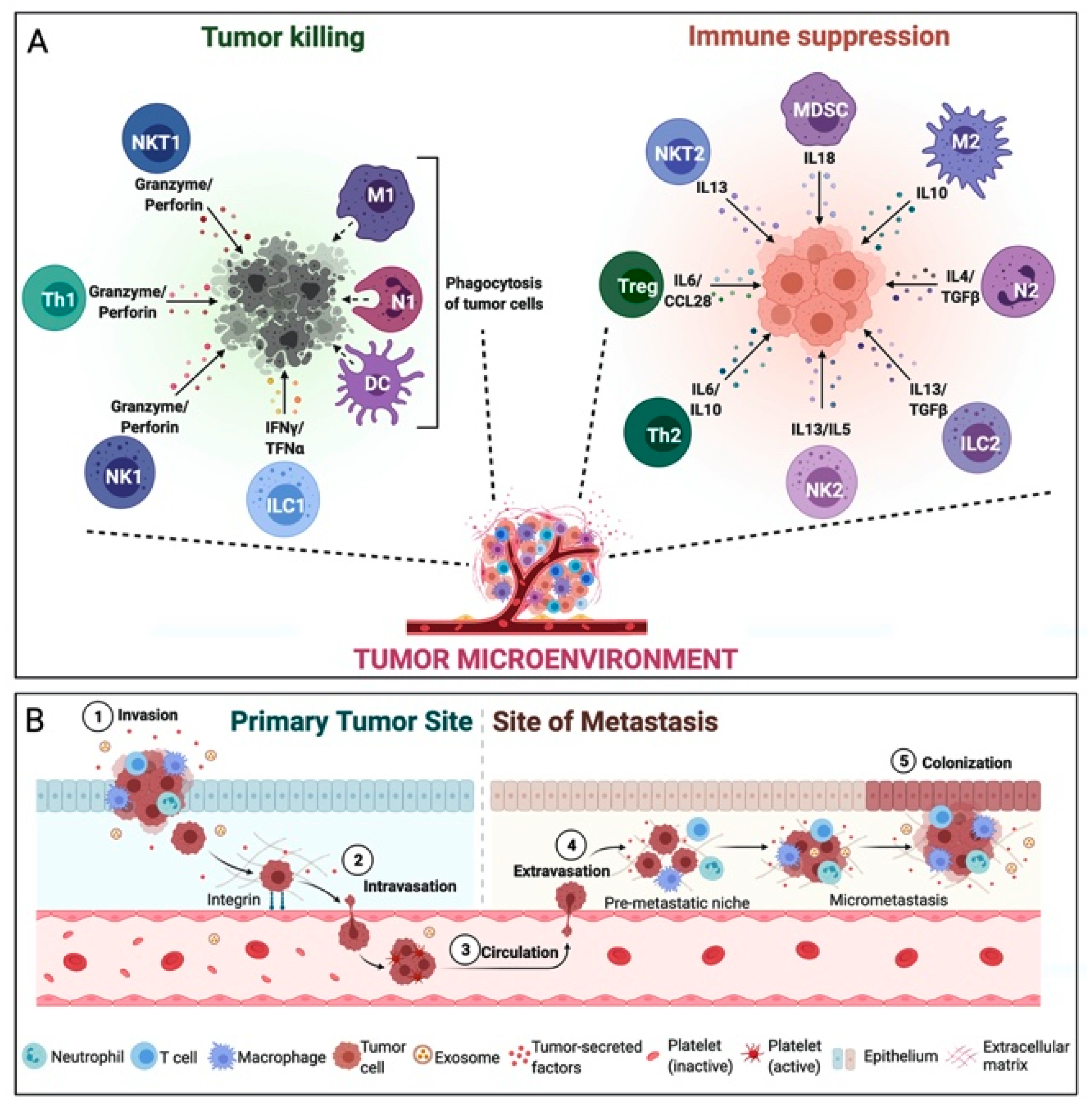
3. Tumor Microenvironment
4. Immunotherapies Targets
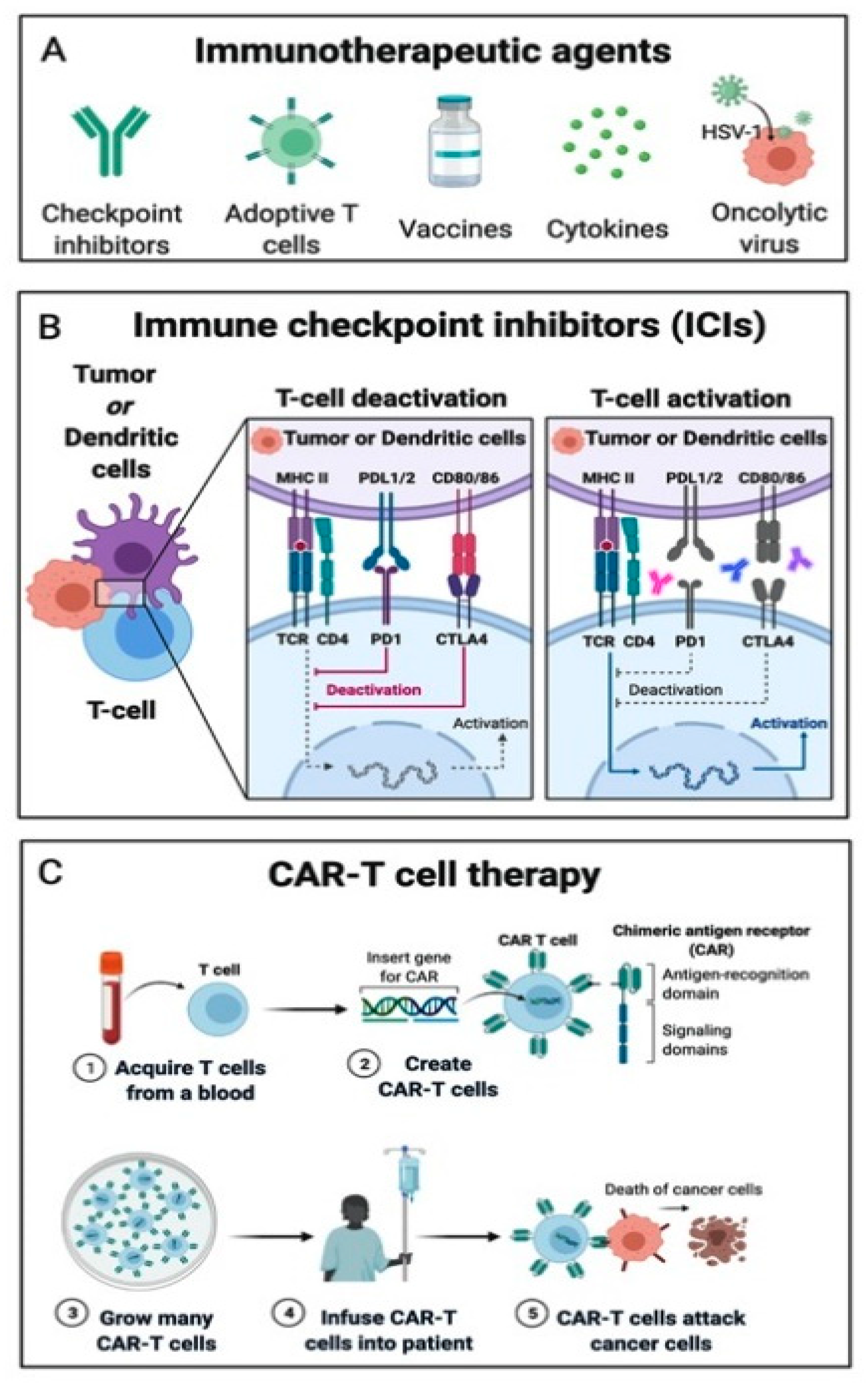
4.1. Immune Checkpoint Inhibitors
4.2. Adoptive Cell Transfer
4.3. Oncolytic Viruses, Cancer Vaccines and Cytokine Therapies
4.4. Open Question on the Use of Immunotherapies Targets
5. Molecular Imaging
5.1. Immuno-PET
5.1.1. Critical Issues in the Development of a Good Radiotracer in Immunotherapy
5.1.2. PD-1/PD-L1 Pathways
Labelled Antibodies
Radiolabeled Small Molecules
Radiolabeled Atezolizumab
Radiolabeled Pembrolizumab
Radiolabeled Nivolumab
5.1.3. Cytotoxic T-lymphocyte–Associated Protein 4 (CTLA-4) Pathway
5.1.4. Adoptive Cell Transfer (ACT) Therapy
5.1.5. Chimeric Antigen Receptor T Cell Therapy (CAR-T)
5.1.6. CD8+ T Lymphocytes
5.1.7. Other Immuno-PET Radiotracers
Tumor Associated Macrophages (TAM)
CD3
CD4
5.2. [18F]FDG PET/CT
6. Artificial Intelligence (AI) in Immunotherapy
6.1. Radiomics Assessing the Response to Immunotherapy and Survival Prediction
6.2. Radiomics Assessing the Molecular Profile
6.3. Deep-Learning Approach
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. J. Clin. Epidemiol. 2009, 62, e1–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nisticò, P.; Signore, A. Relevance of immune cell and tumor microenvironment imaging in the new era of immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Janco, J.M.T.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-Infiltrating Dendritic Cells in Cancer Pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Lecot, P.; Sarabi, M.; Abrantes, M.P.; Mussard, J.; Koenderman, L.; Caux, C.; Bendriss-Vermare, N.; Michallet, M.-C. Neutrophil Heterogeneity in Cancer: From Biology to Therapies. Front. Immunol. 2019, 10, 2155. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J.S.; Sahota, R.A.; Milne, K.; Kost, S.E.; Nesslinger, N.J.; Watson, P.H.; Nelson, B.H. CD20+ Tumor-Infiltrating Lymphocytes Have an Atypical CD27− Memory Phenotype and Together with CD8+ T Cells Promote Favorable Prognosis in Ovarian Cancer. Clin. Cancer Res. 2012, 18, 3281–3292. [Google Scholar] [CrossRef] [Green Version]
- Al-Shibli, K.I.; Donnem, T.; Al-Saad, S.; Persson, M.; Bremnes, R.M.; Busund, L.-T. Prognostic Effect of Epithelial and Stromal Lymphocyte Infiltration in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Bouleau, A.; Lebon, V.; Truillet, C. PET imaging of immune checkpoint proteins in oncology. Pharmacol. Ther. 2021, 222, 107786. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, N.; Mao, Q.; Xia, H. CAR T-cells for cancer therapy. Biotechnol. Genet. Eng. Rev. 2017, 33, 190–226. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia; Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia; Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2016, 374, 998. [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Killock, D. Skin cancer: T-VEC oncolytic viral therapy shows promise in melanoma. Nat. Rev. Clin. Oncol. 2015, 12, 438. [Google Scholar] [CrossRef]
- Kawakami, Y.; Eliyahu, S.; Delgado, C.H.; Robbins, P.F.; Sakaguchi, K.; Appella, E.; Yannelli, J.R.; Adema, G.J.; Miki, T.; Rosenberg, S.A. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. USA 1994, 91, 6458–6462. [Google Scholar] [CrossRef] [Green Version]
- Jandus, C.; Bioley, G.; Dojcinovic, D.; Derré, L.; Baitsch, L.; Wieckowski, S.; Rufer, N.; Kwok, W.W.; Tiercy, J.-M.; Luescher, I.F.; et al. Tumor Antigen–Specific FOXP3+ CD4 T Cells Identified in Human Metastatic Melanoma: Peptide Vaccination Results in Selective Expansion of Th1-like Counterparts. Cancer Res. 2009, 69, 8085–8093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Eynde, B.V.D.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larimer, B.M.; Wehrenberg-Klee, E.; Dubois, F.; Mehta, A.; Kalomeris, T.; Flaherty, K.; Boland, G.; Mahmood, U. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res. 2017, 77, 2318–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [Green Version]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Kluger, H.M.; Zito, C.R.; Turcu, G.; Baine, M.K.; Zhang, H.; Adeniran, A.; Sznol, M.; Rimm, D.L.; Kluger, Y.; Chen, L.; et al. PD-L1 Studies Across Tumor Types, Its Differential Expression and Predictive Value in Patients Treated with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 4270–4279. [Google Scholar] [CrossRef] [Green Version]
- Tunger, A.; Sommer, U.; Wehner, R.; Kubasch, A.S.; Grimm, M.-O.; Bachmann, M.P.; Platzbecker, U.; Bornhäuser, M.; Baretton, G.; Schmitz, M. The Evolving Landscape of Biomarkers for Anti-PD-1 or Anti-PD-L1 Therapy. J. Clin. Med. 2019, 8, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastracci, L.; Fontana, V.; Queirolo, P.; Carosio, R.; Grillo, F.; Morabito, A.; Banelli, B.; Tanda, E.; Boutros, A.; Dozin, B.; et al. Response to ipilimumab therapy in metastatic melanoma patients: Potential relevance of CTLA-4+ tumor infiltrating lymphocytes and their in situ localization. Cancer Immunol. Immunother. 2020, 69, 653–662. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziv, E.; Durack, J.C.; Solomon, S.B. The Importance of Biopsy in the Era of Molecular Medicine. Cancer J. 2016, 22, 418–422. [Google Scholar] [CrossRef] [Green Version]
