
Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis †




1
Department of Chemistry and Biochemistry, University of Wisconsin Milwaukee, Milwaukee, WI 53211, USA
2
RTI International, Center for Drug Discovery, Research Triangle Park, Durham, NC 27709, USA
*
Author to whom correspondence should be addressed.
†
This review is dedicated to Professor Samuel J. Danishefsky for his outstanding contributions to total synthesis and organic chemistry as a whole.
Molecules 2021, 26(11), 3459; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113459
Submission received: 29 April 2021
/
Revised: 24 May 2021
/
Accepted: 27 May 2021
/
Published: 7 June 2021
(This article belongs to the Special Issue Alkaloids in Future Drug Discovery)
Abstract
:Bisindoles are structurally complex dimers and are intriguing targets for partial and total synthesis. They exhibit stronger biological activity than their corresponding monomeric units. Alkaloids, including those containing C-19 methyl-substitution in their monomeric units, their synthetic derivatives, and their mismatched pairs can be attractive targets for synthesis and may unlock better drug targets. We herein discuss the isolation of bisindoles from various Alstonia species, their bioactivity, putative biosynthesis, and synthesis. The total synthesis of macralstonidine, macralstonine, O-acetylmacralstonine, and dispegatrine, as well as the partial synthesis of alstonisidine, villalstonine, and macrocarpamine are also discussed in this review. The completion of the total synthesis of pleiocarpamine by Sato et al. completes the formal synthesis of the latter two bisindoles.
1. Introduction
Nature has been a substantial and sustainable pool of biologically active compounds. Since ancient times natural product extracts (in crude form) have been used in traditional and folk medicines in many countries. In modern times pure (isolated) natural products and their derivatives play an important role in drug discovery, as indicated by their prevalence in approved drugs for clinical use. Out of the 1881 newly FDA-approved drugs over the last four decades (1 January 1981 to 30 September 2019), a significant portion comprising 506 (26.9%) were either natural products or derived from or inspired by natural products [1]. It is expected that the advent of modern and innovative technologies such as computational software, cheminformatics, artificial intelligence, automation, and quantum computing will further boost natural product-based drug discovery. A synergy among these technological milestones would accelerate hit to lead to clinic pathways of drug discovery, and natural products are expected to remain an important source [2]. Moreover, pharmacophores and their unique stereochemical interactions with natural products may stimulate more demanding targets such as protein–protein interactions in the near future and open up a new avenue in modern drug discovery [3]. The majority of biologically active natural products are produced in plants, known traditionally as medicinal plants.
Alstonia, a major genus in the Apocynaceae family of plants, has nearly 155 species and is found all over the world [4]. Robert Brown named it in 1811 in honor of Charles Alston (1685–1760), an eminent botanist at the University of Edinburgh [4]. The Alstonia genus’ trees and shrubs are prevalent in the tropical and subtropical parts of Africa, Asia, and Australia [5]. They contribute significant pharmacological activity, including anticancer, antileishmanial, antimalarial, antitussive, antiviral, antiarthritic, and antibacterial activities [4,5,6,7,8,9]. Several parts of the plants including bark, twigs, and leaves have been traditionally used to treat diseases. Their useful biological activity has aroused profound interest, as indicated by the increasing number of publications and patents each year. To date, more than 300 patents have been filed under the Alstonia genus with various formulations [4]. Notably, 157 different monoterpenoid indole alkaloids have been isolated from this genus between 2010 to 2020 [10].
Alkaloids, the most important class of natural products with structural diversity and significant pharmacological effects, are mainly found in higher plants such as the Apocynaceae, Ranunculaceae, Papaveraceae, and Leguminosae families [11]. These natural products, along with flavonoids, fatty acids, etc., are the major classes of secondary metabolites that are believed to be parts of the plants’ defense mechanism. To date, many monoterpenoid indole and bisindole alkaloids have been found in the Alstonia genus [4]. Modern clinical application of many of these alkaloids are similar to their traditional or folklore applications; for example, cocaine and morphine were used as anesthetics while caffeine and nicotine were used as stimulants [12]. Recently, Fielding et al. illustrated that several anti-coronavirus alkaloids showed potential therapeutic value against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) in their in silico studies [9].
The indole motif is present in many naturally occurring and biologically active compounds. Some of them have been used in the clinic. In addition, there are numerous examples of synthetic compounds with useful medicinal properties that bear the indole moiety. Consequently, the indole scaffold is one of the few “privileged structures” in modern medicinal chemistry and drug discovery [13,14]. The prevalence of bioactivity of indole-containing molecules may be attributed to their similarity to the essential amino acid tryptophan, as well as important biomolecules such as tryptamine and serotonin. Many plant extracts, which likely include alkaloids, have been used from time immemorial in folk medicines for fever, general weakness, dysentery, pain, liver diseases, gastrointestinal diseases, and cancer [15]. Currently, there are many indole alkaloid-based marketed drugs such as sumatriptan for the treatment of migraine; vincristine and vinblastine for the treatment of various cancers, including leukemia and lung cancer; as well as reserpine for the treatment of hypertension and to decrease severe agitation in patients with mental disorders [16].
Bisindole alkaloids are naturally occurring alkaloids containing two indole nuclei. These bisindoles are the products of late-stage biosynthetic processes in higher plants by combining two monomeric units. Depending on the monomeric units involved, bisindoles can be a homo- or heterodimer. As a result, bisindole alkaloids comprise much higher structural complexity than both of the monomeric units that comprise them. As mentioned, bioactive bisindoles are usually more potent than the monomeric units that comprise them, although the mechanism of action is not known [6]. More structural diversity can be achieved via bisindole-based drug discovery by changing both monomeric units to furnish unnatural and pseudo-natural alkaloids. As such, bisindole alkaloids offer a large pool of natural, semi-natural, and chimeric drug candidates that have greater drug-like characteristics. As a result, due to their important biological applications and complex structural features, bisindole alkaloids have engendered the profound interest of synthetic organic and medicinal chemists, computational chemists, and chemical biologists. Compared to the previously published review [6] from this group, the most recent and updated reports on the isolation, important bioactivity, biosynthetic studies, and synthesis of bisindole alkaloids isolated from Alstonia containing either sarpagine, macroline, ajmaline, or pleiocarpamine units form the basis of this review. Moreover, the recent total synthesis of rac-pleiocarpamine (a monomeric unit present in several bisindoles) by Sato et al. [17], which resulted in the formal total synthesis of villalstonine and macrocarpamine, the bioactive C-19 methyl-substituted sarpagine/macroline/ajmaline indole alkaloids, their unnatural enantiomers, and mismatched pairs to form potential bisindoles alkaloids, is also discussed in this review.
2. Occurrence and Isolation of Bisindoles from Alstonia Species
A list of representative families to which the monomeric indole-based units belong, which comprise bisindole alkaloids from Alstonia, is shown in Table 1. In some cases, a monomeric unit itself occurs as an alkaloid, i.e., has been isolated separately and is known by its trivial or given name. Bisindole alkaloids that contain the illustrated monomeric bases are listed in Table 2. Among various species of the Alstonia genus, A. macrophylla and A. angustifolia are the two major sources of bisindole alkaloids discussed herein (vide infra, Table 3). (+)-Alstomacroline 1, a bisindole alkaloid consisting of a macroline and an ajmaline unit, was isolated from the bark of A. macrophylla [18] and the leaves, stem-bark, and root-bark extracts of A. scholaris, A. glaucescens, and A. macrophylla [19,20]. (+)-Alstomacrophylline 2 (macroline–macroline-type) was isolated from the bark of A. macrophylla [18] and the leaves, stem-bark, and root-bark extracts of A. scholaris, A. glaucescens, and A. macrophylla [20]. (-) Alstonisidine 3, which contains a quebrachidine 4 and a macroline 5 unit, was isolated from the bark of A. muelleriana [21,22]. The structure of (-)-alstonisidine 3 was confirmed by X-ray crystallographic data [23]. Yeap et al. recently isolated seven novel bisindoles from the methanol extract of the stem-bark of Malayan A. penangiana [5]. This includes (-)-angustilongine E 6, (-)-angustilongine F 7, (+)-angustilongine G 8, (+)-angustilongine H 9, (+)-angustilongine J 10, (+)-angustilongine K 11, and (-)-angustilongine L 12 (macroline–pleiocarpamine type). (+)-Angustilongine K 11 was converted into (+)-di-O-acetylangustilongine K 13 by stirring it with 10 equivalents of pyridine and 15 equivalents of acetic anhydride for 6 h at room temperature in 95% yield [5]. Among those, angustilongine G 8 and angustilongine H 9 are C-19 methyl substituted [24] bisindoles. The structures of the angustilongines were confirmed by various spectroscopic data, including 1H NMR, 13C NMR, 2D NMR, IR, and HRMS by Yeap et al. [5]. Angustilongine E 6, angustilongine F 7, angustilongine G 8, angustilongine H 9, angustilongine J 10, and angustilongine K 11 are macroline–sarpagine coupled bisindoles. Angustilongine G 8 and angustilongine H 9 differ in stereochemistry only at the C-20 position.
Two macroline units are contained in (-)-lumusidine A 14, (-)-lumusidine B 15, (-)-lumusidine C 16, and (-)-lumusidine D 17 bisindoles. They were isolated from the stem-bark of A. macrophylla and the structures were confirmed via NMR spectroscopy, mass spectrometry, UV spectroscopy, and X-ray crystallography [18]. After isolation, the group of Kam et al. converted oily (-)-lumusidine A 14, (-)-lumusidine B 15, and (-)-lumusidine D 17 into the corresponding crystalline dimethyl diiodide salts (structures not shown) by treatment with an excess of iodomethane for 24 h. The crystalline salts were employed to obtain X-ray crystallographic data to elucidate the exact stereochemical confirmation [18]. (-)-Lumusidine D 17 is also known as thungfaine [25]. (+)-Lumutinine A 18, (-)-lumutinine B 19, (+)-lumutinine C 20, and (+)-lumutinine D 21 are linearly fused bisindoles isolated from the stem-bark of A. macrophylla as a light yellowish oil [26]. (+)-Lumutinine A 18 and (-)-lumutinine B 19 are macroline–macroline-type bisindoles, while (+)-lumutinine C 20, (+)-lumutinine D 21, and (+)-lumutinine E 22 are macroline–sarpagine-type bisindoles. The structures of the lumutinines were elucidated using spectroscopic means including 1D and 2D NMR, IR, as well as mass spectrometric analysis [26]. The structure of (+)-lumutinine D 21 was confirmed by X-ray crystallographic data [27]. (+)-Lumutinine E 22, a macroline–sarpagine-type bisindole, was isolated from the stem-bark of A. angustifolia [28].
(+)-Macralstonidine 23 (macroline–sarpagine-type) was isolated from the bark of A. macrophylla [29,30], as well as from A. somersentenis [29] and A. spectabilis [31]. (+)-Macralstonine 24 was isolated from the leaves, stem-bark, and root-bark extracts of A. scholaris, A. glaucescens, and A. macrophylla extracts [20], A. macrophylla [29,30,32,33,34], A. muelleriana [35], A. angustifolia [36], as well as from A. glabriflora [31]. The structure of (+)-macralstonine 24 was confirmed by various NMR spectroscopy, mass spectrometry, and X-ray crystallography [33]. The (+)-macralstonine 24-related bisindole, (+)-O-acetylmacralstonine 25, was isolated from the leaves, stem-bark, and root-bark extracts of A. scholaris, A. glaucescens, and A. macrophylla [20]. Also, (+)-O-methylmacralstonine 26 was isolated from the leaves, stem-bark, and root-bark of A. scholaris, A. glaucescens, and A. macrophylla extracts [20]. (-)-Anhydromacralstonine 27 was isolated from the stem-bark of A. angustiloba [28] and contains (-)-alstophylline 28 and (+)-macroline 5 as monomeric units. Another (-)-alstophylline 28 and (+)-macroline 5 monomeric bisindole, (+)-Des-N’a-methylanhydromacralstonine 30, was isolated from the bark of A. muelleriana [20,35], the stem-bark of A. angustifolia [33], and A. glabriflora [31]. (+)-Macrocarpamine 31, a hetero-dimeric bisindole containing a (+)-pleiocarpamine 32 and (-)-anhydromacrosalhinemethine 33 monomeric unit was isolated from the leaves, stem-bark, and root-bark of A. scholaris, A. glaucescens, and A. macrophylla [20], as well as the stem-bark of A. angustifolia [28]. 10-Methoxymacrocarpamine 34 and 10-methoxymacrocarpamine N4′-oxide 35 are structurally related bisindoles. These bisindoles were isolated from A. angustifolia leaves [36].
Lim et al. [33] reported the isolation of new macroline–macroline-type bisindoles, (-)-perhentidine A 36, (-)-perhentidine B 37, and (-)-perhentidine C 38 from the ethanolic extract of the stem-bark of Malayan A. macrophylla and A. angustifolia. The structurally related bisindole, (-)-perhentinine 39 (macroline–macroline-type), was isolated from the stem-bark and leaves of A. macrophylla and the leaves of A. angustifolia [18]. The exact structure of (-)-perhentinine 39 was confirmed by X-ray crystallography, converting it into the dimethyl diiodide salt by treating it with an excess of iodomethane. The X-ray crystallographic data of (-)-perhentinine 39 also helped in the structural characterizations of perhentidines A–C (36–38) [33]. Tan et al. isolated three macroline–sarpagine-type bisindoles: (+)-perhentisine A 40, (-)-perhentisine B 41, and (+)-perhentisine C 42 from the stem-bark of A. angustifolia as a light yellow-colored oil together with other bisindoles [28]. The structures of these bisindoles were also elucidated using various NMR and MS techniques [28]. (+)-Villastonine 43, a macroline–pleiocarpamine-type bisindole, was isolated from the stem-bark, root-bark, and leaves of various Alstonia species, including A. spectabilis [29] and A. muelleriana [37] by LeQuesne et al., A. macrophylla [30,32,38], and A. angustifolia [36,39]. Schmid et al. elucidated the structure of (+)-villalstonine 43 by spectroscopic means, accompanied by degradation, and Nordman et al. confirmed the structure by X-ray crystallography [38,40].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 3.
Isolation and plant morphology of bisindole alkaloids from Alstonia species.
| Bisindoles | Types of Monomeric Units Present in Bisindoles | Alstonia Species | Morphology and References |
|---|---|---|---|
| (+)-Alstomacroline 1 | Macroline-sarpagine | A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark, and root-bark [19,20] |
| A. macrophylla | Bark [18] | ||
| (+)-Alstomacrophylline 2 | Macroline-macroline | A. macrophylla | Bark [19] |
| A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark, and root-bark [20] | ||
| (−) Alstonisidine 3 | Ajmaline-macroline | A. muelleriana | Bark [21,22] |
| (−)-Angustilongine E 6 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (−)-Angustilongine F 7 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (+)-Angustilongine G 8 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (+)-Angustilongine H 9 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (+)-Angustilongine J 10 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (+)-Angustilongine K 11 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (−)-Angustilongine L 12 | Macroline-pleiocarpamine | A. penangiana | Stem-bark [5] |
| (−)-Anhydromacralstonine 27 | Macroline-macroline | A. angustifolia | Stem-bark [28] |
| (+)-Des-N’a-Methylanhydromacralstonine 30 | Macroline-macroline | A. muelleriana | Bark [20] |
| A. angustifolia | Stem-bark [33] | ||
| A. muelleriana | Leaves, stem-bark and root-bark [20,35] | ||
| (−)-Lumusidine A 14 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| (−)-Lumusidine B 15 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| (−)-Lumusidine C 16 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| (−)-Lumusidine D 17 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| (+)-Lumutinine A 18 | Macroline-macroline | A. macrophylla | Stem-bark [26] |
| (−)-Lumutinine B 19 | Macroline-macroline | A. macrophylla | Stem-bark [26] |
| (+)-Lumutinine C 20 | Macroline-sarpagine | A. macrophylla | Stem-bark [26] |
| (+)-Lumutinine D 19 | Macroline-sarpagine | A. macrophylla | Stem-bark [26] |
| (+)-Lumutinine E 21 | Macroline-sarpagine | A. angustifolia | Stem-bark [28] |
| (+)-Macralstonidine 23 | Macroline-sarpagine | A. macrophylla | Bark [29,30] |
| A. somersentenis | Bark [29] | ||
| A. spectabilis | Bark [31] | ||
| (+)-Macralstonine 24 | Macroline-macroline | A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark and root-bark [19] |
| A. macrophylla | [29,30,32,33,34] | ||
| A. angustifolia | [36] | ||
| A. muelleriana | [35] | ||
| A. glabriflora Mgf. | [31] | ||
| (+)-O-Acetylmacralstonine 25 | Macroline-macroline | A. scholaris, A. glaucescens, and A. macrophylla extracts | [20] |
| (+)-O-Methylmacralstonine 26 | Macroline-macroline | A. scholaris, A. glaucescens, and A. macrophylla extracts | [20] |
| (+)-Macrocarpamine 31 | Macroline-pleiocarpamine | A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark, and root-bark [20] |
| A. angustifolia | Stem-bark [28] | ||
| 10-Methoxy macrocarpamine 34 | Macroline-pleiocarpamine | A. angustifolia | Leaves [41] |
| 10-Methoxy macrocarpamine 4′-N-oxide 35 | Macroline-pleiocarpamine | A. angustifolia | Leaves [41] |
| (−)-Perhentidine A 36 | Macroline-macroline | A. macrophylla and A. angustifolia | Stem-bark [28,33] |
| (−)-Perhentidine B 37 | Macroline-macroline | A. macrophylla and A. angustifolia | Stem-bark [33] |
| (−)-Perhentidine C 38 | Macroline-macroline | A. macrophylla and A. angustifolia | Stem-bark [28,33] |
| (−)-Perhentinine 39 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| A. angustifolia | Leaves [18] | ||
| A. macrophylla | Leaves [18] | ||
| (+)-Perhentisine A 40 | Macroline-sarpagine | A. angustifolia | Stem-bark [28] |
| (−)-Perhentisine B 41 | Macroline-sarpagine | A. angustifolia | Stem-bark [28] |
| (+)-Perhentisine C 42 | Macroline-sarpagine | A. angustifolia | Stem-bark [28] |
| A. muelleriana | Leaves and stem-bark [37] | ||
| (+)-Villalstonidine A 47 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| (+)-Villalstonidine B 48 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| A. macrophylla | Stem-bark [27] | ||
| (+)-Villalstonidine C 49 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| (+)-Villalstonidine D 50 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| (+)-Villalstonidine E 51 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| (+)-Villalstonidine F 52 | Macroline-pleiocarpamine | A. macrophylla | Stem-bark [27] |
| (+)-Villalstonine 43 | Macroline-pleiocarpamine | A. angustifolia | Leaves and stem-bark [28,36,39,41] |
| A. macrophylla | [41] | ||
| A. villosa | [41] | ||
| A. verticillosa | [41] | ||
| A. somersentensis | [41] | ||
| Villalstonine N(4)-oxide 44 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark, and root-bark [20,30,32,38] | ||
| (+)-10-Methoxy villalstonine 45 | Macroline-pleiocarpamine | A. angustifolia | Leaves [41] |
| 10-Methoxy villalstonine 4′-N-oxide 46 | Macroline-pleiocarpamine | A. angustifolia | Leaves [41] |
Villalstonine N(4)-oxide 44 was isolated from the stem-bark of A. angustifolia [28]. It was also isolated from the leaves, stem-bark, and root-bark of A. scholaris, A. glaucescens, and A. macrophylla extracts [20]. Moreover, two villalstonine 44-related bisindoles, (+)-10-methoxy villalstonine 45 and 10-methoxy villalstonine N(4)-oxide 46, were isolated from the leaves of A. angustifolia [41]. Villalstonidine A 47, villalstonidine B 48, villalstonidine C 49, villalstonidine D 50, villalstonidine E 51, (+)-villalstonidine F 52, and villalstonine N(4)-oxide 44 are macroline–pleiocarpamine-type bisindoles and are close in structure to villalstonine 43. Villalstonidines A–D (47–50) were isolated from the stem-bark of A. angustifolia [28]. Additionally, (+)-villalstonidine F 52 {N(1)-demethylderivative of villalstonine 43} and (+)-villalstonidine B 48 were isolated from the stem-bark of A. macrophylla [27].