- McQuerry, J.A.; Chang, J.T.; Bowtell, D.D.L.; Cohen, A.; Bild, A.H. Mechanisms and clinical implications of tumor heterogeneity and convergence on recurrent phenotypes. J. Mol. Med. 2017, 95, 1167–1178. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès–Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Zou, Y.; Hu, X.; Zheng, S.; Yang, A.; Li, X.; Tang, H.; Kong, Y.; Xie, X. Discordance of immunotherapy response predictive biomarkers between primary lesions and paired metastases in tumours: A multidimensional analysis. EBioMedicine 2021, 63, 103137. [Google Scholar] [CrossRef]
- De Jong, M.; Essers, J.; Van Weerden, W.M. Imaging preclinical tumour models: Improving translational power. Nat. Rev. Cancer 2014, 14, 481–493. [Google Scholar] [CrossRef]
- Graham, M.M. Clinical Molecular Imaging with Radiotracers: Current Status. Med. Princ. Pract. 2012, 21, 197–208. [Google Scholar] [CrossRef]
- Sharma, R.; Aboagye, E. Development of radiotracers for oncology-the interface with pharmacology. Br. J. Pharmacol. 2011, 163, 1565–1585. [Google Scholar] [CrossRef] [Green Version]
- Abousaway, O.; Rakhshandehroo, T.; Abbeele, A.D.V.D.; Kircher, M.F.; Rashidian, M. Noninvasive Imaging of Cancer Immunotherapy. Nanotheranostics 2021, 5, 90–112. [Google Scholar] [CrossRef] [PubMed]
- Meidenbauer, N.; Marienhagen, J.; Laumer, M.; Vogl, S.; Heymann, J.; Andreesen, R.; Mackensen, A. Survival and tumor localization of adoptively transferred Melan-A-specific T cells in melanoma patients. J. Immunol. 2003, 170, 2161–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, A.; Pandey, M.K.; Demirhan, Y.E.; Nesbitt, J.J.; Crespo-Diaz, R.J.; Terzic, A.; Behfar, A.; DeGrado, T.R. Novel 89Zr cell labeling approach for PET-based cell trafficking studies. EJNMMI Res. 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weist, M.R.; Starr, R.; Aguilar, B.; Chea, J.; Miles, J.K.; Poku, E.; Gerdts, E.; Yang, X.; Priceman, S.J.; Forman, S.J.; et al. PET of Adoptively Transferred Chimeric Antigen Receptor T Cells with 89Zr-Oxine. J. Nucl. Med. 2018, 59, 1531–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurebayashi, Y.; Choyke, P.L.; Sato, N. Imaging of cell-based therapy using 89Zr-oxine ex vivo cell labeling for positron emission tomography. Nanotheranostics 2021, 5, 27–35. [Google Scholar] [CrossRef]
- Severin, G.W.; Engle, J.W.; Barnhart, T.E.; Nickles, R.J. 89Zr Radiochemistry for Positron Emission Tomography. Med. Chem. 2011, 7, 389–394. [Google Scholar] [CrossRef]
- McCarthy, C.E.; White, J.M.; Viola, N.T.; Gibson, H.M. In vivo Imaging Technologies to Monitor the Immune System. Front. Immunol. 2020, 11, 1067. [Google Scholar] [CrossRef]
- Xenaki, K.T.; Oliveira, S.; Henegouwen, P.M.P.V.B.E. Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors. Front. Immunol. 2017, 8, 1287. [Google Scholar] [CrossRef]
- Thurber, G.M.; Zajic, S.C.; Wittrup, K.D. Theoretic Criteria for Antibody Penetration into Solid Tumors and Micrometastases. J. Nucl. Med. 2007, 48, 995–999. [Google Scholar] [CrossRef] [Green Version]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Li, A.; Liu, Q.; Yuan, X.; Xu, H.; Jiao, D.; Pestell, R.G.; Han, X.; Wu, K. Recent advances of bispecific antibodies in solid tumors. J. Hematol. Oncol. 2017, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Lesniak, W.G.; Miller, M.S.; Lisok, A.; Sikorska, E.; Wharram, B.; Kumar, D.; Gabrielson, M.; Pomper, M.G.; Gabelli, S.B.; et al. Rapid PD-L1 detection in tumors with PET using a highly specific peptide. Biochem. Biophys. Res. Commun. 2017, 483, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; Van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 2018, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bensch, F.; Van Der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; Van Der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Pandit-Taskar, N.; Postow, M.A.; Hellmann, M.D.; Harding, J.J.; Barker, C.A.; O’Donoghue, J.A.; Ziolkowska, M.; Ruan, S.; Lyashchenko, S.K.; Tsai, F.; et al. First-in-Humans Imaging with 89Zr-Df-IAB22M2C Anti-CD8 Minibody in Patients with Solid Malignancies: Preliminary Pharmacokinetics, Biodistribution, and Lesion Targeting. J. Nucl. Med. 2020, 61, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Mayer, A.T.; Xu, L.; Reeves, R.E.; Gano, J.; Gambhir, S.S. Novel Radiotracer for ImmunoPET Imaging of PD-1 Checkpoint Expression on Tumor Infiltrating Lymphocytes. Bioconjug. Chem. 2015, 26, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, A.; Nedrow, J.R.; Park, S.; Banerjee, S.R.; Rittenbach, A.; Jammes, F.; Tsui, B.; Sgouros, G. Imaging, Biodistribution, and Dosimetry of Radionuclide-Labeled PD-L1 Antibody in an Immunocompetent Mouse Model of Breast Cancer. Cancer Res. 2016, 76, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Veen, E.L.; Suurs, F.V.; Cleeren, F.; Bormans, G.; Elsinga, P.H.; Hospers, G.A.P.; Hooge, M.N.L.-D.; De Vries, E.G.E.; De Vries, E.F.J.; Antunes, I.F. Development and Evaluation of Interleukin-2–Derived Radiotracers for PET Imaging of T Cells in Mice. J. Nucl. Med. 2020, 61, 1355–1360. [Google Scholar] [CrossRef]
- Li, M.; Ehlerding, E.B.; Jiang, D.; E Barnhart, T.; Chen, W.; Cao, T.; Engle, J.W.; Cai, W. In vivo characterization of PD-L1 expression in breast cancer by immuno-PET with 89Zr-labeled avelumab. Am. J. Transl. Res. 2020, 12, 1862–1872. [Google Scholar]
- Kumar, D.; Lisok, A.; Dahmane, E.; McCoy, M.; Shelake, S.; Chatterjee, S.; Allaj, V.; Sysa-Shah, P.; Wharram, B.; Lesniak, W.G.; et al. Peptide-based PET quantifies target engagement of PD-L1 therapeutics. J. Clin. Investig. 2019, 129, 616–630. [Google Scholar] [CrossRef] [Green Version]
- Bao, R.; Wang, Y.; Lai, J.; Zhu, H.; Zhao, Y.; Li, S.; Li, N.; Huang, J.; Yang, Z.; Wang, F.; et al. Enhancing Anti-PD-1/PD-L1 Immune Checkpoint Inhibitory Cancer Therapy by CD276-Targeted Photodynamic Ablation of Tumor Cells and Tumor Vasculature. Mol. Pharm. 2019, 16, 339–348. [Google Scholar] [CrossRef]
- Kikuchi, M.; Clump, D.A.; Srivastava, R.M.; Sun, L.; Zeng, D.; Diaz-Perez, J.A.; Anderson, C.J.; Edwards, W.B.; Ferris, R.L. Preclinical immunoPET/CT imaging using Zr-89-labeled anti-PD-L1 monoclonal antibody for assessing radiation-induced PD-L1 upregulation in head and neck cancer and melanoma. Oncoimmunology 2017, 6, e1329071. [Google Scholar] [CrossRef] [Green Version]
- Christensen, C.; Kristensen, L.K.; Alfsen, M.Z.; Nielsen, C.H.; Kjaer, A. Quantitative PET imaging of PD-L1 expression in xenograft and syngeneic tumour models using a site-specifically labelled PD-L1 antibody. Eur. J. Nucl. Med. Mol. Imaging 2019, 47, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Lv, G.; Sun, X.; Qiu, L.; Sun, Y.; Li, K.; Liu, Q.; Zhao, Q.; Qin, S.; Lin, J. PET Imaging of Tumor PD-L1 Expression with a Highly Specific Nonblocking Single-Domain Antibody. J. Nucl. Med. 2019, 61, 117–122. [Google Scholar] [CrossRef]
- Rubins, D.J.; Meng, X.; McQuade, P.; Klimas, M.; Getty, K.; Lin, S.-A.; Connolly, B.M.; O’Malley, S.S.; Haley, H.; Purcell, M.; et al. In Vivo Evaluation and Dosimetry Estimate for a High Affinity Affibody PET Tracer Targeting PD-L1. Mol. Imaging Biol. 2020, 23, 241–249. [Google Scholar] [CrossRef]
- De Silva, R.A.; Kumar, D.; Lisok, A.; Chatterjee, S.; Wharram, B.; Rao, K.V.; Mease, R.; Dannals, R.F.; Pomper, M.G.; Nimmagadda, S. Peptide-Based 68Ga-PET Radiotracer for Imaging PD-L1 Expression in Cancer. Mol. Pharm. 2018, 15, 3946–3952. [Google Scholar] [CrossRef] [Green Version]
- Lesniak, W.G.; Mease, R.C.; Chatterjee, S.; Kumar, D.; Lisok, A.; Wharram, B.; Kalagadda, V.R.; Emens, L.A.; Pomper, M.G.; Nimmagadda, S. Development of [18F]FPy-WL12 as a PD-L1 Specific PET Imaging Peptide. Mol. Imaging 2019, 18, 1536012119852189. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.T.; Natarajan, A.; Gordon, S.R.; Maute, R.L.; McCracken, M.N.; Ring, A.M.; Weissman, I.L.; Gambhir, S.S. Practical Immuno-PET Radiotracer Design Considerations for Human Immune Checkpoint Imaging. J. Nucl. Med. 2017, 58, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Truillet, C.; Oh, H.L.J.; Yeo, S.P.; Lee, C.Y.; Huynh, L.T.; Wei, J.; Parker, M.F.L.; Blakely, C.; Sevillano, N.; Wang, Y.H.; et al. Imaging PD-L1 Expression with ImmunoPET. Bioconjug. Chem. 2018, 29, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Han, Y.; Liu, G.; Xu, Y.; Duan, D.; Liu, H.; Du, F.; Luo, P.; Liu, Z. Preclinical Study of a Fully Human Anti-PD-L1 Antibody as a Theranostic Agent for Cancer Immunotherapy. Mol. Pharm. 2018, 15, 4426–4433. [Google Scholar] [CrossRef] [PubMed]
- Wissler, H.L.; Ehlerding, E.B.; Lyu, Z.; Zhao, Y.; Zhang, S.; Eshraghi, A.; Buuh, Z.Y.; McGuth, J.C.; Guan, Y.; Engle, J.W.; et al. Site-Specific Immuno-PET Tracer to Image PD-L1. Mol. Pharm. 2019, 16, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Cheng, S.; Zou, S.; Zhu, D.; Zhu, T.; Wang, P.; Zhu, X. Immuno-PET Imaging of 89Zr Labeled Anti-PD-L1 Domain Antibody. Mol. Pharm. 2018, 15, 1674–1681. [Google Scholar] [CrossRef]
- Li, D.; Zou, S.; Cheng, S.; Song, S.; Wang, P.; Zhu, X. Monitoring the Response of PD-L1 Expression to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Nonsmall-Cell Lung Cancer Xenografts by Immuno-PET Imaging. Mol. Pharm. 2019, 16, 3469–3476. [Google Scholar] [CrossRef]
- Miao, Y.; Lv, G.; Chen, Y.; Qiu, L.; Xie, M.; Lin, J. One-Step Radiosynthesis and Initial Evaluation of a Small Molecule PET Tracer for PD-L1 Imaging. Bioorg. Med. Chem. Lett. 2020, 30, 127572. [Google Scholar] [CrossRef]
- Vento, J.; Mulgaonkar, A.; Woolford, L.; Nham, K.; Christie, A.; Bagrodia, A.; De Leon, A.D.; Hannan, R.; Bowman, I.; McKay, R.M.; et al. PD-L1 detection using 89Zr-atezolizumab immuno-PET in renal cell carcinoma tumorgrafts from a patient with favorable nivolumab response. J. Immunother. Cancer 2019, 7, 144. [Google Scholar] [CrossRef]
- Van Der Veen, E.L.; Giesen, D.; Jong, L.P.-D.; Jorritsma-Smit, A.; Vries, E.G.E.D.; Hooge, M.N.L.-D. 89Zr-pembrolizumab biodistribution is influenced by PD-1-mediated uptake in lymphoid organs. J. Immunother. Cancer 2020, 8, e000938. [Google Scholar] [CrossRef]
- Li, W.; Wang, Y.; Rubins, D.; Bennacef, I.; Holahan, M.; Haley, H.; Purcell, M.; Gantert, L.; Hseih, S.C.; Judo, M.; et al. PET/CT Imaging of 89Zr-N-sucDf-Pembrolizumab in Healthy Cynomolgus Monkeys. Mol. Imaging Biol. 2020, 23, 250–259. [Google Scholar] [CrossRef]
- Cole, E.L.; Kim, J.; Donnelly, D.J.; Smith, R.A.; Cohen, D.; Lafont, V.; Morin, P.E.; Huang, R.Y.-C.; Chow, P.L.; Hayes, W.; et al. Radiosynthesis and preclinical PET evaluation of 89Zr-nivolumab (BMS-936558) in healthy non-human primates. Bioorg. Med. Chem. 2017, 25, 5407–5414. [Google Scholar] [CrossRef]