3. Bioactivity of Bisindole Alkaloids from Alstonia Species
Studies from various groups have shown that bisindoles have anticancer activity in different cell lines, including HT-29 and vincristine-resistant KB/VJ300 cells. Bisindoles are also reported to have other biological activities, including antiprotozoal activity against Plasmodium falciparum and antileishmanial activity against promastigotes of Entamoeba histolytica. The reported biological activity of bisindoles from various Alstonia species including the semi-synthetic derivatives reviewed herein are listed in Table 4. (+)-Alstomacroline 1 and (+)-alstomacrophylline 2 bisindoles were active against the K1 (multi-drug resistant) strain of P. falciparum with an IC50 1.12 ± 0.35 μM and IC50 1.10 ± 0.30 μM, respectively [42]. Newly isolated macroline-sarpagine-type bisindoles, (−)-angustilongines E 6, (-)-angustilongine F 7, (+)-angustilongine G 8, (+)-angustilongine H 9, (+)-angustilongine J 10, and (+)-angustilongine K 11 showed in vitro growth inhibitory activity against human cancer cell lines, inclusive of KB, vincristine-resistant strains of KB, HCCT 116, PC-3, MDA-MB-231, LNCaP, MCF7, HT-29, and A549 cells with IC50 values ranging from 0.02 to 9.0 μM in the study from the group of Kam [5]. Kam et al. [18] also reported the anticancer activity of (−)-lumusidine A 14, (−)-lumusidine B 15, (−)-lumusidine C 16, and (−)-lumusidine D 17. These bisindoles were cytotoxic against KB/VJ300 cells ranging from IC50 0.16 to 5.03 μg/mL (μM) values with 0.12 μM vincristine added [18]. Lumutinine A 18, lumutinine B 19, lumutinine C 20, lumutinine D 21, and lumutinine E 22 exhibited moderate anticancer activity against KB/VJ300 cells ranging in IC50 value from 0.10 to 4.61 μg/mL (μM) again with 0.12 μM vincristine added in the studies from the same group (Kam) [18].
(+)-Macralstonine 24 was active against the K1 strain of P. falciparum (IC50 8.92 ± 2.95 μM). Notably, the derivatives of (+)-macralstonine 24 were more active than (+)-macralstonine 24 itself. (+)-O-methyl macralstonine 26 and (+)-O-acetyl macralstonine 25 demonstrated more potent activity against the K1 strain of P. falciparum with IC50 values of 0.85 ± 0.20 μM and IC50 0.53 ± 0.09 μM, respectively [20]. Likely, the functionalization facilitated the transportation of these bisindoles through the cell membranes of parasites and red blood cells, which would have enhanced the activity as lipophilicity rose [20]. (+)-O-acetyl macralstonine 25 and (+)-alstomacroline 1 were also somewhat active against the T9-96 strain of P. falciparum with IC50 values of 12.4 and 10.2 μM, respectively [20]. Heterodimeric alkaloid (−)-anhydromacralstonine 27 showed moderate cytotoxicity (IC50 value of 0.44 μg/mL (μM) in KB/VJ300 cells with 0.12 μM of vincristine added [18]). Another semisynthetic analog of macralstonine 24, the related O-acetyl-E-seco-macralstonine 53, showed strong anticancer activity. It was prepared by the reaction of macralstonine 24 with acetic anhydride/pyridine in DCM [18]. It demonstrated potent activity against vincristine-resistant KB/VJ300 cells with an IC50 value of 0.27 μg/mL (μM), with 0.12 μM of vincristine added to the assay [18]. (+)-Macralstonidine 23 was found to exhibit moderately active anticancer activity. It was active against KB/VJ300 cells with an IC50 value of 0.13 μg/mL (μM) [18].
(−)-Macrocarpamine 31 exhibited antiprotozoal and anticancer activity in various studies. It showed significant antiprotozoal activity in vitro in studies from Wright et al. against E. histolytica and P. falciparum with ED50 values of 8.12 (95% C.I.) μM and 9.36 (95% C.I.) μM, respectively [43]. Keawpradub et al. reported significant activity of (−)-macrocarpamine 31 against the K1 strain of P. falciparum with an IC50 value of 0.36 μM [20]. Furthermore, (−)-macrocarpamine 31 showed antimalarial activity against the T9-96 strain of P. falciparum with an IC50 > 39 μM. The ancient folklore use of the extracts from A. angustifolia in Malaya for treatment of malaria and dysentery is supported by these in vitro studies [44]. (−)-Macrocarpamine 31 was strongly cytotoxic in KB/VJ300 cells with an IC50 value of 0.53 μg/mL (μM) [18]. (−)-Perhentidine A 36 and (−)-perhentidine B 37 showed strong cytotoxicity against KB/VJ300 cells with IC50 values of 2.29 and 0.84 μg/mL (μM), respectively [18]. O-Acetylperhentidine A 54 and O-acetylperhentidine B 55 also exhibited strong cytotoxicity against KB/VJ300 cells with IC50 values of 0.36 and 0.28 μg/mL (μM), respectively [18]. (−)-Perhentidine A 36 and (−)-perhentidine B 37 were treated individually by dropwise addition of acetic anhydride in a py/DCM solution, followed by stirring at room temperature for 2 h to furnish the semisynthetic (−)-O-acetylperhentidine A 54, and (−)-O-acetylperhentidine B 55, respectively [18]. (−)-Perhentinine 39 and O-acetylperhentinine 56 were cytotoxic against KB/VJ300 cells with IC50 values of 0.52 and 0.30 μg/mL (μM), respectively, in the studies by Kam et al. [18].
(+)-Villalstonine 43 has demonstrated various biological activities including anticancer, antimalarial, and antiamoebic activity. (+)-Villastonine 43 was 1/15th as potent as chloroquine (antimalarial drug) against malaria [45]. It exhibited potent antiplasmodial activity against the multidrug-resistant K1 strain of P. falciparum with an IC50 value of 0.27 μM [20]. Wright et al. tested this compound for antiamoebic activity against E. histolytica [43]. (+)-Villalstonine 43 showed six times less activity (ED50 11.8 μM) than the antiamoebic drug emetine (ED50 2.04 μM). These results also concur with the use of various parts of the A. angustifolia plant from ancient times to treat malaria and amoebic dysentery [43]. Moreover, (+)-villalstonine 43 was cytotoxic against KB cells with an ED50 value of 11.6 μM [43]. (+)-Villalstonine 43 was also potent against the T9-96 strain of P. falciparum with an IC50 value of 0.94 μM. It also showed anticancer activity against the HT-29 cell line with an ED50 value of 8.0 μM (paclitaxel was the positive control). Also, it was cytotoxic against vincristine-resistant KB/VJ300 cells with an IC50 value of 0.42 μg/mL (μM) [18]. On the other hand, the derivative of (+)-villalstonine 43, villalstonine N(4)-oxide 44 was less potent (IC50 10.7 ± 1.9) than (+)-villalstonine 43 itself. The increase in the ionic charge (decrease in lipophilicity) might have reduced the ability of villalstonine N(4)-oxide 44 to cross through the cell membranes of red blood cells or the parasites, which if correct, would explain the weaker activity [20]. Villalstonine N(4)-oxide 44 is also an antileishmanial bisindole. It was active against promastigotes of Leishmania mexicana with an IC50 value of 80.3 μM [39]. (+)-Villalstonine 43-related alkaloids (+)-villalstonidine B 48 and (+)-villalstonidine F 52 were found to be strongly cytotoxic against KB/VJ300 cells with IC50 values of 0.35 and 5.64 μg/mL (μM), respectively [18]. (+)-Villalstonidine D 50 exhibited antileishmanial activity. It was active against promastigotes of L. mexicana with an IC50 value of 120.4 μM in a study from Pan et al. [39]. Another (+)-villalstonine 43-related alkaloid, (+)-villalstonidine E 51, demonstrated anticancer and antimalarial activity. It was cytotoxic against the HT-29 cell lines with an ED50 value of 6.5 μM and was active against promastigotes of L. mexicana with an IC50 value of 78 μM in the study from the same group [39]. Many bisindoles are yet to be screened for their activity because of the paucity of material; however, their significant role in future drug discovery should be considered.
4. Biosynthesis
4.1. Proposed Biogenetic Pathway toward Angustilongines (6–9)
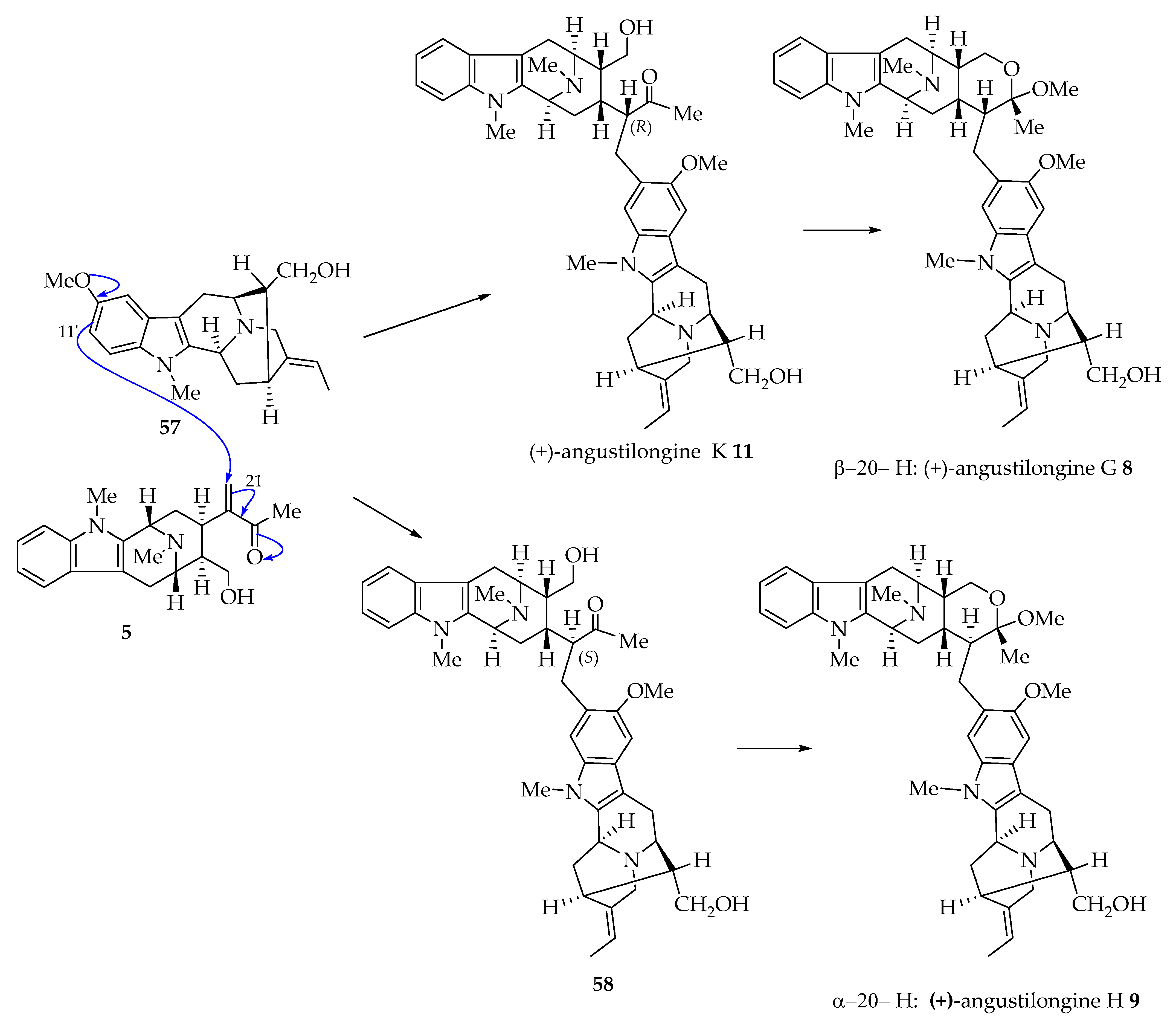
Yeap et al. put forward a plausible biogenetic pathway to the angustilongines [5]. The nucleophilic C-11′ of 10-methoxyaffinisine 57 undergoes conjugate addition onto C-21 of the hypothetical macroline unit 5 to give (+)-angustilongine K 11 and 58 (Figure 1). The (+)-angustilongine K 11 and 58, after the E-ring closes to the hemiacetal, would give (+)-angustilongine G 8 and (+)-angustilongine H 9, respectively. Since methanol was used during the extraction of the alkaloids, there could be the possible formation of bisindoles 8 and 9 as artifacts [5]. Similarly, nucleophilic addition of C-9′ of 10-methoxyaffinisine 57 by conjugate addition (Michael reaction) onto C-21 of the macroline unit 5 would give angustilongine J 10. The E-ring closure of 10 gives angustilongine F 7 (Figure 2). The 10-methoxyaffinisine 57 through its nucleophilic C-11′ carbon atom addition to the C-19 of an E-seco-talcarpine derivative 59 results in angustilongine E 6 via hydroxy aldehyde 60 [5]. Another likely mechanism is a Friedel–Crafts alkylation of the oxonium ion of corresponding macroline units (cyclization followed by loss of water) to the respective sarpagine units as suggested by Fukuyama to furnish angustilongines (mechanism not shown for angustilongines 6–11 see Section 4.4 with representative examples of (+)-macralstonine 24 and (+)-lumutinine A 18) [6].