- England, C.G.; Jiang, D.; Ehlerding, E.B.; Rekoske, B.T.; Ellison, P.A.; Hernandez, R.; Barnhart, T.E.; McNeel, D.G.; Huang, P.; Cai, W. 89Zr-labeled nivolumab for imaging of T-cell infiltration in a humanized murine model of lung cancer. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Ehlerding, E.B.; Lee, H.J.; Jiang, D.; A Ferreira, C.; Zahm, C.D.; Huang, P.; Engle, J.W.; McNeel, D.G.; Cai, W. Antibody and fragment-based PET imaging of CTLA-4+ T-cells in humanized mouse models. Am. J. Cancer Res. 2019, 9, 53–63. [Google Scholar]
- Ehlerding, E.B.; England, C.G.; Majewski, R.L.; Valdovinos, H.F.; Jiang, D.; Liu, G.; McNeel, D.G.; Nickles, R.J.; Cai, W. ImmunoPET Imaging of CTLA-4 Expression in Mouse Models of Non-small Cell Lung Cancer. Mol. Pharm. 2017, 14, 1782–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.; Laureano, Á.M.; Silla, L.M.D.R.; Lee, D.A. Natural killer cell adoptive immunotherapy: Coming of age. Clin. Immunol. 2017, 177, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Stringaris, K.; Davidson-Moncada, J.K.; Reger, R.; Adler, S.S.; Dunbar, C.; Choyke, P.L.; Childs, R.W. In Vivo Tracking of Adoptively Transferred Natural Killer Cells in Rhesus Macaques Using 89Zirconium-Oxine Cell Labeling and PET Imaging. Clin. Cancer Res. 2020, 26, 2573–2581. [Google Scholar] [CrossRef]
- Shaffer, T.M.; Aalipour, A.; Schürch, C.M.; Gambhir, S.S. PET Imaging of the Natural Killer Cell Activation Receptor NKp30. J. Nucl. Med. 2020, 61, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.R.; Soria, B.; Capilla-Gonzalez, V. Therapeutic Potential of Mesenchymal Stem Cells for Cancer Therapy. Front. Bioeng. Biotechnol. 2020, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Belderbos, S.; González-Gómez, M.A.; Cleeren, F.; Wouters, J.; Piñeiro, Y.; Deroose, C.M.; Coosemans, A.; Gsell, W.; Bormans, G.; Rivas, J.; et al. Simultaneous in vivo PET/MRI using fluorine-18 labeled Fe3O4@Al(OH)3 nanoparticles: Comparison of nanoparticle and nanoparticle-labeled stem cell distribution. EJNMMI Res. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Keu, K.V.; Witney, T.H.; Yaghoubi, S.; Rosenberg, J.; Kurien, A.; Magnusson, R.; Williams, J.; Habte, F.; Wagner, J.R.; Forman, S.; et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci. Transl. Med. 2017, 9, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpe, A.; Lang, C.; Lim, L.; Man, F.; Kurtys, E.; Ashmore-Harris, C.; Johnson, P.; Skourti, E.; de Rosales, R.T.M.; Fruhwirth, G.O. Spatiotemporal PET Imaging Reveals Differences in CAR-T Tumor Retention in Triple-Negative Breast Cancer Models. Mol. Ther. 2020, 28, 2271–2285. [Google Scholar] [CrossRef]
- Minn, I.; Huss, D.J.; Ahn, H.H.; Chinn, T.M.; Park, A.; Jones, J.; Brummet, M.; Rowe, S.P.; Sysa-Shah, P.; Du, Y.; et al. Imaging CAR T cell therapy with PSMA-targeted positron emission tomography. Sci. Adv. 2019, 5, eaaw5096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, C.; Feldmann, A.; Koristka, S.; Schäfer, M.; Bergmann, R.; Mitwasi, N.; Berndt, N.; Bachmann, D.; Kegler, A.; Schmitz, M.; et al. A theranostic PSMA ligand for PET imaging and retargeting of T cells expressing the universal chimeric antigen receptor UniCAR. Oncoimmunology 2019, 8, 1659095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Singh, S.K.; Rana, B.; Rana, A. Tumor-infiltrating CD8+ T cell antitumor efficacy and exhaustion: Molecular insights. Drug Discov. Today 2021. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Gooden, M.J.M.; De Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Tavaré, R.; McCracken, M.N.; Zettlitz, K.A.; Knowles, S.M.; Salazar, F.B.; Olafsen, T.; Witte, O.N.; Wu, A.M. Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.K.; Fröhlich, C.; Christensen, C.; Melander, M.C.; Poulsen, T.T.; Galler, G.R.; Lantto, J.; Horak, I.D.; Kragh, M.; Nielsen, C.H.; et al. CD4+ and CD8a+ PET imaging predicts response to novel PD-1 checkpoint inhibitor: Studies of Sym021 in syngeneic mouse cancer models. Theranostics 2019, 9, 8221–8238. [Google Scholar] [CrossRef]
- Griessinger, C.M.; Olafsen, T.; Mascioni, A.; Jiang, Z.K.; Zamilpa, C.; Jia, F.; Torgov, M.; Romero, J.M.; Marchioni, F.; Satpayev, D.; et al. The PET-Tracer 89Zr-Df-IAB22M2C Enables Monitoring of Intratumoral CD8 T-cell Infiltrates in Tumor-Bearing Humanized Mice after T-cell Bispecific Antibody Treatment. Cancer Res. 2020, 80, 2903–2913. [Google Scholar] [CrossRef]
- Kristensen, L.K.; Christensen, C.; Alfsen, M.Z.; Cold, S.; Nielsen, C.H.; Kjaer, A. Monitoring CD8a+ T Cell Responses to Radiotherapy and CTLA-4 Blockade Using [64Cu]NOTA-CD8a PET Imaging. Mol. Imaging Biol. 2020, 22, 1021–1030. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.W.; Tavaré, R.; Mahakian, L.M.; Silvestrini, M.T.; Tam, S.; Ingham, E.S.; Salazar, F.B.; Borowsky, A.D.; Wu, A.M.; Ferrara, K.W. CD8+ T-Cell Density Imaging with 64Cu-Labeled Cys-Diabody Informs Immunotherapy Protocols. Clin. Cancer Res. 2018, 24, 4976–4987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashidian, M.; Ingram, J.R.; Dougan, M.; Dongre, A.; Whang, K.A.; LeGall, C.; Cragnolini, J.J.; Bierie, B.; Gostissa, M.; Gorman, J.; et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J. Exp. Med. 2017, 214, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; LaFleur, M.W.; Verschoor, V.L.; Dongre, A.; Zhang, Y.; Nguyen, T.H.; Kolifrath, S.; Aref, A.R.; Lau, C.J.; Paweletz, C.P.; et al. Immuno-PET identifies the myeloid compartment as a key contributor to the outcome of the antitumor response under PD-1 blockade. Proc. Natl. Acad. Sci. USA 2019, 116, 16971–16980. [Google Scholar] [CrossRef] [Green Version]
- Namavari, M.; Chang, Y.F.; Kusler, B.; Yaghoubi, S.; Mitchell, B.S.; Gambhir, S.S. Synthesis of 2′-Deoxy-2′-[18F]Fluoro-9-β-D-Arabinofuranosylguanine: A Novel Agent for Imaging T-Cell Activation with PET. Mol. Imaging Biol. 2011, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Levi, J.; Lam, T.; Goth, S.R.; Yaghoubi, S.; Bates, J.; Ren, G.; Jivan, S.; Huynh, T.L.; Blecha, J.E.; Khattri, R.; et al. Imaging of Activated T Cells as an Early Predictor of Immune Response to Anti-PD-1 Therapy. Cancer Res. 2019, 79, 3455–3465. [Google Scholar] [CrossRef] [Green Version]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nat. Cell Biol. 1999, 397, 263–266. [Google Scholar] [CrossRef]
- Xiao, Z.; Mayer, A.T.; Nobashi, T.W.; Gambhir, S.S. ICOS Is an Indicator of T-cell–Mediated Response to Cancer Immunotherapy. Cancer Res. 2020, 80, 3023–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opitz, C.A.; Patterson, L.F.S.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Li, R.; Ng, T.S.C.; Courties, G.; Rodell, C.B.; Prytyskach, M.; Kohler, R.H.; Pittet, M.J.; Nahrendorf, M.; Weissleder, R.; et al. Quantitative Imaging of Tumor-Associated Macrophages and Their Response to Therapy Using 64Cu-Labeled Macrin. ACS Nano 2018, 12, 12015–12029. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [Green Version]
- Nigam, S.; McCarl, L.; Kumar, R.; Edinger, R.S.; Kurland, B.F.; Anderson, C.J.; Panigrahy, A.; Kohanbash, G.; Edwards, W.B. Preclinical ImmunoPET Imaging of Glioblastoma-Infiltrating Myeloid Cells Using Zirconium-89 Labeled Anti-CD11b Antibody. Mol. Imaging Biol. 2020, 22, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.A.; Kossatz, S.; Carter, L.M.; Pirovano, G.; Brand, C.; Guru, N.; Pérez-Medina, C.; Lewis, J.S.; Mulder, W.J.M.; Reiner, T. An 89Zr-HDL PET Tracer Monitors Response to a CSF1R Inhibitor. J. Nucl. Med. 2020, 61, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Goggi, J.L.; Hartimath, S.V.; Hwang, Y.; Tan, Y.X.; Khanapur, S.; Ramasamy, B.; Jiang, L.; Yong, F.F.; Cheng, P.; Tan, P.W.; et al. Examining Immunotherapy Response Using Multiple Radiotracers. Mol. Imaging Biol. 2020, 22, 993–1002. [Google Scholar] [CrossRef]
- Larimer, B.M.; Wehrenberg-Klee, E.; Caraballo, A.; Mahmood, U. Quantitative CD3 PET Imaging Predicts Tumor Growth Response to Anti-CTLA-4 Therapy. J. Nucl. Med. 2016, 57, 1607–1611. [Google Scholar] [CrossRef] [Green Version]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-Cell Engaging Antibodies for Cancer Therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef] [Green Version]
- Kujawski, M.; Li, L.; Bhattacharya, S.; Wong, P.; Lee, W.-H.; Williams, L.; Li, H.; Chea, J.; Poku, K.; Bowles, N.; et al. Generation of dual specific bivalent BiTEs (dbBIspecific T-cell engaging antibodies) for cellular immunotherapy. BMC Cancer 2019, 19, 882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, T.R.; Castro, I. Interleukin-2 Receptor Signaling: At the Interface between Tolerance and Immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gialleonardo, V.; Signore, A.; Glaudemans, A.W.J.M.; Dierckx, R.A.J.O.; De Vries, E.F.J. N-(4-18F-Fluorobenzoyl)Interleukin-2 for PET of Human-Activated T Lymphocytes. J. Nucl. Med. 2012, 53, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Hartimath, S.V.; Draghiciu, O.; Van De Wall, S.; Manuelli, V.; Dierckx, R.A.J.O.; Nijman, H.W.; Daemen, T.; De Vries, E.F.J. Noninvasive monitoring of cancer therapy induced activated T cells using [18F]FB-IL-2 PET imaging. Oncoimmunology 2017, 6, e1248014. [Google Scholar] [CrossRef] [Green Version]
- Van Der Veen, E.L.; Antunes, I.F.; Maarsingh, P.; Hessels-Scheper, J.; Zijlma, R.; Boersma, H.H.; Jorritsma-Smit, A.; Hospers, G.A.P.; De Vries, E.G.E.; Hooge, M.N.L.-D.; et al. Clinical-grade N-(4-[18F]fluorobenzoyl)-interleukin-2 for PET imaging of activated T-cells in humans. EJNMMI Radiopharm. Chem. 2019, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, M. Tapping CD4 T Cells for Cancer Immunotherapy: The Choice of Personalized Genomics. J. Immunol. 2015, 194, 2049–2056. [Google Scholar] [CrossRef] [Green Version]
- Tavaré, R.; McCracken, M.N.; Zettlitz, K.A.; Salazar, F.B.; Olafsen, T.; Witte, O.N.; Wu, A.M. Immuno-PET of Murine T Cell Reconstitution Postadoptive Stem Cell Transplantation Using Anti-CD4 and Anti-CD8 Cys-Diabodies. J. Nucl. Med. 2015, 56, 1258–1264. [Google Scholar] [CrossRef] [Green Version]
- Freise, A.C.; Zettlitz, K.A.; Salazar, F.B.; Lu, X.; Tavaré, R.; Wu, A.M. ImmunoPET Imaging of Murine CD4+ T Cells Using Anti-CD4 Cys-Diabody: Effects of Protein Dose on T Cell Function and Imaging. Mol. Imaging Biol. 2017, 19, 599–609. [Google Scholar] [CrossRef]