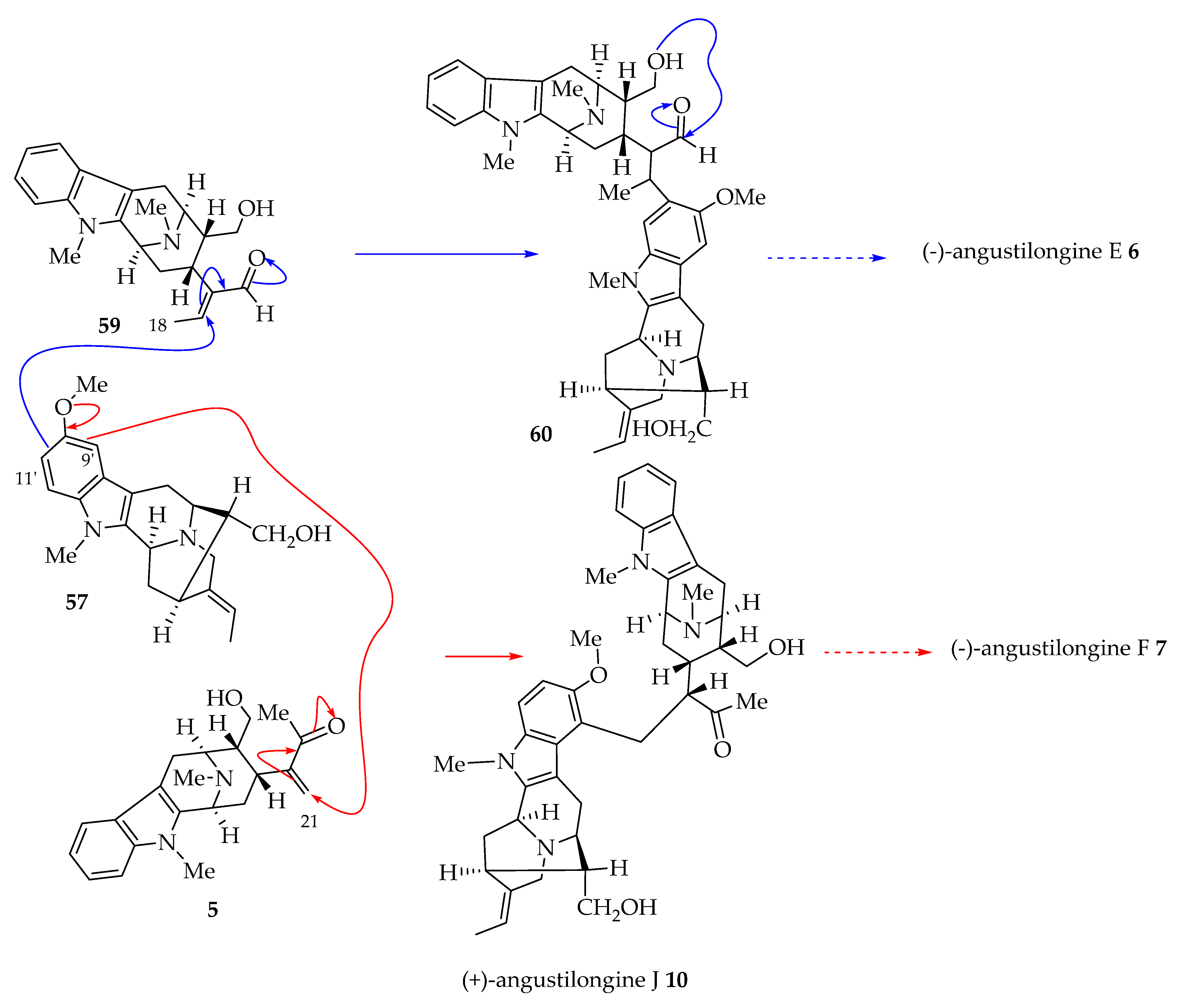
4.2. Plausible Biogenetic Pathway to (-)-Lumusidines A-D (14–17)
The conjugate addition of the C-10′ carbon atom of alstophylline 28 by a Michael reaction onto the α, β unsaturated aldehyde 59 (E-ring opened talcarpine derivative) could potentially give hydroxy ketone bisindole alkaloid 61 (Figure 3). Alcohol 61 on subsequent closing of the E-ring to form a hemiacetal gives (-)-lumusidine B 15, which after dehydration gives (-)-lumusidine A 14. Similarly, the C-12′ carbon atom of alstophylline monomer 28 could add to the α, β unsaturated macroline counterpart 5 to furnish a hydroxy ketone (not shown). The cyclization of the hydroxy ketone in a similar way to give a hemiacetal 62 would follow. Subsequently, the dehydration of hemiacetal 62 would likely form lumusidine D 17. The lumusidine C 16 is assumed to be an artifact formed from hemiacetal 63 (a closed form of (-)-perhentinine 39) as ethanol was used during the extraction [18]. However, again this biosynthetic pathway could occur just as likely via a Friedel–Crafts alkylation of the oxonium ion of 59 (cyclization followed by loss of water) to the olefin of precursor 28 (see Section 4.4 with representative examples of (+)-macralstonine 24 and (+)-lumutinine A 18) [6].
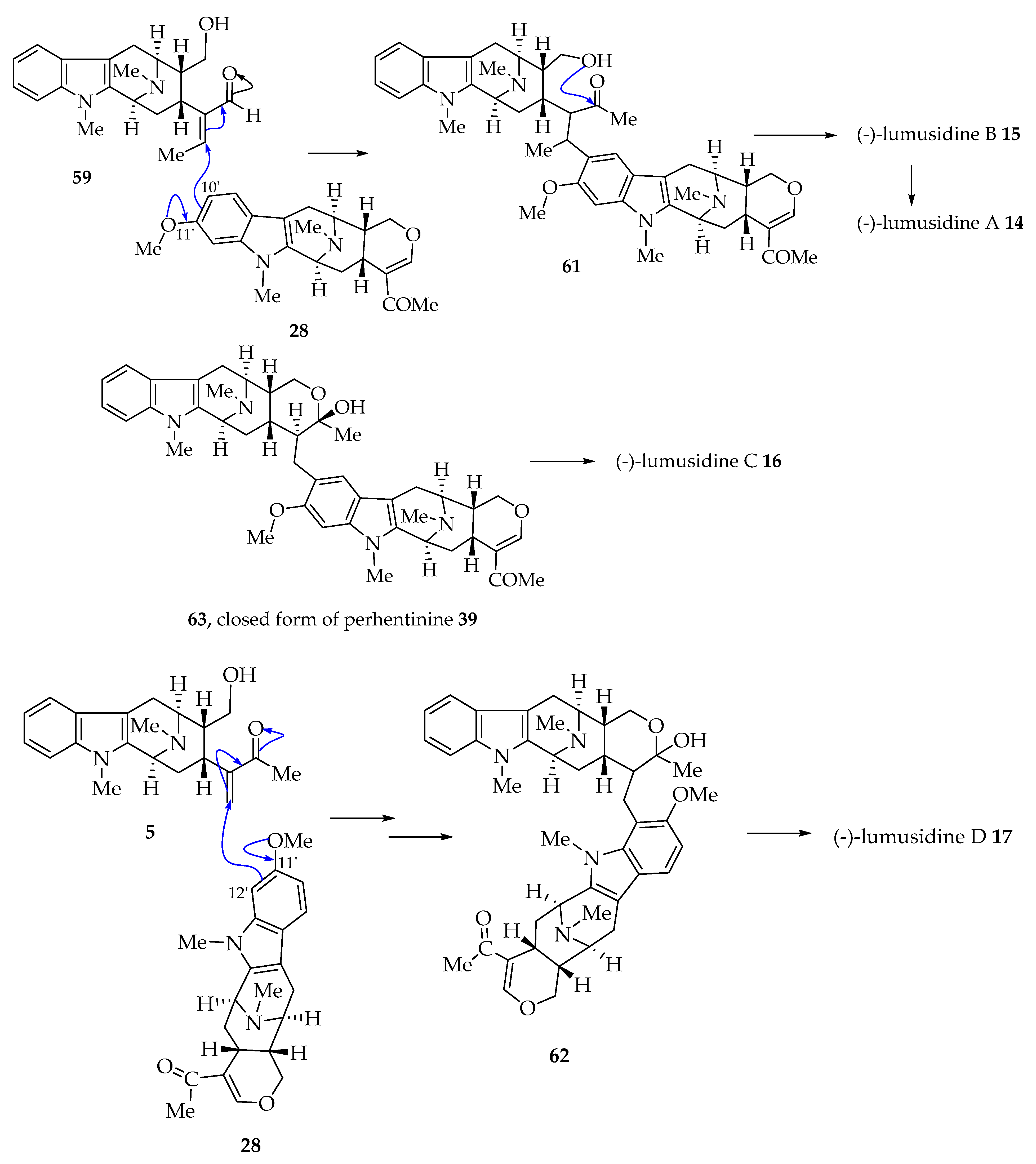
4.3. Proposed Biogenetic Pathway to Lumutinines A–D (16–19)
The hydroxy ketone 64 could be formed by Michael addition of the C-10’ carbon atom of alstophylline 65 onto the olefinic moiety of macroline 5 (Figure 4) [26]. The cyclization of the hydroxy ketone would give the hemiketal 66, which on ketalization furnishes (+)-lumutinine A 18. Likewise, Michael addition by C-12’ of 67, C-9’ of 68, and C-11’ of 69 (Figure 4) onto macroline 5 yields (-)-lumutinine B 19, (+)-lumutinine C 20, and (+)-lumutinine D 21, respectively [26]. Again, the Friedel–Crafts alkylation of the oxonium ion of 65 (cyclization followed by loss of water) is just as likely (see Section 4.4 with representative examples of (+)-macralstonine 24 and (+)-lumutinine A 18) [6].
4.4. Possible Alternative Mechanism of Bisindole Formation of (+)-Lumutinine A 16 and (+)-Macralstonine 24 as Representative Examples via a Friedel–Crafts Alkylation Process as Suggested by Fukuyama
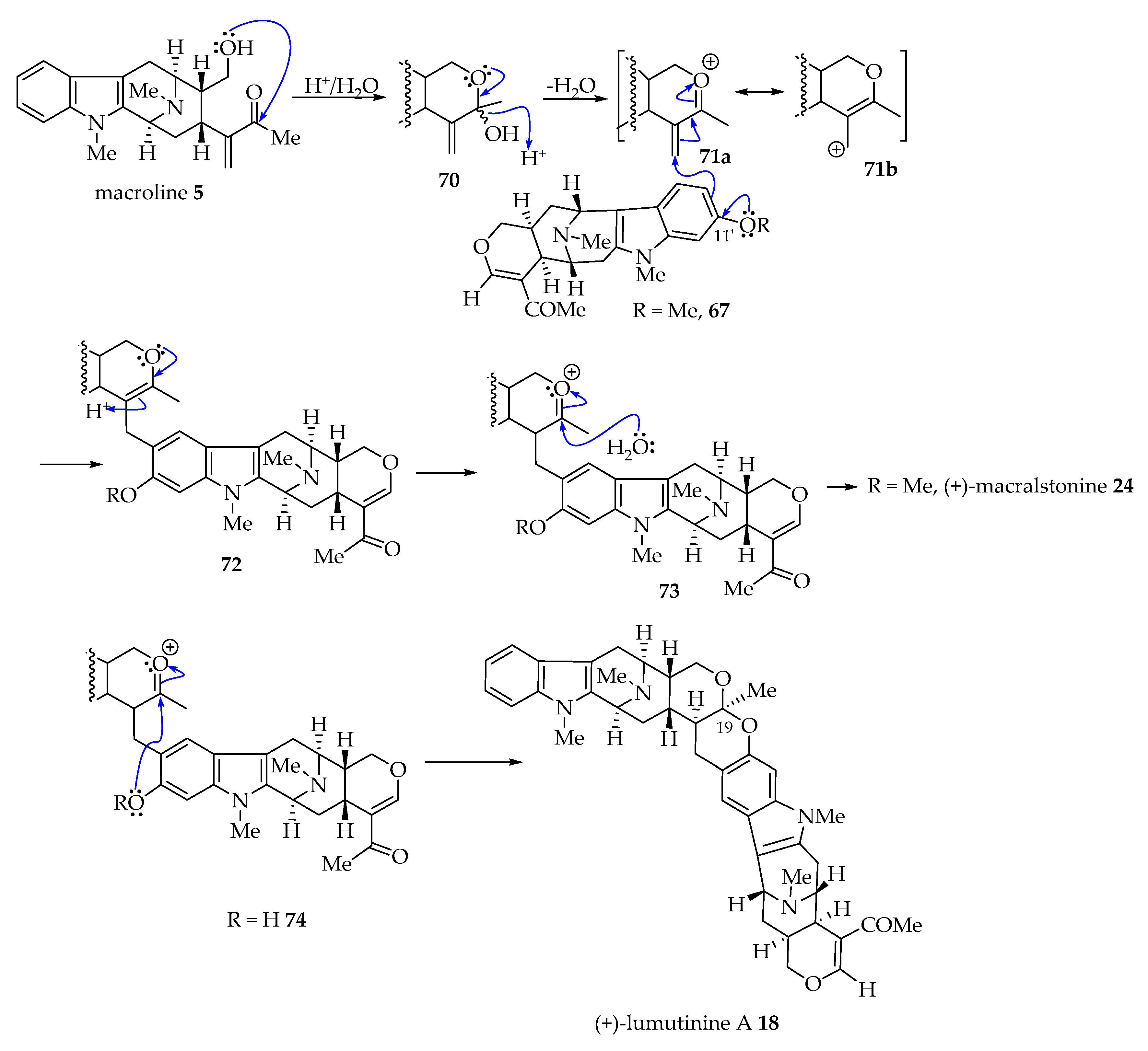
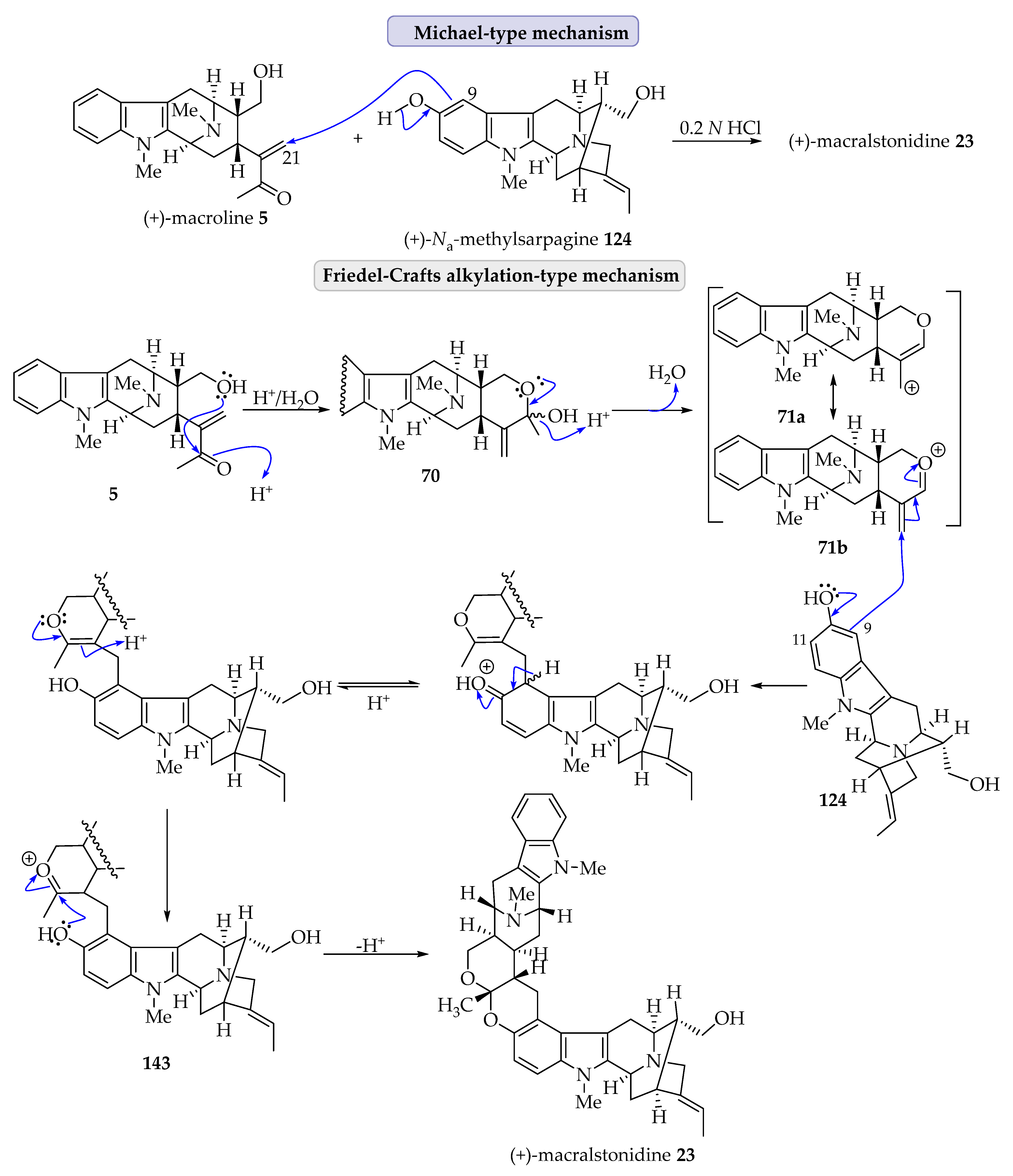
In addition to the Michael addition process described above, there is another potential mechanism for the coupling of ring-A oxygenated macroline-type alkaloids with macroline 5 that involves a Friedel–Crafts alkylation process stabilized by an oxonium ion [6]. As shown in the figure below (Figure 5), an acid-catalyzed intramolecular cyclization of macroline 5 by nucleophilic attack of the C-19 hydroxyl function onto the carbonyl carbon atom in 1, 2-addition fashion would furnish the cyclic hemiacetal 70. The loss of a water molecule would generate an oxonium ion species (71a or 71b). At this stage, an electrophilic attack of the electron-rich aromatic ring of 11-methoxy macroline 67 via the C-10 carbon atom onto the olefin in 71a would generate a cyclic enol ether 72. This cyclic hemiacetal could undergo isomerization, generating an oxonium ion intermediate 73 only to be attacked by water to generate the cyclic ether (as in macralstonine 24). Alternatively, the phenolic hydroxy group at the C-10 carbon atom in 10-hydroxy macroline (see 74) could attack the oxonium ion to form a cyclic ketal (as in lumutinine A 16). This type of mechanism was suggested by Fukuyama for some bisindoles isolated from Alstonia species [6].