- Kaira, K.; Kuji, I.; Kagamu, H. Value of 18F-FDG-PET to predict PD-L1 expression and outcomes of PD-1 inhibition therapy in human cancers. Cancer Imaging 2021, 21, 762. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Huff, D.T.; Jeraj, R.; Albertini, M.R. FDG PET/CT for Assessment of Immune Therapy: Opportunities and Understanding Pitfalls. Semin. Nucl. Med. 2020, 50, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Ayati, N.; Sadeghi, R.; Kiamanesh, Z.; Lee, S.T.; Zakavi, S.R.; Scott, A.M. The value of 18F-FDG PET/CT for predicting or monitoring immunotherapy response in patients with metastatic melanoma: A systematic review and meta-analysis. Eur. J. Nucl. Med. Mol. Imaging 2020, 48, 428–448. [Google Scholar] [CrossRef] [PubMed]
- Iravani, A.; Hicks, R.J. Imaging the Cancer Immune Environment and Its Response to Pharmacologic Intervention, Part 1: The Role of 18F-FDG PET/CT. J. Nucl. Med. 2020, 61, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Beer, L.; Hochmair, M.; Haug, A.R.; Schwabel, B.; Kifjak, D.; Wadsak, W.; Fuereder, T.; Fabikan, H.; Fazekas, A.; Schwab, S.; et al. Comparison of RECIST, iRECIST, and PERCIST for the Evaluation of Response to PD-1/PD-L1 Blockade Therapy in Patients With Non–Small Cell Lung Cancer. Clin. Nucl. Med. 2019, 44, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Mulkey, F.; Theoret, M.R.; Keegan, P.; Pazdur, R.; Sridhara, R. Comparison of iRECIST versus RECIST V.1.1 in patients treated with an anti-PD-1 or PD-L1 antibody: Pooled FDA analysis. J. Immunother. Cancer 2020, 8, e000146. [Google Scholar] [CrossRef] [Green Version]
- Tazdait, M.; Mezquita, L.; Lahmar, J.; Ferrara, R.; Bidault, F.; Ammari, S.; Balleyguier, C.; Planchard, D.; Gazzah, A.; Soria, J.C.; et al. Patterns of responses in metastatic NSCLC during PD-1 or PDL-1 inhibitor therapy: Comparison of RECIST 1.1, irRECIST and iRECIST criteria. Eur. J. Cancer 2018, 88, 38–47. [Google Scholar] [CrossRef]
- Cheson, B.D.; Ansell, S.; Schwartz, L.; Gordon, L.I.; Advani, R.; Jacene, H.A.; Hoos, A.; Barrington, S.F.; Armand, P. Refinement of the Lugano Classification lymphoma response criteria in the era of immunomodulatory therapy. Blood 2016, 128, 2489–2496. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Teng, R.; Schöder, H.; Humm, J.L.; Ni, A.; Michaud, L.; Nakajima, R.; Yamashita, R.; Wolchok, J.D.; Weber, W.A. 18F-FDG PET/CT for Monitoring of Ipilimumab Therapy in Patients with Metastatic Melanoma. J. Nucl. Med. 2019, 60, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Anwar, H.; Sachpekidis, C.; Winkler, J.; Kopp-Schneider, A.; Haberkorn, U.; Hassel, J.C.; Dimitrakopoulou-Strauss, A. Absolute number of new lesions on 18F-FDG PET/CT is more predictive of clinical response than SUV changes in metastatic melanoma patients receiving ipilimumab. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 376–383. [Google Scholar] [CrossRef]
- Hodi, F.S.; Ballinger, M.; Lyons, B.; Soria, J.C.; Nishino, M.; Tabernero, J.; Powles, T.; Smith, D.; Hoos, A.; McKenna, C.; et al. Immune-Modified Response Evaluation Criteria In Solid Tumors (imRECIST): Refining Guidelines to Assess the Clinical Benefit of Cancer Immunotherapy. J. Clin. Oncol. 2018, 36, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the Evaluation of Immune Therapy Activity in Solid Tumors: Immune-Related Response Criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.B.; Queiroz, M.A.; Barbosa, F.G.; Nunes, R.F.; Zaniboni, E.C.; Ruiz, M.M.; Jardim, D.; Marin, J.F.G.; Cerri, G.G.; Buchpiguel, C.A. Reassessing Patterns of Response to Immunotherapy with PET: From Morphology to Metabolism. Radiographics 2021, 41, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, K.W.; Pyo, J.; Suh, C.H.; Yoon, S.; Hatabu, H.; Nishino, M. Incidence of Pseudoprogression during Immune Checkpoint Inhibitor Therapy for Solid Tumors: A Systematic Review and Meta-Analysis. Radiology 2020, 297, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Seban, R.D.; Nemer, J.S.; Marabelle, A.; Yeh, R.; Deutsch, E.; Ammari, S.; Moya-Plana, A.; Mokrane, F.Z.; Gartrell, R.D.; Finkel, G.; et al. Prognostic and theranostic 18F-FDG PET biomarkers for anti-PD1 immunotherapy in metastatic melanoma: Association with outcome and transcriptomics. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Laudicella, R.; Comelli, A.; Stefano, A.; Szostek, M.; Crocè, L.; Vento, A.; Spataro, A.; Comis, A.D.; La Torre, F.; Gaeta, M.; et al. Artificial Neural Networks in Cardiovascular Diseases and its Potential for Clinical Application in Molecular Imaging. Curr. Radiopharm. 2020, 13. [Google Scholar] [CrossRef]
- Lambin, P.; Leijenaar, R.T.H.; Deist, T.M.; Peerlings, J.; De Jong, E.E.C.; Van Timmeren, J.; Sanduleanu, S.; Larue, R.T.H.M.; Even, A.J.G.; Jochems, A.; et al. Radiomics: The Bridge between Medical Imaging and Personalized Medicine. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/?term=Lambin++Radiomics%3A+the+bridge+between+medical+imaging+and+personalized+medicine (accessed on 7 February 2020).
- Mayerhoefer, M.E.; Materka, A.; Langs, G.; Häggström, I.; Szczypiński, P.; Gibbs, P.; Cook, G. Introduction to Radiomics. J. Nucl. Med. 2020, 61, 488–495. [Google Scholar] [CrossRef]
- Lee, D.Y.; Kim, Y.I. Prognostic Value of Maximum Standardized Uptake Value in 68Ga-Somatostatin Receptor Positron Emission Tomography for Neuroendocrine Tumors: A Systematic Review and Meta-analysis. Clin. Nucl. Med. 2019, 44, 777–783. [Google Scholar] [CrossRef]