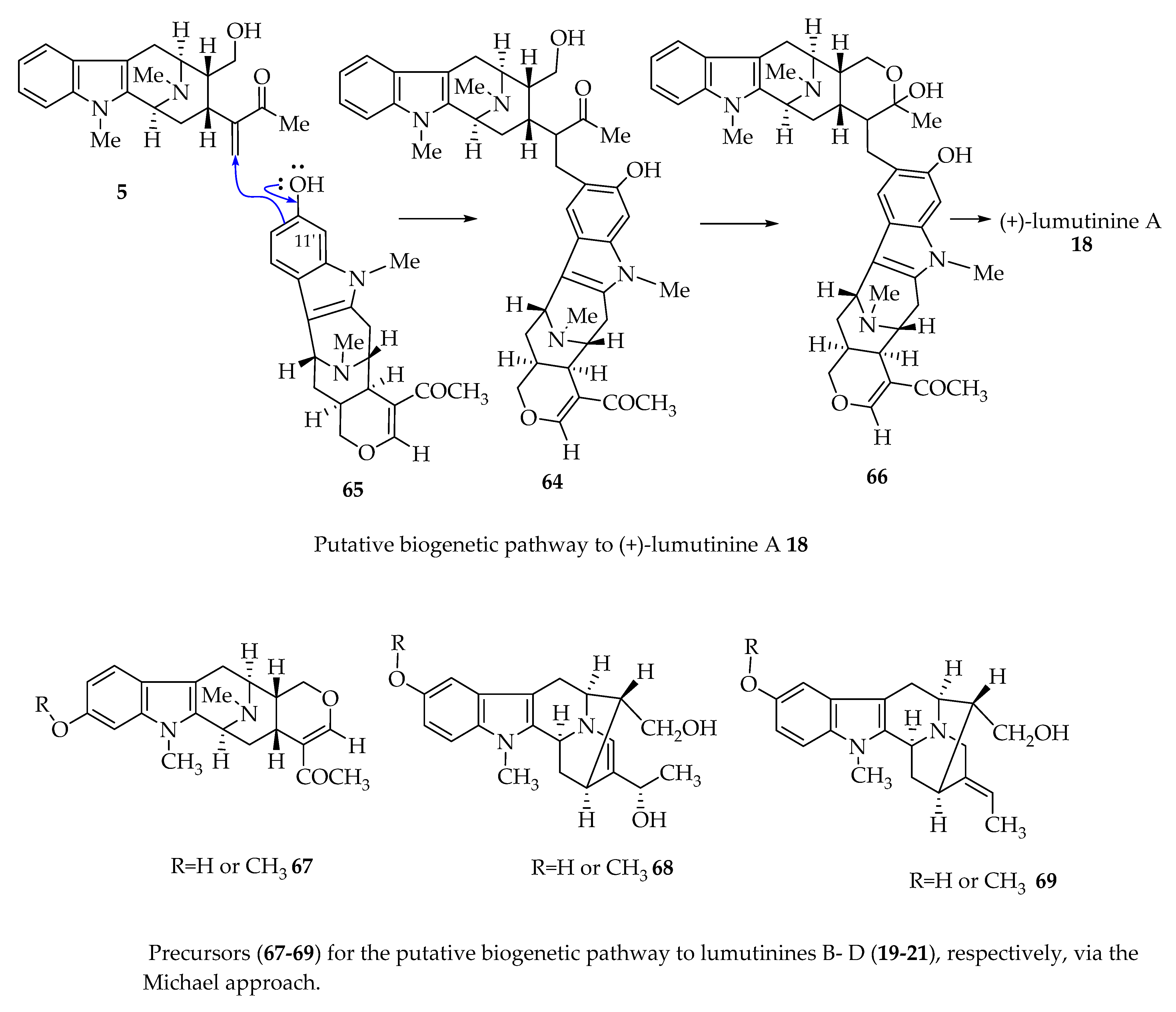
4.5. Proposed Biogenetic Pathway to (-)-Macrocarpamine 31
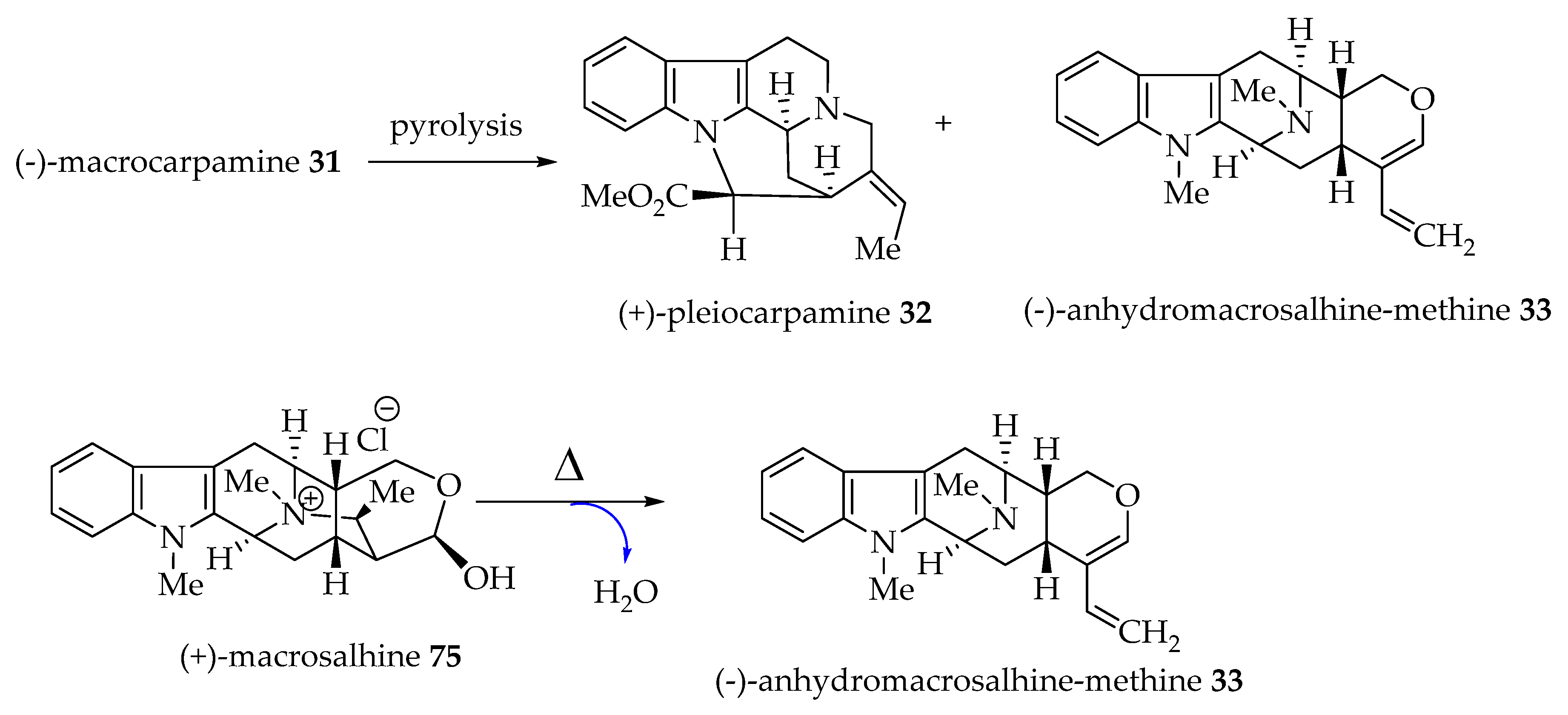
(-)-Macrocarpamine 31, a bisindole alkaloid comprised of two Alstonia monomeric units (+)-pleiocarpamine 32 and (-)-anhydromacrosalhine-methine 33, was proposed to originate from (-)-anhydromacrosalhine-methine 33 and (+)-pleiocarpamine 32 by Hesse, Schmid et al. [46]. (-)-Anhydromacrosalhine-methine 33 and (+)-pleiocarpamine 32 are the degradation products of (-)-macrocarpamine 31. (-)-Anhydromacrosalhine-methine 33 was also obtained by dehydration of (+)-macrosalhine 75 in the study by Khan et al. (Figure 6) [6,47].
5. Synthesis of Representative Monomeric Units of Bisindole Alkaloids from Alstonia Species Discussed Herein, Required for Bisindole Synthesis
5.1. The Total Synthesis of (±)-Pleiocarpamine 32
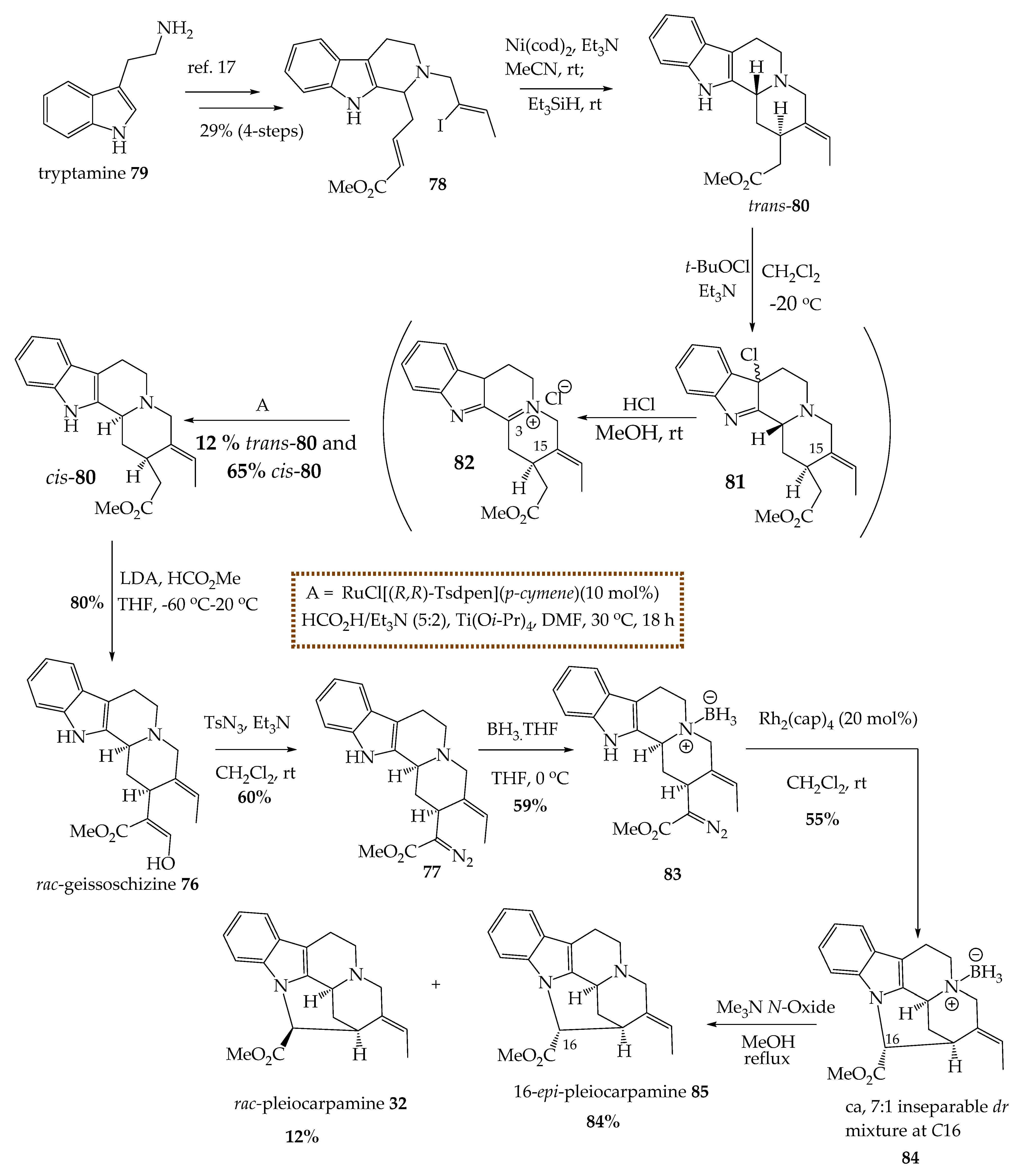
(+)-Pleiocarpamine 32 has been isolated from various species of Alstonia [6,32,36,48,49,50,51]. The unprecedented (+)-bipleiophylline (not shown) was found to contain two pleiocarpamine 32 units connected by utilizing an aromatic spacer (pyrocatechuic acid). The homo-dimer (+) bipleiophylline was potentially active against drug-sensitive, vincristine-resistant human KB and Jurkat cells, while its monomeric precursor (+)-pleiocarpamine 32 was not very active [52]. These findings again demonstrate the increased activity of dimeric alkaloids as compared to their monomeric counterparts. The (+)-pleiocarpamine 32 was thought to be formed biosynthetically by a ring-closing reaction between N1 and C16 of geissoschizine 76 [17]. Recently Sato et al. reported the total synthesis of the C-mavacurine type indole alkaloid, (±)-pleocarpamine 32 via a biomimetic synthesis [17]. A critical step, the direct cyclization between N1 and C16, was thought to be challenging because of the strain generated in the corynanthe pentacyclic framework. This may likely be the reason for the time it took to achieve the total synthesis of pleiocarpamine 32 by many groups since its isolation. In fact, (±)-2,7-dihydropleiocarpamine (not shown) was synthesized in 1993 by Jiménez et al. [53]. However, Sato et al. successfully cyclized the strained structure of 77 by a carbene N-H insertion reaction between the N1 and C16 bonds (Scheme 1) [17].
The ester 78, was prepared in four steps from tryptamine 79 in 29% overall yield [17,54]. The subsequent reductive cyclization of ester 78 onto cis-ester 80 was carried out using Ni(cod)2. However, at this step, the desired cyclized cis-ester 80 was formed as a minor product (31%) with the undesired trans-ester 80 (55%) predominating [17]. The undesired trans-ester 80 was first treated with tert-butyl hypochlorite and triethylamine at –20 °C to obtain intermediate 81, which on further reaction with hydrochloric acid in methanol at room temperature furnished iminium intermediate 82 [17]. Sato et al. screened several reducing agents and reaction conditions to reduce the iminium intermediate 82 into the cis-ester 80. The optimized condition for the reduction of the iminium intermediate employed by Sato et al. was a Noyori catalyst and formic acid/triethylamine (5:2) mixture with titanium tetra-isopropoxide (at 30 °C) to yield the desired cis-ester 80 as a major product (cis/trans ratio 5:3.1) [17]. The cis-ester 83 was converted into (±)-geissoschizine 76 in 80% yield using an excess of lithium diisopropylamine and methyl formate. The treatment of (±)-geissoschizine 79 with tosyl azide and triethylamine furnished diazo compound 77 in 60% yield [17].
While exploring a suitable catalyst for the metal carbenoid cyclization reaction, the Rh2(cap)4 and Rh2(esp)2 catalyzed reactions lead to decomposition of substrate 77 [17]. Only Rh2(OAc)4 furnished the distinguishable but unanticipated product (not shown) via a D-ring expansion reaction [17]. At room temperature, no reaction happened while employing the rhodium complex (PPh3)3RhCl or the copper catalyst Cu(MeCN).BF4 [17]. Moreover, the use of the JohnPhosAu(MeCN)SbF6 catalyst was also unsuccessful because it formed the D-ring expanded product (not shown) with β-hydride elimination (not shown) [17]. Sato et al. predicted that a modification of the lone pair on the N(4) of the compound 77 would form a cis-quinolizidine ring making cyclization more favorable by decreasing the distance between N1 and C16. This was the key to the first total synthesis of (±)-pleiocarpamine 32. Thus, substrate 77 was modified to borane salt 83 by using the BH3–THF complex to react at the Nb nitrogen atom [17]. Finally, cyclization via a carbene N–H insertion was accomplished by treatment of cyclization precursor 83 with Rh2(cap)4 (20 mol %) at room temperature, and the products so obtained were an inseparable diastereomeric mixture 84 at C16 (ca,7:1) in 55% yield [17]. To remove the borane protecting group, the diastereomeric mixture 84 was refluxed with trimethylamine N-oxide in methanol, which furnished (±)-pleiocarpamine 32 and epi-pleiocarpamine 85 [17].