- Valentinuzzi, D.; Vrankar, M.; Boc, N.; Ahac, V.; Zupancic, Z.; Unk, M.; Skalic, K.; Zagar, I.; Studen, A.; Simoncic, U.; et al. [18F]FDG PET immunotherapy radiomics signature (iRADIOMICS) predicts response of non-small-cell lung cancer patients treated with pembrolizumab. Radiol. Oncol. 2020, 54, 285–294. [Google Scholar] [CrossRef]
- Polverari, G.; Ceci, F.; Bertaglia, V.; Reale, M.L.; Rampado, O.; Gallio, E.; Passera, R.; Liberini, V.; Scapoli, P.; Arena, V.; et al. 18F-FDG Pet Parameters and Radiomics Features Analysis in Advanced Nsclc Treated with Immunotherapy as Predictors of Therapy Response and Survival. Cancers 2020, 12, 1163. [Google Scholar] [CrossRef]
- Mu, W.; Tunali, I.; Gray, J.E.; Qi, J.; Schabath, M.B.; Gillies, R.J. Radiomics of 18F-FDG PET/CT images predicts clinical benefit of advanced NSCLC patients to checkpoint blockade immunotherapy. Eur. J. Nucl. Med. Mol. Imaging 2019, 47, 1168–1182. [Google Scholar] [CrossRef]
- Park, S.; Ha, S.; Lee, S.-H.; Paeng, J.C.; Keam, B.; Kim, T.M.; Kim, D.-W.; Heo, D.S. Intratumoral heterogeneity characterized by pretreatment PET in non-small cell lung cancer patients predicts progression-free survival on EGFR tyrosine kinase inhibitor. PLoS ONE 2018, 13, e0189766. [Google Scholar] [CrossRef]
- Parvez, A.; Tau, N.; Hussey, D.; Maganti, M.; Metser, U. 18F-FDG PET/CT metabolic tumor parameters and radiomics features in aggressive non-Hodgkin’s lymphoma as predictors of treatment outcome and survival. Ann. Nucl. Med. 2018, 32, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Aide, N.; Fruchart, C.; Nganoa, C.; Gac, A.C.; Lasnon, C. Baseline 18F-FDG PET radiomic features as predictors of 2-year event-free survival in diffuse large B cell lymphomas treated with immunochemotherapy. Eur. Radiol. 2020, 30, 4623–4632. [Google Scholar] [CrossRef] [PubMed]
- Prigent, K.; Lasnon, C.; Ezine, E.; Janson, M.; Coudrais, N.; Joly, E.; Césaire, L.; Stefan, A.; Depontville, M.; Aide, N. Assessing immune organs on 18F-FDG PET/CT imaging for therapy monitoring of immune checkpoint inhibitors: Inter-observer variability, prognostic value and evolution during the treatment course of melanoma patients. Eur. J. Nucl. Med. Mol. Imaging 2021. [Google Scholar] [CrossRef]
- Basler, L.; Gabryś, H.S.; Hogan, S.A.; Pavic, M.; Bogowicz, M.; Vuong, D.; Tanadini-Lang, S.; Förster, R.; Kudura, K.; Huellner, M.W.; et al. Radiomics, Tumor Volume, and Blood Biomarkers for Early Prediction of Pseudoprogression in Patients with Metastatic Melanoma Treated with Immune Checkpoint Inhibition. Clin. Cancer Res. 2020, 26, 4414–4425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyasu, S.; Nishio, M.; Isoda, H.; Nakamoto, Y.; Togashi, K. Usefulness of gradient tree boosting for predicting histological subtype and EGFR mutation status of non-small cell lung cancer on 18F FDG-PET/CT. Ann. Nucl. Med. 2020, 34, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, X.; Zhao, Y.; Zhang, J.; Zhang, Z.; Wang, J.; Wang, Y.; Dai, M.; Han, J. Value of pre-therapy 18F-FDG PET/CT radiomics in predicting EGFR mutation status in patients with non-small cell lung cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yin, G.; Zhang, Y.; Dai, D.; Liu, J.; Chen, P.; Zhu, L.; Ma, W.; Xu, W. Predictive Power of a Radiomic Signature Based on 18F-FDG PET/CT Images for EGFR Mutational Status in NSCLC. Front. Oncol. 2019, 9, 1062. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhang, Y.; Xu, J.; Ji, M.; Guo, Y.; Guo, Y.; Xiao, J.; Yao, X.; Shi, H.; Zeng, M. Assessing EGFR gene mutation status in non-small cell lung cancer with imaging features from PET/CT. Nucl. Med. Commun. 2019, 40, 842–849. [Google Scholar] [CrossRef]
- Jiang, M.; Sun, D.; Guo, Y.; Guo, Y.; Xiao, J.; Wang, L.; Yao, X. Assessing PD-L1 Expression Level by Radiomic Features From PET/CT in Nonsmall Cell Lung Cancer Patients: An Initial Result. Acad. Radiol. 2020, 27, 171–179. [Google Scholar] [CrossRef]
- Park, C.; Na, K.J.; Choi, H.; Ock, C.Y.; Ha, S.; Kim, M.; Park, S.; Keam, B.; Kim, T.M.; Paeng, J.C.; et al. Tumor immune profiles noninvasively estimated by FDG PET with deep learning correlate with immunotherapy response in lung adenocarcinoma. Theranostics 2020, 10, 10838–10848. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of cancer-associated immune cells in human solid tumors. Elife 2018, 7, e36967. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, V.; Cesano, A.; Marincola, F.; Kather, J.N. Next Generation Imaging Techniques to Define Immune Topographies in Solid Tumors. Front. Immunol. 2021, 11, 3519. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Radionuclide | Half-life | Type of Emission | Energy of Emission (keV) | Particle Maximum Range in Water |
|---|---|---|---|---|
| [11C] (carbon) | 20.4 min | β+ | 970 | 3.67 mm |
| [68Ga] (gallium) | 67.7 min | β+ | 1900 | 9.06 mm |
| [18F] (fluorine) | 109.7 min | β+ | 640 | 2.16 mm |
| [64Cu] (copper) | 12.7 h |
β+ (19%)
β− (38%) γ (43%) |
657
141 511 and 1346 | ≃ 2 mm |
| [89Zr] (zirconium) | 3.27 d | β+ | 909 | ≃ 3 mm |
| Targeting Molecule | Agent | Molecule Type | Tumor Model | Stage | Reference |
|---|---|---|---|---|---|
| PD-L1 | [18F]BMS-986192 | adnectin | NSCLC patients | clinical | [60] |