5.2. Biosynthetic Relations among the Sarpagine/Macroline/Ajmaline Family of Alkaloids
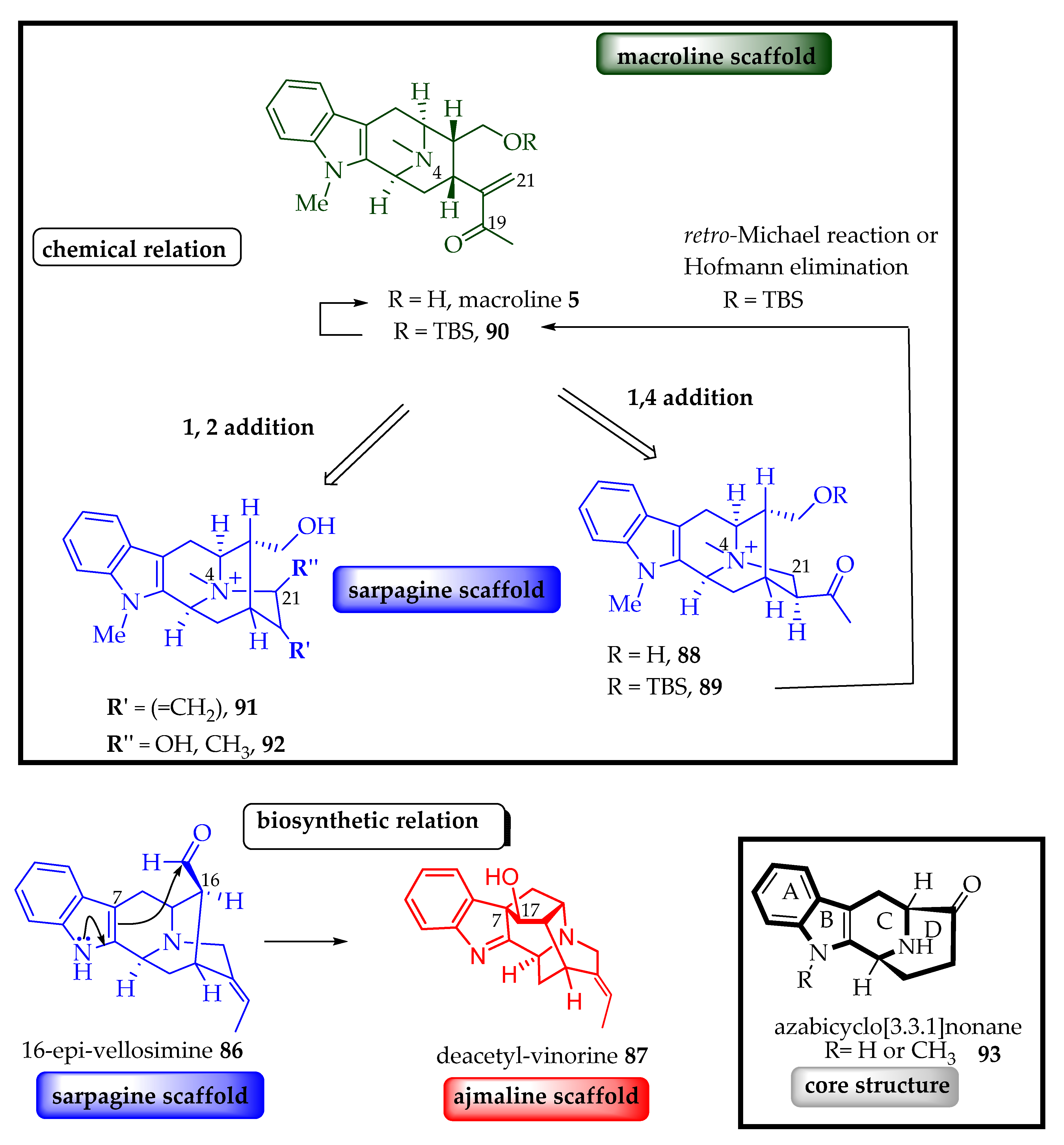
Sarpagine, macroline, and ajmaline indole alkaloids are biosynthetically and structurally related (Figure 7) [13]. We herein follow the biogenetic numbering system of LeMen and Taylor to discuss their potential formation [55]. The sarpagine series consists of attachment of the C-21 carbon atom to the N(4) bond, while the macroline series lack this bond. The stereocenters in the sarpagine alkaloids at C-3 (S), C-5 (R), C-15 (R), and C-16 (R) are identical to the macroline series at the respective carbon atoms. The ajmaline series also contain a C-7 and C-17 carbon atom linkage. In comparison with the ajmaline series, the sarpagine family at C-16 is R but antipodal in the ajmaline series. Both the sarpagine and the ajmaline series consist of the characteristic quinuclidine ring and the C-5 and C-16 carbon atom linkage. Sarpagine and ajmaline alkaloids both contain the characteristic C-21 and N(4) bond [13,56]. Stӧckigt et al. reported the conversion of 16-epi-vellosimine 86 (sarpagine) into the vinorine 87 (ajmaline) structure (Figure 7) by employing the acetyl CoA-dependent vinorine synthase enzyme [57,58,59,60]. The macroline and sarpagine alkaloids are biosynthetically related, as shown in Figure 7. Either of the β-keto quaternary ammonium intermediates 88 or 89 can be furnished by 1, 4 Michael addition of the N(4) nitrogen atom of macroline onto the α, β-unsaturated carbonyl system 5 or 90 at C-21. Similarly, α-hydroxy quaternary ammonium compounds 91 and 92 can be formed by direct 1, 2 addition of the N(4) nitrogen atom to the C-19 carbonyl carbon of 5 and 90 [13]. In contrast, the retro-Michael reaction of TBS protected intermediate 89, followed by removal of the silyl protecting group converted sarpagine 89 into the macroline unit 5 in the studies from LeQuesne et al. [61] and Cook et al. [62]. The sarpagine/macroline/ajmaline group of indole alkaloids have been isolated principally from higher plants, including the Apocynaceae family and the genera Alstonia and Rauwolfia [13]. The intriguing molecular complexity, interconversions, and their divergent biological activity prompted the successful syntheses of several of these indole alkaloids [7,63] and bisindole alkaloids [45,64,65] as mentioned earlier.
5.3. Synthesis of Monomeric Macroline Units Employed for the Synthesis of Bisindole Alkaloids
5.3.1. An Improved Total Synthesis of (-)-Alstophylline 28
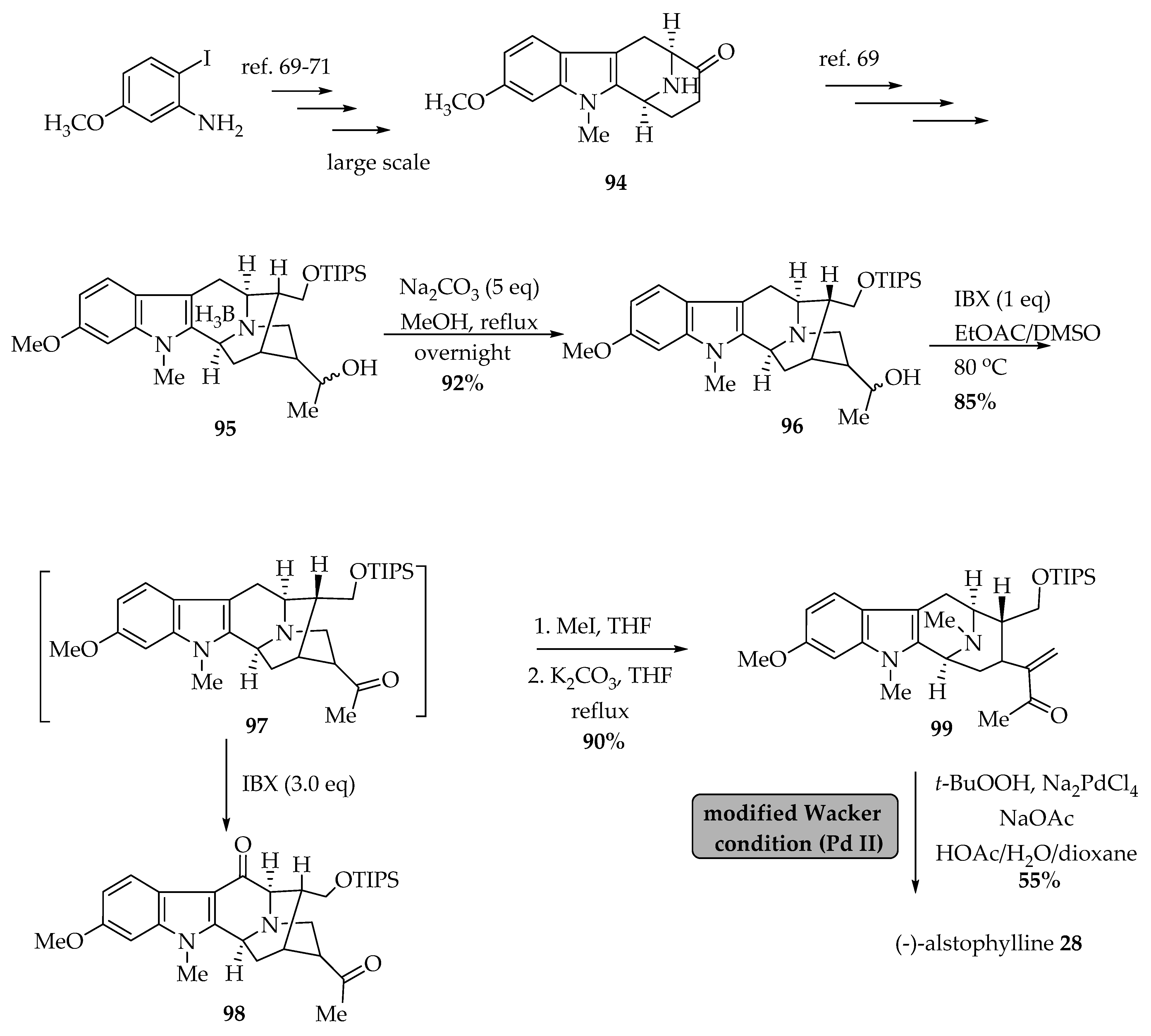
Liao et al. [66] improved the synthesis of (-)-Alstophylline 28 which, has been isolated from various Alstonia species including A. angustifolia [36], A. glabriflora Mgf. [31], and A. macrophylla [30,67,68]. Liu et al. accomplished a large-scale enantiospecific total synthesis of intermediates, which later resulted in the total synthesis of (-)-alstophylline 28 [69]. Later, Liao et al. improved the total synthesis of (-)-alstophylline 28 employing modified Wacker reaction conditions, which provided a faster route for synthesis of macroline-related indole alkaloids (Scheme 2) [66]. The 6-methoxy tetracyclic ketone 94 was synthesized employing the method using the published procedures [69,70,71], which later was converted into the Nb-BH3 adduct 95 using the method developed by Liu et al. earlier [69]. The Nb-BH3 adduct 95 so formed was refluxed with sodium bicarbonate in methanol overnight to get alcohol 96 in 92% yield. One equivalent of the 2-iodozybenzoic acid (IBX) converted (at high temperature) the alcohol functional group in 96 into the ketone 97 in 85% yield. The addition of three more equivalents of IBX at reflux permitted the formation of the new alkaloid 6-oxoalstophylline 98. The Nb-quaternization of ketone 97 was carried out using MeI/THF, which was followed by a retro-Michael reaction in the presence of K2CO3/THF to furnish α,β-unsaturated ketone 99 in 90% yield. Finally, the (-)-alstophylline 28 was obtained using the Wacker-related process (Pd II) modified by Tsuji et al. [72] and Cook et al. [66] from ketone 99 in 55% yield. Importantly, the modified Wacker-related process is also an improved route to several indole alkaloids with the enone system including alstonerine 100 [6]. In addition, the key oxidation reaction provides a shorter route from 97 to 6-oxoalstophylline 98.
5.3.2. Enantiospecific Total Synthesis of (-)-Anhydromacrosalhine-methine 33 via the Asymmetric Pictet–Spengler Reaction
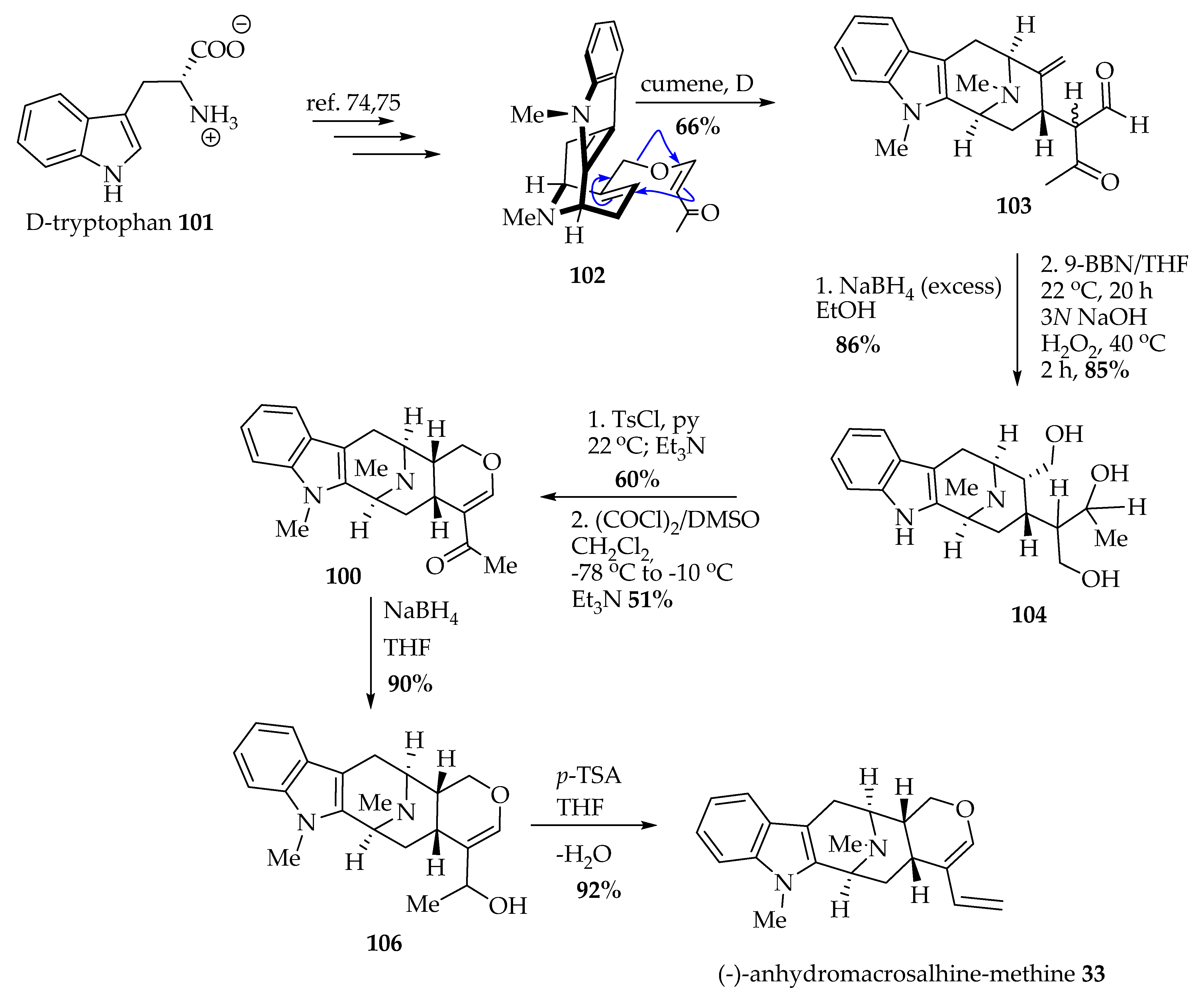
In a partial synthesis, Gan et al. [73] prepared (-)-anhydromacrosalhine-methine 33 via degradation of natural (+)-ajmaline by following the improved procedure of Sakai et al. [73]. In addition to the semisynthesis of (-)-anhydromacrosalhine-methine 33 for comparison purposes, Gan et al. also accomplished the total synthesis of 33 starting from D-tryptophan 101 via the asymmetric Pictet–Spengler reaction (>98% ee) (Scheme 3) [44]. The enone ether 102 was synthesized employing the procedures developed in Milwaukee [74,75]. The β-dicarbonyl compound 103 was prepared in 66% yield from a stereoselective (α-face) Claisen rearrangement of olefin 102 at high temperature (175 °C) [44]. The reduction of the β-dicarbonyl compound 103 that resulted with NaBH4 provided the diol (not shown), which subsequently underwent a stereospecific hydroboration–oxidation reaction and was converted into triol 104. (-)-Alstonerine 100 was furnished by the regioselective cyclization reaction of the monotosylate of triol 104, which was followed by the improved Swern oxidation in 51% yield, accompanied by dihydroalstonerine 105 (not shown in Scheme 3) in 31% yield [44,75]. (-)-Alstonerine 100, upon reduction with NaBH4, furnished allylic alcohol 106 in 90% yield, which later was dehydrated in the presence of p-toluenesulfonic acid to afford (-)-anhydromacrosalhine-methine 33 in 92% yield [44].
5.3.3. Improved Synthesis of (+)-Macroline 5
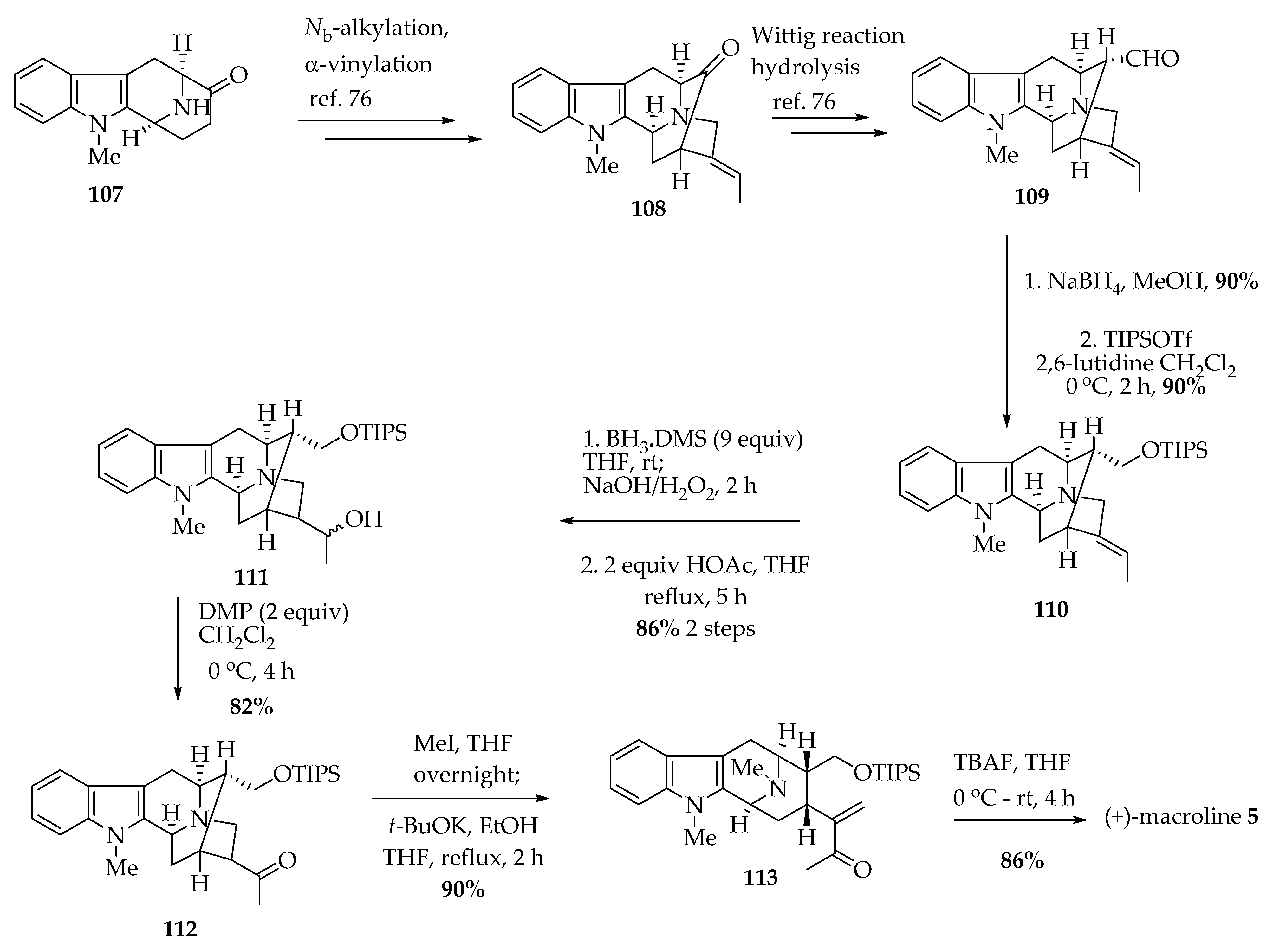
Alkaloids such as (-)-alstonerine 100, (+)-alstonisidine 3, and (-)-alstophylline 28 are believed to originate from (+)-macroline 5 as their biogenetic precursor unit; however, the isolation of (+)-macroline 5 has not yet been reported [6]. The synthesis of (+)-macroline 5 was improved by Liao et al. [76], whereas the first biomimetic synthesis of (+)-macroline 5 was carried out by LeQuesne et al. in 1980 [61], following the procedure of Gorman et al. [77]. Neukomm et al. elucidated the absolute configuration of macroline 5 with X-ray crystallographic data [78]. Bi et al. accomplished the enantiospecific total synthesis of (+)-macroline 5 [79], which was indistinguishable from the spectral data compared to the natural one [80]. The formal synthesis of (+)-macroline 5 was also reported by Kwon utilizing a phosphine-catalyzed annulation process (not shown) [81].
Later, Liao et al. improved the total synthesis of (+)-macroline 5 in higher overall yield and fewer steps. The tetracyclic ketone 107, obtained on the large scale by following the procedure developed in Milwaukee, which underwent Nb-alkylation with (Z)-1-bromo-2-iodo-2-butene (not shown), was followed by a palladium catalyzed stereospecific α-vinylation to provide pentacyclic ketone 108 (Scheme 4) [76]. The ketone 108 underwent the usual one-carbon homologation via a Wittig olefination, and this was followed by acidic hydrolysis to obtain the pentacyclic aldehyde 109. The aldehyde functional group in 109 was reduced using sodium borohydride to provide the first enantiospecific synthesis of (+)-affinisine (not shown) in 90% yield. Then triisopropylsilyl (TIPS) triflate and 2,6-lutidine were used to protect the primary alcohol of (+)-affinisine as a TIPS ether 110 in 90% yield. The TIPS ether 110 was employed for the hydroboration–oxidation reaction, which furnished the desired secondary alcohol 111 in 86% yield. The alcohol 111, so formed, was oxidized to a ketone 112 using the Dess-Martin periodinane reagent in 82% yield. The ketone 112 was quaternized at the Nb-nitrogen atom by treatment with iodomethane in THF at 0 °C. The stable macroline equivalent 113 was obtained after treatment of Nb-methyl iodide salt with potassium tert-butoxide in ethanol, after which it was treated with tetrabutylammonium fluoride to furnish macroline 5 in 86% yield (99% ee) after removal of the TIPS group [76].
5.3.4. Stereospecific Access to the Macroline Core 5
Recently, Kadam et al. developed another route to the pentacyclic core of macroline 5-related alkaloids. Starting from commercially available L-tryptophan (not shown), the core architecture of macroline 5 was accessed via an Ireland–Claisen rearrangement (ICR)/Pictet–Spengler cyclization protocol (Scheme 5) [82]. The L-tryptophan derivative 114 was subjected to Arndt–Eistert conditions to implement a one-carbon homologation to furnish acid 115. The acid 115 was subsequently coupled with allylic alcohol 116 under standard amide coupling conditions to furnish the allylic ester 117, the substrate for the Ireland–Claisen rearrangement. The allylic ester 117 produced the corresponding carboxylic acids (not shown) under ICR as an inseparable mixture that was converted into the corresponding esters 118A/A’ by treating the ICR mixture. The esters were isolated in a 9:1 ratio of diastereomers that was separable. The ester function in 118A was reduced by DIBAL-H, and the so formed primary alcohol was protected with a TBS group to provide silyl ether 119. The olefin 119 was subjected to a hydroboration–oxidation sequence and this was followed by oxidation of the so formed alcohol with Dess-Martin periodinane to furnish aldehyde 120. Upon desilylation with TBAF, the alcohol reacted intramolecularly with the proximate aldehyde function forming the corresponding hemiacetal (not shown). Subsequent tetra-propylammonium perruthenate (TPAP)/N-methylmorpholine N-oxide (NMO) mediated oxidation of the hemiacetal furnished δ-lactone 121. Deprotection of the Boc protecting group and hydrolysis of the acetal was done under acidic conditions with TFA setting up the conditions for an intramolecular Pictet–Spengler cyclization that furnished the pentacyclic core structure 122 of the macroline alkaloids. The exomethylene was introduced by reacting 122 with the formaldehyde equivalent in the presence of 1,8-di-azabicyclo[5.4.0]undec-7-ene (DBU). This was followed by the addition of MeLi to furnish macroline 5. On the other hand, alstomicine 123 was achieved by treating the lactone 122 with MeLi [82].
5.3.5. Enantiomeric Total Synthesis of (+)-Na-Methylsarpagine 124
The alkaloid (+)-Na-methylsarpagine 124 was isolated from the bark of A. spectabilis [31,41], which is present as a monomeric unit in the (+)-macralstonidine 23 bisindole. The first total synthesis of (+)-Na-methylsarpagine 124 was accomplished by Zhao et al. [83,84]. The 5-methoxy-Na-methyl d-(+)-tryptophan ethyl ester 125 was prepared on a large scale starting from commercially available p-anisidine 126 (Scheme 6). The 5-methoxy tetracyclic ketone 127 was furnished in high overall yield from ester 125 employing chemical conversions such as, Nb-benzylation, the asymmetric Pictet–Spengler reaction, diastereospecific Dieckmann cyclization, decarboxylation reaction, and catalytic debenzylation [6,83]. Furthermore, 5-methoxy tetracyclic ketone 127 upon further usual transformations including Nb-alkylation, the Wittig olefination, hydrolysis/epimerization gave majvinine 128 in 98% ee [83,84]. The 10-hydroxy indole 129 was furnished after the treatment of C-10 methoxy intermediate (+)-majvinine 128 employing six equivalents of BBr3 in DCM at −78 °C. The subsequent reduction of the aldehyde group in intermediate 129 was effectively carried out using sodium borohydride in ethanol to furnish (+)-Na-methylsarpagine 124 in 90% yield [83].
6. The Synthesis of Bisindole Alkaloids
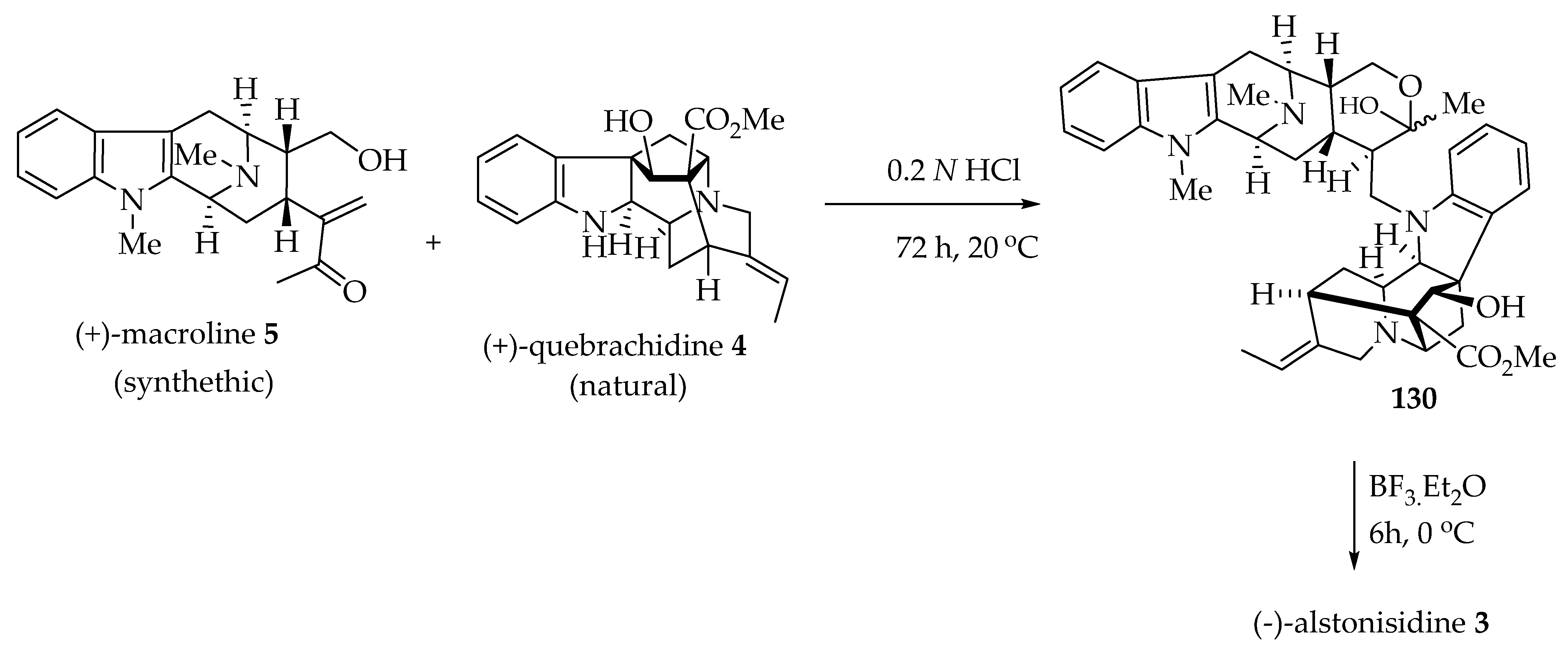
6.1. Biomimetic Partial Synthesis of (-)-Alstonisidine 3
(-)-Alstonisidine 3 contains a (+)-quebrachidine 4 (ajmaline-type) and a (+)-macroline 5 piece as monomeric units. Both monomeric alkaloids were employed in the biomimetic partial synthesis of (-)-alstonisidine 3 by LeQuesne, Burke, and Cook [85]. The (+)-macroline 5 used in this partial synthesis was obtained after the degradation of villamine (not shown) by the method of Schmid et al. [80].
The hemiketal 130 was formed after the Michael reaction between the Na-H function of a (+)-quebrachidine 4 and (+)-macroline 5 in the presence of 0.2 N HCl at 20 °C for 3 days (Scheme 7). The hemiketal 130 underwent an electrophilic substitution reaction (ortho to the amino group of (+)-quebrachidine 4) after the treatment with BF3 etherate at low temperature (0 °C) to furnish (-)-alstonisidine 3. The spectral data of (-)-alstonisidine 3 were identical to those of naturally isolated (-)-alstonisidine 3 [37,51].
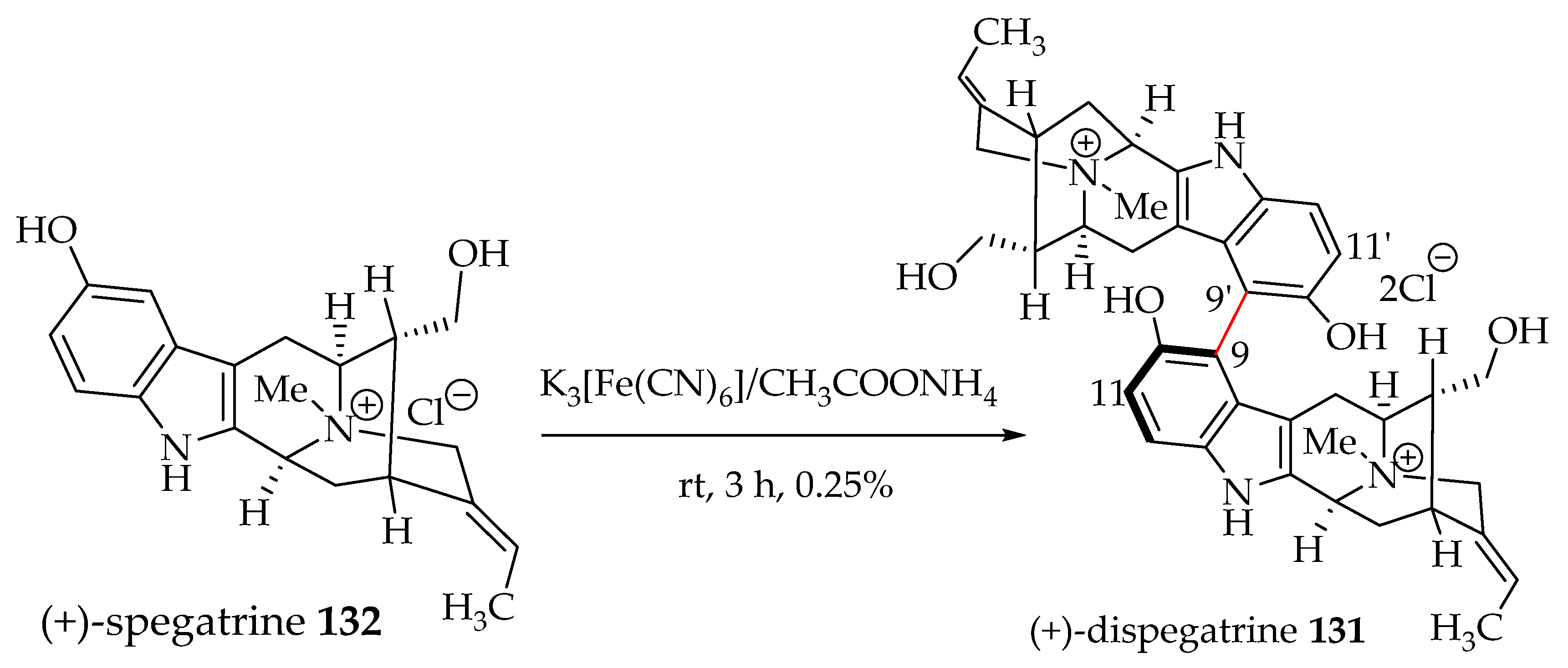
6.2. Biomimetic Partial Synthesis of (+)-Dispegatrine 131
Although the focus of this review is bisindoles from Alstonia species, dispegatrine 131, a hypotensive bisindole, is comprised of two sarpagine units. It is discussed here because some of the monomers such as sarpagine are commonly found in many Alstonia species. In the biomimetic partial synthesis of (+)-dispegatrine 131 by Yu et al., the monomer spegatrine 132 was subjected to an oxidative phenolic coupling (Scheme 8). Although the yield of this coupling was very poor, it served as proof of the concept that dispegatrine 131 may actually originate biosynthetically via oxidative phenolic coupling, although the chemical yield was 0.25%. This partial synthesis confirmed the structure of (+)-dispegatrine 131 [86]. However, no other diastereomer was observed, and the axial chirality across C9-C9′ could not be confirmed. In addition, due to the lack of X-ray crystals, the isolation chemists could not assign the important axial chirality at C9-C9′ [86].
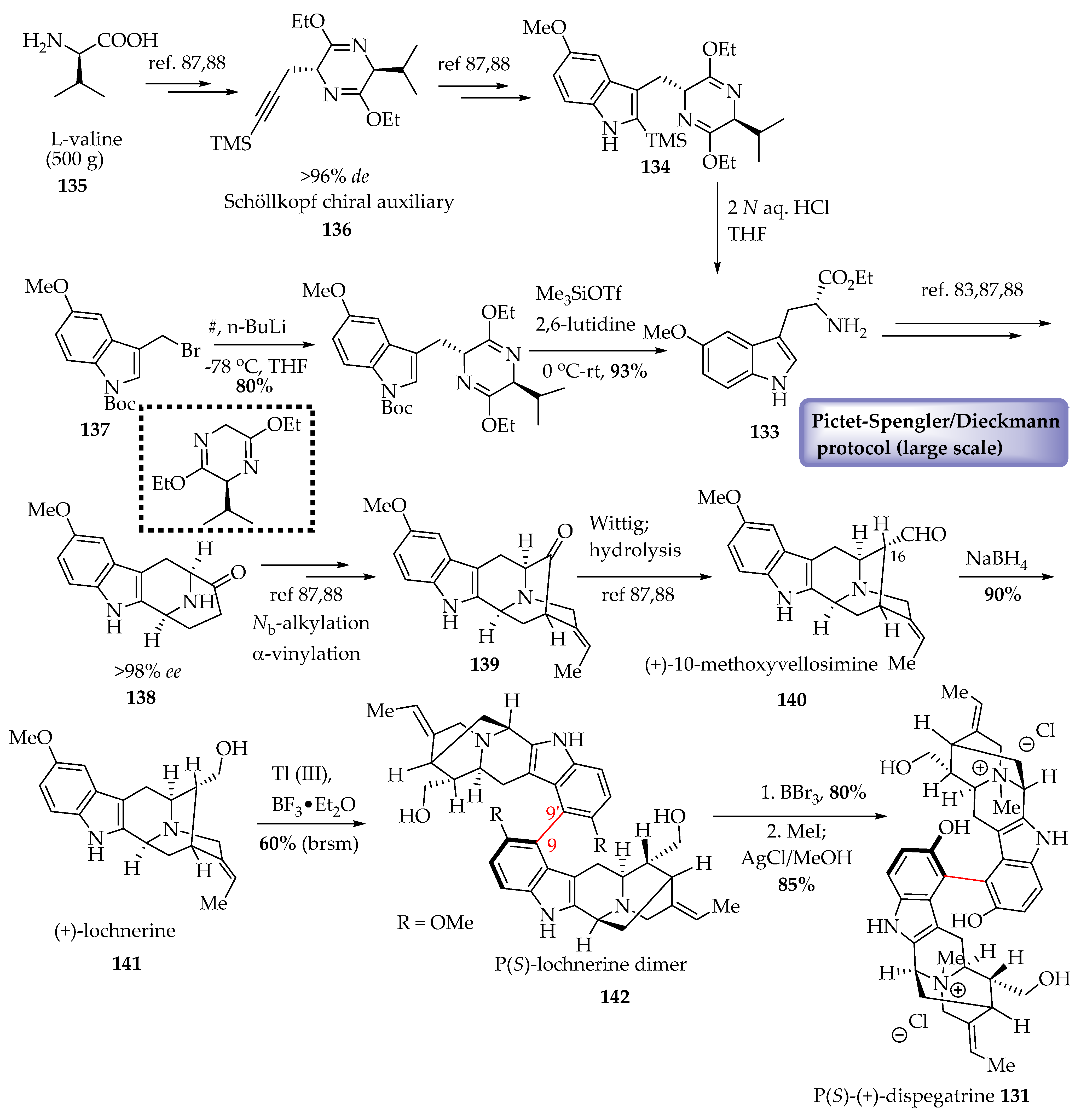
Enantiospecific Total Synthesis of (+)-dispegatrine 131 in >98% ee
It was suggested that internal chiral induction was responsible for the atroposelectivity, which was exploited as an advantage in the first enantiospecific and convergent total synthesis of (+)-dispegatrine 131 from spegatrine 132 by Edwankar et al. [87,88]. The total synthesis of dispegatrine 131 required the 5-methoxy tryptophan unit 133, which was accessed via the general strategy for the synthesis of ring-A oxygenated D-tryptophan unit 134 previously developed in Milwaukee (Scheme 9). L-Valine 135 served as the source of chirality in the Schöllkopf chiral auxiliary 136 and was employed in a large-scale reaction (500 g). After obtaining the required chiral starting material, D-tryptophan ethyl ester 133 (which could also be obtained from the Boc protected 5-methoxy indole 137 (Scheme 9)), the large-scale Pictet–Spengler/Dieckmann protocol developed in Milwaukee was employed to gain access to the key tetracyclic ketone 138 in excellent yield and >98% enantiomeric excess. This tetracyclic ketone 138 furnished the sarpagine system 139 after Nb-alkylation and α-vinylation, both of which could also be performed on large scales. The pentacyclic ketone 139 containing the sarpagine architecture was further functionalized. A one-carbon homologation via a Wittig olefination, followed by hydrolysis under acidic conditions, furnished 10-methoxy vellosimine 140 for the first time (>98% ee). Reduction of the C-16 formyl function with NaBH4 furnished (+)-lochnerine 141 in excellent yield. A thallium (III)-mediated oxidative coupling facilitated by borane trifluoride diethyl etherate as the Lewis acid provided the dimerized lochnerine 142 as the P(S)-atropodiastereomer. The absolute configuration was confirmed by X-ray analysis with a MoKα source at low temperature. At the conclusion of this total synthesis, the final assault, demethylation of the two methoxy groups followed by quaternization of the Nb-nitrogen atoms was performed. Treatment of the lochnerine dimer 142 with boron tribromide etherate furnished the bis-phenolic dimer (not shown) in 80% yield. Quaternization was executed by treating the tertiary amine functions with excess iodomethane, and this was followed by exchange of iodide with chloride as the counter anion with the help of a AgCl additive. This furnished the first and biomimetic total synthesis of the antihypertensive bisindole, the P(S)-atropodiastereomer of (+)-dispegatrine 131 [87,88].