| [89Zr]-labeled atezolizumab | antibody IgG1 | metastatic bladder cancer, NSCLC, or triple-negative breast cancer | clinical | [61] | |
| PD-1 | [89Zr]nivolumab | antibody IgG4 | NSCLC patients | clinical | [60] |
| CD8+ | [89Zr]Df-IAB22M2C | minibody | melanoma, lung cancer, and hepatocellular carcinoma | clinical | [62] |
| Radiological Response Criteria | irRC | imRECIST | |
| Complete response (CR) | Disappearance of all target lesions. Determined by two observations not less than 4 weeks apart. | Disappearance of all target and non-target lesions, without any new lesions. Any pathological lymph nodes must have reduction in short axis to <10 mm. Determined by two observations not less than 4 weeks apart. | |
| Partial response (PR) | Sum of product of all lesions decreased by >50% for at least 4 weeks; no new lesions; no progression of any lesions. | At least a 30% decrease of the sum of maximum diameters of target lesions; no new lesions; no progression of disease. | |
| Stable disease (SD) | Sum of product of all lesions decreased by <50% or increased by <25% in the size of one or more lesions. | Does not meet the criteria for CR, PR or PD, taking as reference the smallest sum of maximum diameters of target lesions. | |
| Progressive disease (PD) | A single lesion increased by >25% (over the smallest measurement achieved for the single lesion) or the appearance of new lesions, that has to be confirmed in 2 consecutive observations at least 4 weeks apart. | Sum of the maximum diameter of lesions increased by >20% over the smallest achieved sum of maximum diameter. The appearance of new lesions and/or progression of non-target lesions are considered iUPD and must be confirmed 4–8 weeks later as iCPD. Progression is not confirmed in case of shrinkage of these lesions at 4–8 weeks and evaluation must be reset. | |
| Functional response criteria | imPERCIST | PERCIMT | LYRIC |
| Complete metabolic response (CMR) | Complete resolution of [18F]FDG uptake within all lesions, to a level of less than or equal to that of the mean liver activity and indistinguishable from the background (blood pool uptake). | Complete resolution of [18F]FDG uptake within all lesions, to a level of less than or equal to that of the mean liver activity and indistinguishable from the background (blood pool uptake). | PET Deauville score * = 1, 2, or 3, with or without a residual mass on CT, target nodes/nodal masses must regress to <1.5 cm in longest diameter. |
| Partial metabolic response (PMR) | Reduction of at least 30% in the sum of SULpeak of all target lesions detected at baseline and an absolute drop of 0.8 SULpeak units. | Reduction of at least 30% in the sum of SULpeak of all target lesions detected at baseline and an absolute drop of 0.8 SULpeak units. | PET Deauville score * = 4 or 5 with reduced uptake compared with baseline and residual mass(es) of any size or on CT > 50% decrease in SPD of up to 6 target measurable nodes and extranodal sites. |
| Stable metabolic disease (SMD) | Does not meet the criteria for CR, PR or PD. | Does not meet the criteria for CR, PR or PD. | Does not meet the criteria for CR, PR or PD. |
| Progressive metabolic disease (PMD) | Increased of at least 30% in the sum of SULpeak of all target lesions detected at baseline or new FDG-avid lesions are considered UPMD and must be confirmed 4–8 weeks later as CPMD. Progression is not confirmed in case of PMR or SMD at 4–8 weeks and evaluation must be reset. | Progressive disease if: ≥4 new lesions (<1 cm in functional diameter); ≥3 new lesions (>1 cm in functional diameter); ≥2 new lesions (>1.5 cm in functional diameter). | PET Deauville score * = 4 or 5 with an increase in intensity of uptake from baseline and/or new FDG-avid foci consistent with lymphoma at interim or end of-treatment assessment or on CT > 50% increase in SPD of target measurable nodes and extranodal sites. Immune response exception (IR): - IR1 > 50% increase in SPD in first 12 weeks; - IR2 < 50% increase in SPD with new lesion(s) or >50% increase in PPD of a lesion or set of lesions at any time during treatment; - IR3 = increase in FDG uptake without a concomitant increase in lesion size meeting criteria for PD. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liberini, V.; Laudicella, R.; Capozza, M.; Huellner, M.W.; Burger, I.A.; Baldari, S.; Terreno, E.; Deandreis, D. The Future of Cancer Diagnosis, Treatment and Surveillance: A Systemic Review on Immunotherapy and Immuno-PET Radiotracers. Molecules 2021, 26, 2201. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082201
Liberini V, Laudicella R, Capozza M, Huellner MW, Burger IA, Baldari S, Terreno E, Deandreis D. The Future of Cancer Diagnosis, Treatment and Surveillance: A Systemic Review on Immunotherapy and Immuno-PET Radiotracers. Molecules. 2021; 26(8):2201. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082201
Chicago/Turabian StyleLiberini, Virginia, Riccardo Laudicella, Martina Capozza, Martin W. Huellner, Irene A. Burger, Sergio Baldari, Enzo Terreno, and Désirée Deandreis. 2021. "The Future of Cancer Diagnosis, Treatment and Surveillance: A Systemic Review on Immunotherapy and Immuno-PET Radiotracers" Molecules 26, no. 8: 2201. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26082201