6.3. Biomimetic Total Synthesis of (+)-Macralstonidine 23
(+)-Macralstonidine 23 is composed of a macroline 5 and a Na-methyl sarpagine 124 unit. (+)-Na-Methyl sarpagine 124 (sarpagine unit) was isolated from A. spectabilis [31] and its total synthesis was accomplished by Zhao et al., as discussed earlier [83,84]. In a chemical sense, (+)-macralstonidine 23 should arise from the acid-mediated condensation of macroline 5 with (+)-Na-methyl sarpagine 124. When macroline 5 was stirred with (+)-Na-methyl sarpagine 124 under mildly acidic conditions, the so formed cyclic ketal was identical to the authentic sample of macralstonidine 23 [31]. In this case too, the condensation followed by cyclic ketal formation can be explained by two different mechanisms. The straightforward Michael-type cyclization, which was discussed previously, is shown below (Scheme 10). On the other hand, the Friedel–Crafts alkylation-type mechanism that involved stabilization of the macroline enone with an oxonium anion is somewhat more probable from a chemical sense. As shown in Scheme 10, after initial intramolecular reaction of the C-17 hydroxyl function with the macroline ketone 5, followed by acid-catalyzed dehydration, this process produced the oxonium species, which existed in two resonance stabilized forms (71a and 71b). The electron-rich phenolic sarpagine 124 carbon atom could attack this electrophile from the C-9 position, which was ortho to the phenolic group. No alkylation at the C-11 position was observed. The so formed enol ether underwent acid-mediated rearrangement forming an oxonium intermediate 143, which made it susceptible to nucleophilic attack by the phenolic oxygen atom furnishing the cyclic ketal present in macralstonidine 23. Since macroline 5 and Na-methyl sarpagine 124 have both been synthesized in Milwaukee, this constitutes an enantiospecific total synthesis of (+)-macralstonidine 23 [76,79].
6.4. Biomimetic Total Synthesis of (+)-Macralstonine 24
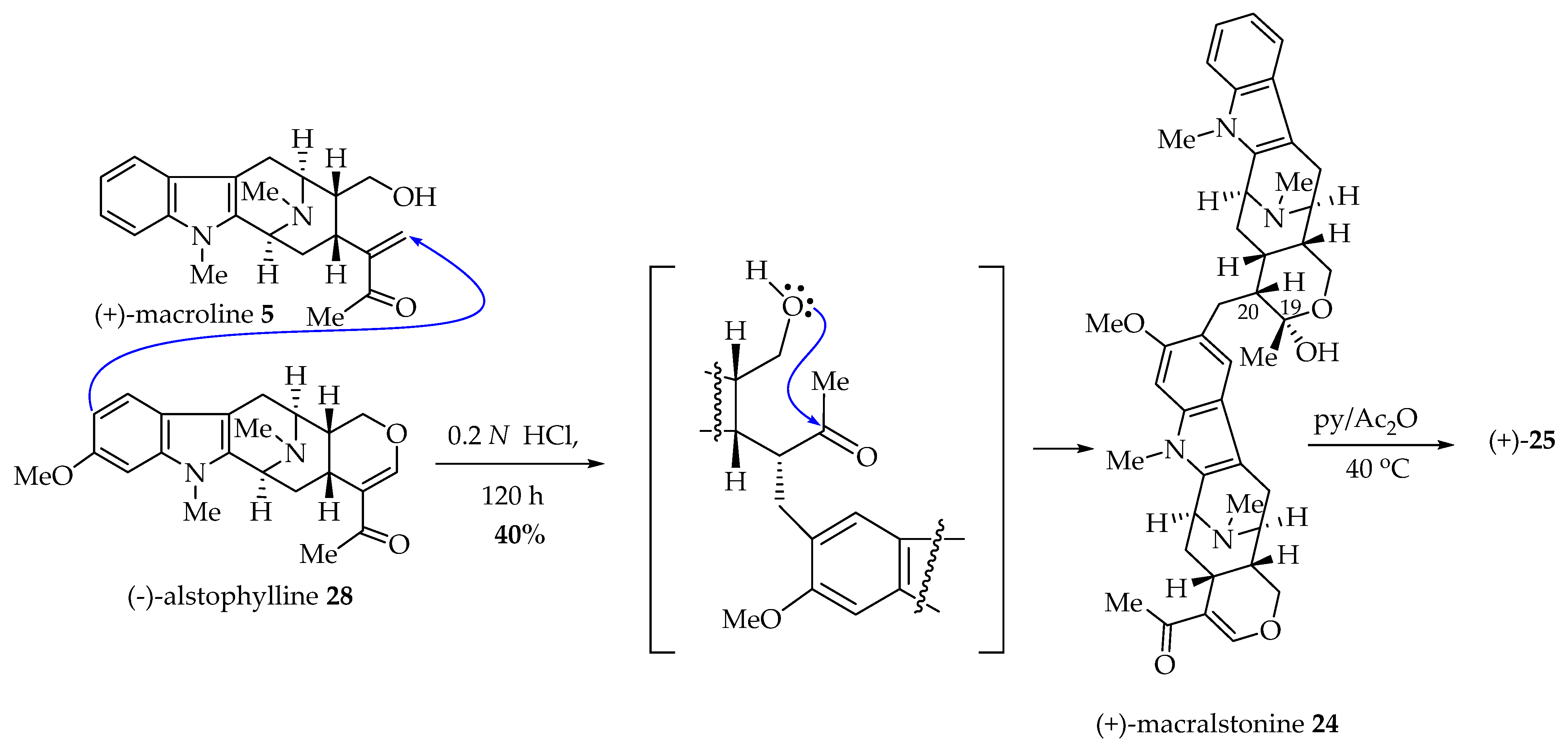
LeQuesne et al. [89] coupled natural (-)-alstophylline 28 and an excess of (+)-macroline 5 in aqueous 0.2N HCl solution at 20 °C for 120 h. The bisindole, (+)-macralstonine 24, was obtained in 40% yield as a mixture of C-20 epimers, which later was converted into O-acetyl macralstonine 25 as the sole product after the treatment with acetic anhydride and pyridine [89]. LeQuesne et al. [89] confirmed the biosynthetic route (Scheme 11) of macralstonine 24 proposed by Schmid et al.; however the Friedel–Crafts alkylation process, as discussed above (Figure 5) is possible as well. Since the total synthesis of both macroline 5 and alstophylline 28 were accomplished by Liao et al., this constitutes the total synthesis of macralstonine 24, and O-acetylmacaralstonine 25 by LeQuesne, Liao, Burke, and Cook et al. [76,89].
6.5. Partial Synthesis of (-)-Macrocarpamine 31
Gan et al. accomplished the partial synthesis of (-)-macrocarpamine 31 in stereospecific and enantiospecific fashion via the coupling reaction between the protonated natural (+)-pleiocarpamine 32 and synthetic (-)-anhydromacrosalhine-methine 33 (Scheme 12) [44]. The portion-wise addition of six equivalents of (-)-anhydromacrosalhine-methine 33 to (+)-pleiocarpamine 32 in 0.2 N dry HCl/THF, followed by basic workup (10% NH4OH), furnished (-)-macrocarpamine 31 in 75% yield [65]. It is important to stir pleiocarpamine 32 in acid to form the iminium ion first and then add (-)-33 to it. Initial treatment of (-)-anhydromacrosalhine-methine 33 and (+)-pleiocarpamine 32 in aqueous and mild acidic conditions (0.2 N HCl) converted enol 33 into its hydrated product, inhibiting the desired product 144 formation. This was reported to be the first coupling reaction between an iminium ion and vinylogous enol ether of such type (33) and the first proton-mediated coupling reaction to obtain a bisindole via the coupling of two monomeric units from the Alstonia genus [65]. The total synthesis of (-)-macrocarpamine 31 has not been reported yet. Since the total synthesis of both of its monomeric units are now successfully completed, the formal total synthesis of (-)-macrocarpamine 31 is completed [17,44].
6.6. Partial Synthesis of (+)-Villalstonine 43
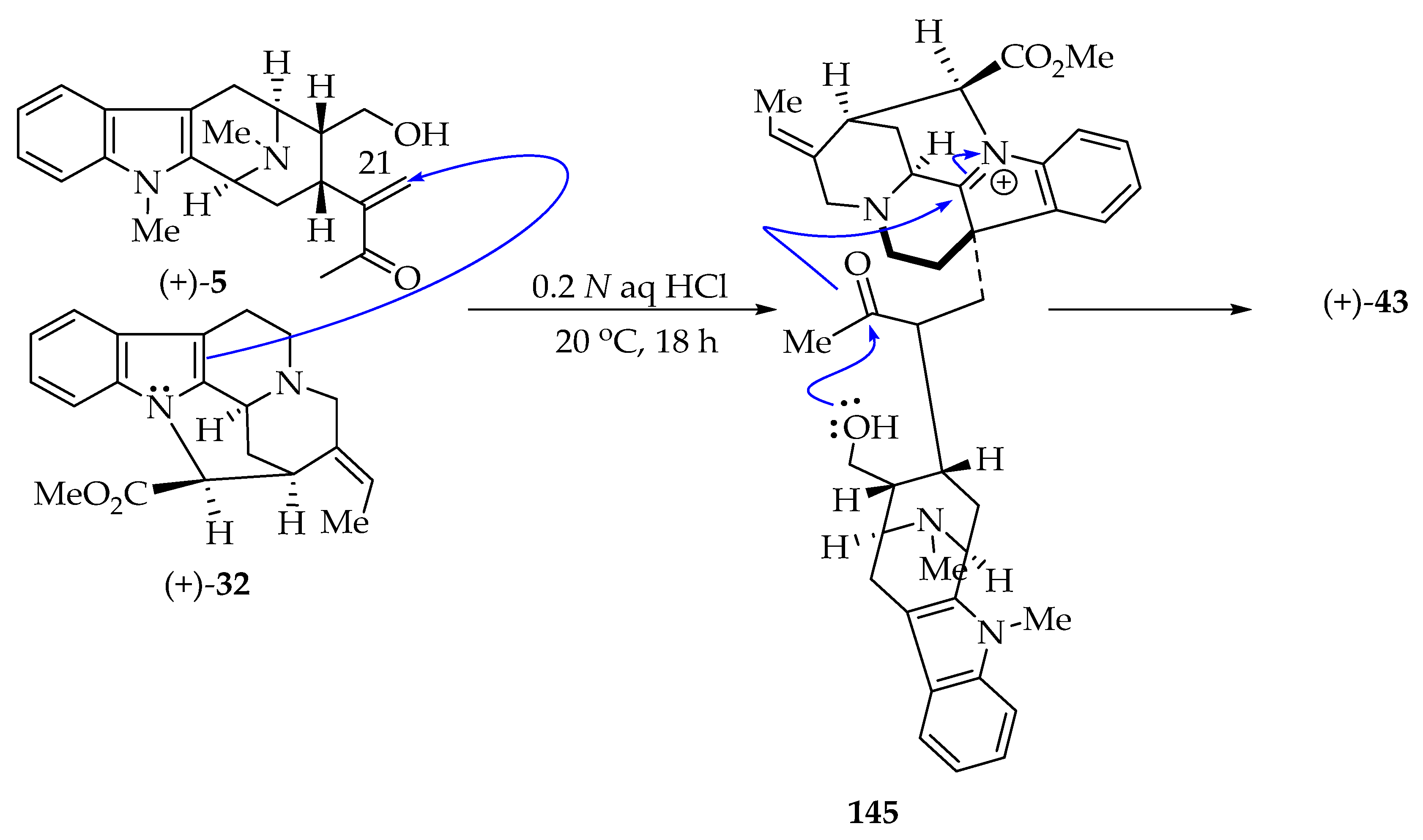
Schmid et al. described the degradation of (+)-villalstonine 43 into two monomeric indole alkaloids, (+)-pleiocarpamine 32 and the macroline base 5 [38]. Furthermore, Schmid anticipated forming a new acetal–amino acetal function in (+)-villalstonine 43 by electrophilic addition of the enone function of macroline 5 to the (+)-pleiocarpamine 32, followed by ring closure. The formal total synthesis of (+)-villalstonine 43 has recently been accomplished, since Sato et al. [17] completed the total synthesis of rac-pleiocarpamine 32 and Liao, Bi, and Zhang prepared macroline 5 [45,90,91]. Initially, LeQuesne, Burke and Cook accomplished the partial synthesis of (+)-villalstonine 43 by a biomimetic-type coupling reaction, which was reported as the first among bisindole alkaloids from the Alstonia genus (Scheme 13 [51]. LeQuesne et al. treated (+)-pleiocarpamine 32 with (+)-macroline 5 in the presence of 0.2 N aqueous hydrochloric acid at 20 °C for 18 h to furnish (+)-villalstonine 43 as a single product on TLC [51]. The macroline 5 has a limited shelf life because it cyclizes intramolecularly to give the more stable dihydroalstonerine 105 (Scheme 14) [6].
The biomimetic synthesis of (+)-villalstonine 43 is thought to begin via electrophilic addition of the macroline enone 5 onto pleiocarpamine 32 from the α-face (less hindered) to give the intermediate 145 (Scheme 13) [51]. The intermediate 145 then forms a hemiacetal, after which the newly formed acetal sequentially attacks the iminium ion of the indoline from the less hindered side to furnish the acetal–amino acetal function in (+)-villalstonine 43 [51].
6.7. Improved Partial Synthesis of (+)-Villalstonine 43
Because macroline 5 under acidic conditions forms part of the very stable dihydroalstonerine 105 (Scheme 14), the process was later modified by using the more stable macroline equivalent 146 [45]. The alcohol functional group in the macroline equivalent 5 was protected as its TBDMS ether 146 [79]. The natural (+)-pleiocarpamine 32 was received from Professor LeQuesne and then treated with the more stable macroline equivalent 146 in the presence of 0.2 N aqueous hydrochloric acid and fluoride ion (tert-butylammonium fluoride) for 24 h at 22 °C (Scheme 15). This procedure was followed by a basic workup with aqueous NH4OH to obtain (+)-villalstonine 43 as an oil in 60% condensation yield. Traces of (+)-pleiocarpamine 32 remained on TLC. The desilylation might occur before condensation to give macroline 146, or the condensation may take place first as shown in Scheme 15 [6]. The spectroscopic data, including IR and NMR, TLC, and mass spectrometry of synthetic (+)-villalstonine 43, were identical when compared to the authentic sample separately isolated from A. muelleriana by LeQuesne and Schmid et al. [38,51]. Since the total synthesis of both of its monomeric units are now successfully completed, the formal total synthesis of (+)-villalstonine 43 is completed [47,78].
7. C-19 Methyl-Substituted Sarpagine/Macroline/Ajmaline Alkaloids
The C-19 methyl-substituted sarpagine/macroline/ajmaline-type alkaloids are an emerging group of structurally and biosynthetically related alkaloids. To date, more than 70 monomeric indole alkaloids that belong to this sub-family have been isolated. In addition to the monomeric bases, several bisindole alkaloids consisting of at least one monomeric unit with a C-19 methyl group as their obligate constituent have been found in various medicinal plants worldwide (vide infra, Table 2). Bioactive C-19 methyl-substituted sarpagine/macroline bases and related alkaloids occur in various Alstonia species. The reported bioactivity of representative examples is listed herein (vide infra, Figure 8). It can be seen in Figure 8 that these alkaloids contain a stereogenic methyl function at the C-19 position and presumably originate from common or similar biosynthetic precursors. As a result, some common intermediates or core architecture should enable the total synthesis of these alkaloids. As illustrated earlier in Figure 7, macroline, sarpagine, and ajmaline alkaloids are essentially biosynthetic relatives, and they contain the azabicyclo[3.3.1]nonane core 93 present in their structures. Thus, any effective and divergent synthetic strategy should involve robust access to the common intermediates, preferably containing the azabicyclo[3.3.1]nonane core system 93. The total synthesis of bisindoles containing the C-19 methyl function has not previously been reported. On the other hand, there has been notable progress in developing synthetic strategies for practical access to this important class of bioactive indole alkaloids [7,62].
Enantiospecific Total Synthesis of C-19 Methyl-Substituted Sarpagine/Macroline Alkaloids via the General Strategy Developed in Milwaukee
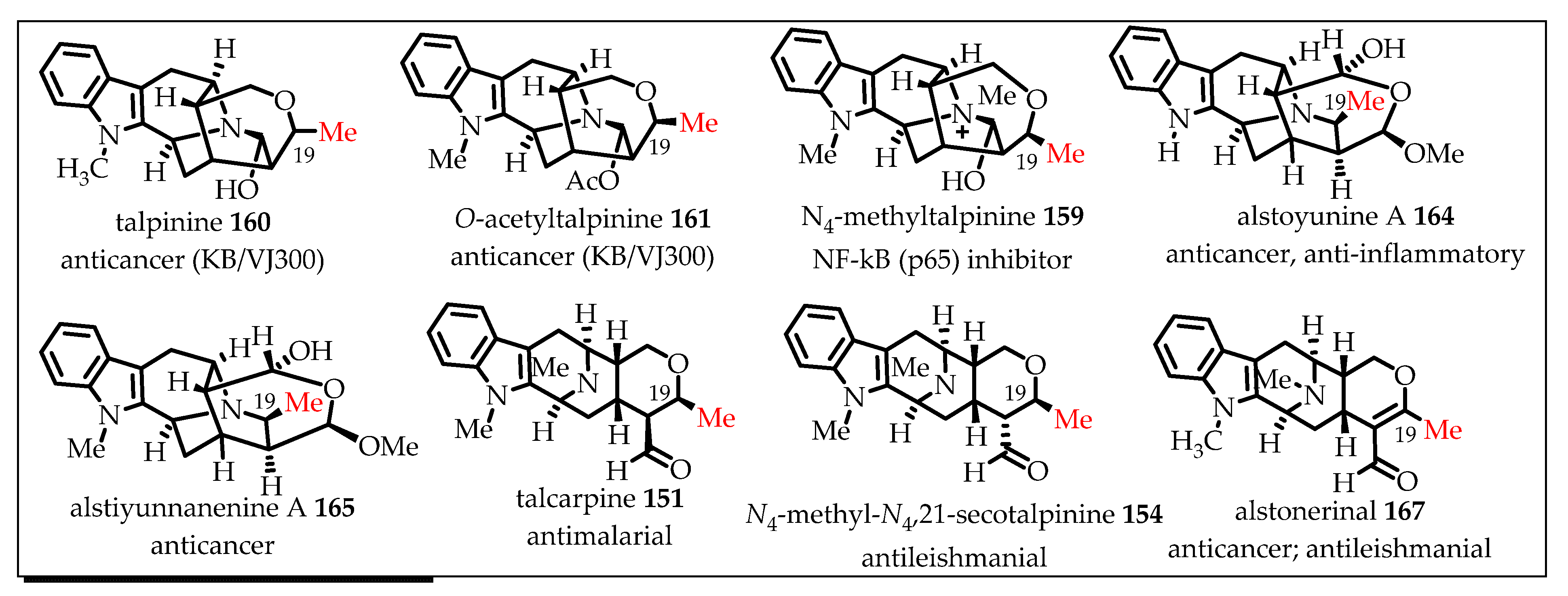
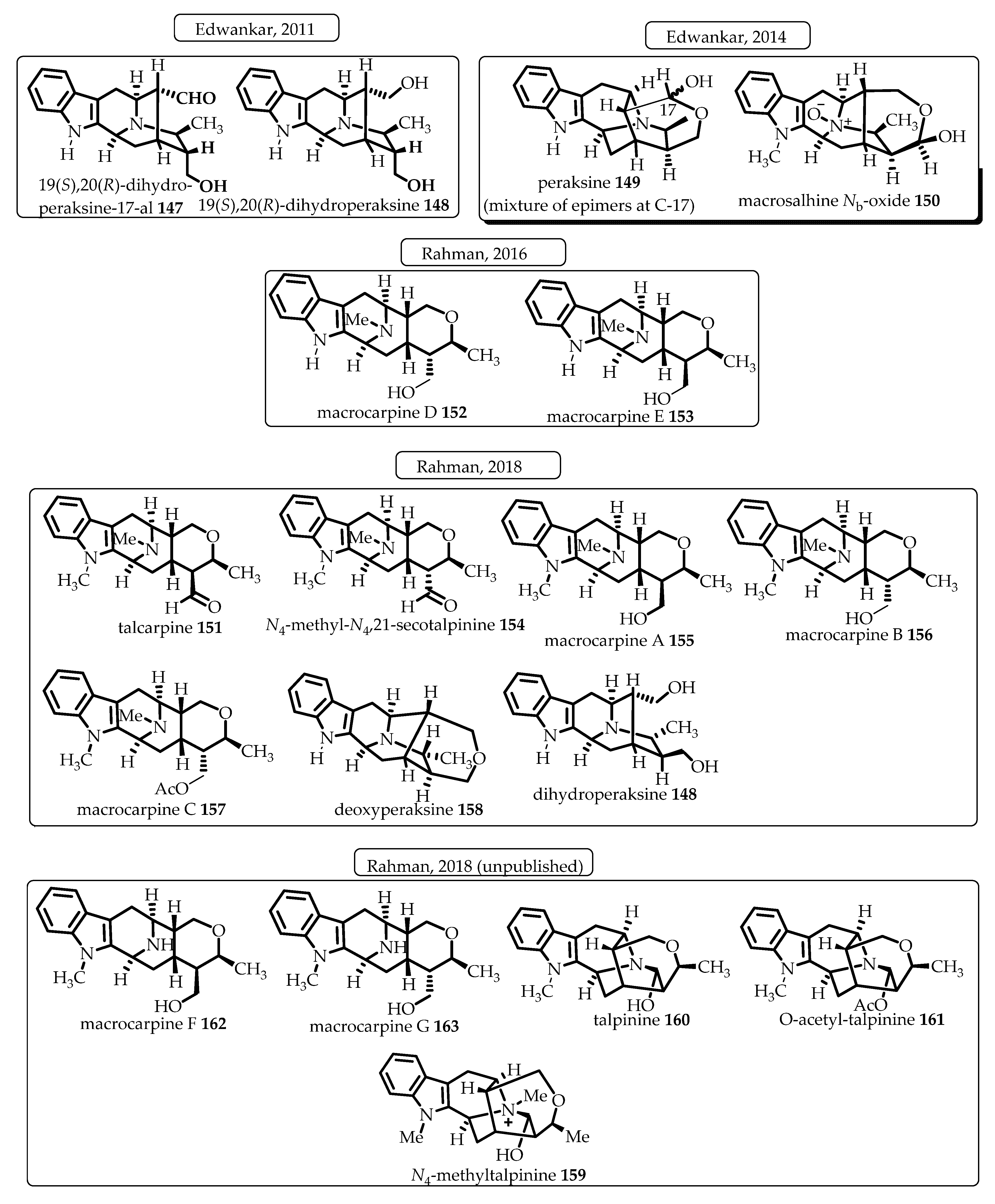
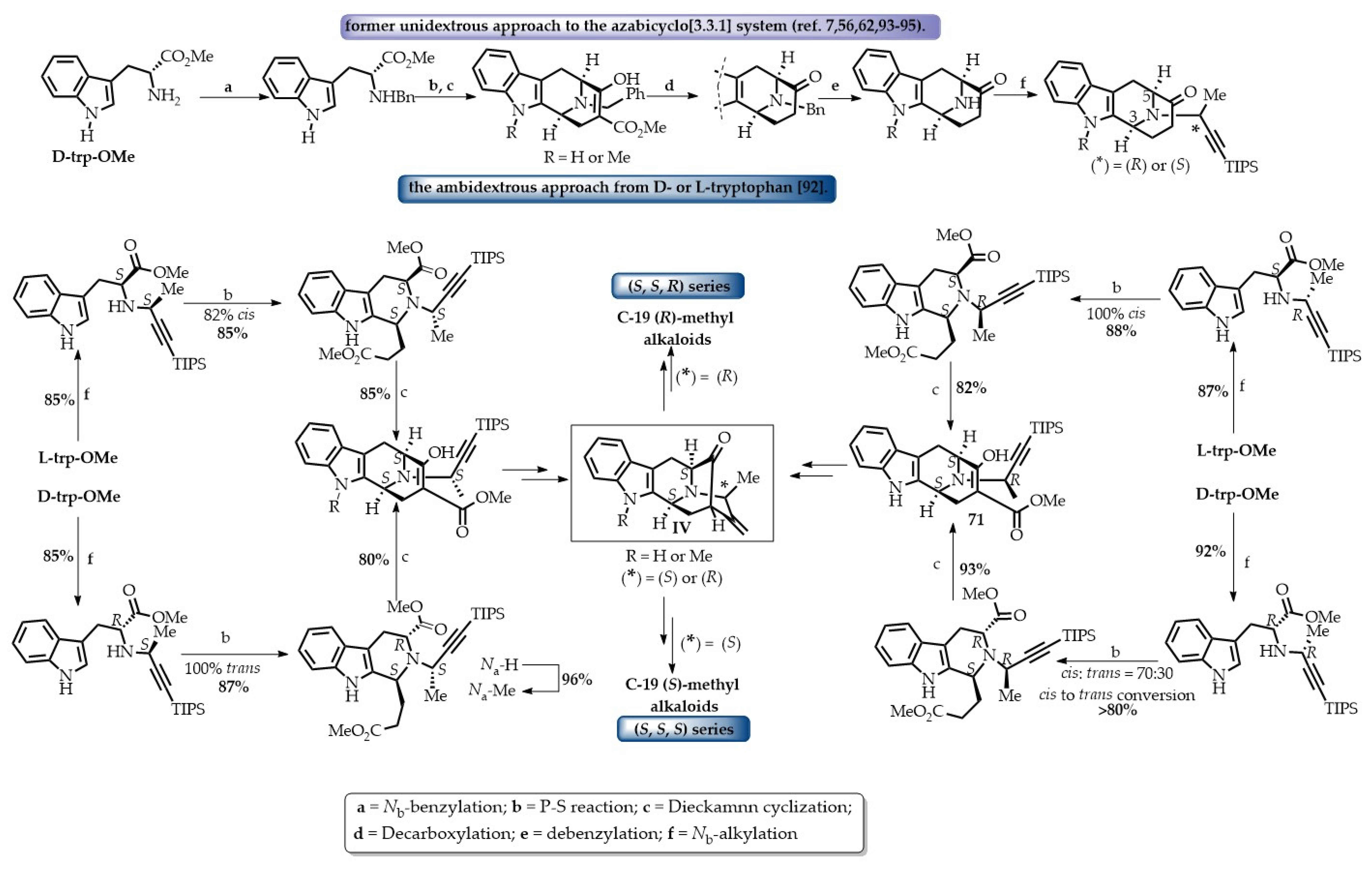
During the development of an improved and shorter access to the tetracyclic ketone containing the azabicyclo[3.3.1]nonane core architecture, Rahman et al. developed an ambidextrous Pictet–Spengler (P-S)/Dieckmann protocol that provided efficient stereospecific access to the desired common intermediates towards the C-19 methyl-substituted sarpagine/macroline-type monomeric indole alkaloids [92]. The key tetracyclic intermediates for access to both the C-19 (S) or (R)-methyl-substituted alkaloids can now be synthesized either from D- or from L-tryptophan, at will, albeit at improved yield and in a route two steps shorter than the former strategy (Scheme 16). Further studies in this vein resulted in an illustration of the applicability of the so developed ambidextrous P-S strategy by accessing both enantiomers (individually) of the azabicyclo[3.3.1]nonane core structures, while starting from the same tryptophan auxiliary. One of the important implications of this ambidextrous P-S/Dieckmann protocol is that not only are the natural alkaloids containing both (S) and (R) C-19 methyl-containing alkaloids available but their enantiomers (unnatural alkaloids) can also be accessed using this strategy from the same chiral auxiliary (Scheme 17). To date, this unified general strategy has been illustrated in the total synthesis of more than a dozen alkaloids from this sub-family (Figure 9) [7,56,62,93,94,95]. A detailed discussion of the reported total synthesis of the individual monomeric alkaloids from this sub-family is out of the scope of this review. The enantiospecific total synthesis of the first example of sarpagine-type alkaloids isolated from the root-culture of R. serpentina that belong to this group was reported by Edwakar et al. [94]. These two monoterpenoid sarpagine alkaloids, 19(S),20(R)-dihydroperaksine-17-al 147 and 19(S),20(R)-dihydroperaksine 148 (Figure 9) represented a novel subgroup of sarpagine alkaloids, identified by Stöckigt et al. [96]. Afterwards, several other alkaloids including peraksine 149, macrosalhine-Nb-oxide 150, and talcarpine 151 (formal synthesis) from this group were reported by employing the general strategy developed in Milwaukee [93]. Later, as a result of continued studies in this vein, Rahman et al. employed an efficient copper-mediated enolate-driven cross-coupling process that replaced the previous palladium-catalyzed coupling to gain an improved access to the core architecture of this subgroup of alkaloids and illustrated the process in the total synthesis of two monomeric macroline alkaloids, macrocarpines D 152 and E 153 [62]. In 2018, Rahman et al. reported an improved strategy for a two-step shorter and higher overall yield to achieve access to the key tetracyclic ketone intermediates. The total synthesis of a number of sarpagine and macroline alkaloids, including talcarpine 151, N4-methyl-N4,21-secotalpinine 154, macrocarpines A–C (155–157), dihydroperaksine 148, and deoxyperaksine 158, was completed and reported in the same paper. This also corrected the reported optical rotations of N4-methyl-N4,21-secotalpinine 154 and macrocarpine A 155 [7]. Afterwards, the focus turned toward the development of the ambidextrous Pictet–Spengler approach for accessing natural alkaloids either from D- or L-tryptophan, at will. Besides, both natural and unnatural enantiomers would be accessible from the same chiral starting material. Several more alkaloids from this subgroup, including the potent NF-kB inhibitor N4-methyltalpinine 159, anticancer alkaloids talpinine 160 and O-acetyl talpinine 161, as well as the macrocarpines F-G (162–163), have been synthesized and will be reported in due course (Figure 9) [97].
Any member of this group would be accessible employing this general strategy, albeit most of these alkaloids await their total synthesis. In addition, no bisindole alkaloid from this group has been synthesized, to date, but the building blocks are now in place. The important bioactivity of a number of alkaloids from this group or their unnatural enantiomers (both monomeric and bisindole) by combining different bioactive monomeric units might result in novel chemical entities, which are unavailable in normal biosynthetic processes. In this respect, the enantiomers of the bioactive monomeric C-19 methylated alkaloids would also be available from either of the tryptophan esters at will. Enantiomers may have similar or even better properties depending on their metabolism, and this holds true for bisindoles, as well as their mismatched pairs.
8. Unnatural (Enantiomers or Synthetic Derivatives) Alkaloids in Drug Discovery
Natural molecules or compounds are usually chiral molecules that are the secondary metabolites used in plants or organisms, perhaps as defense mechanisms. The plants or organisms have evolved to produce only particular enantiomers, which results in the isolation and screening of only particular enantiomers, i.e., the natural enantiomers. Optically pure drugs from natural sources exhibit desired biological activity usually in particular enantiomeric forms, for example, morphine and reserpine [98]. Anticipated pharmacological effects can be achieved by applying absolute stereochemistry or enantioselectivity of compounds in many enzymatic reactions [98]. Logically, the unnatural enantiomers might be as good as the natural enantiomers or even better in activity and toxicity profile, depending on the metabolism. For example, a drug in WHO’s essential list, lamivudine, an unnatural deoxycytidine analog, has saved millions of lives after its approval by the FDA in 1995 [99]. The unnatural L-nucleoside-based drug, emtricitabine, exhibited 100 times greater potency and longer duration of action than its natural D-nucleoside analog [92,99,100]. Among 185 novel small molecules approved by the FDA since 1981 for treatment of cancer, 120 (64.9%) are from a natural source, natural product mimetic, or natural product pharmacophore-inspired synthetic drug [1]. Notably, several C’-20 urea derivates of the robust clinical drug vinblastine are reported to be 10–200 times more active than the natural vinblastine analogs themselves against various human tumor cell lines [101]. When compared to the original compound, unnatural/synthetic drug candidates such as analogs of bryostatin, vincristine, and vinblastine might exhibit excellent biological activity [102,103,104]. The modified vinca alkaloids (fluorine substitution at C10′) 10′-fluorovinblastine and 10′-fluorovincristine exhibited substantially stronger activity than the natural microtubule inhibitors vinblastine and vincristine against the sensitive and vinblastine-resistant tumor cell lines, in the study from the Boger group [105]. Thus, it is crucial to discover the pharmacological effects of derivatives and unnatural enantiomers of natural products.
9. Conclusions
Natural products and nature-inspired molecules are expected to continue playing an indispensable role in modern drug discovery. Compared to their monomeric units, bisindoles are more potent in many cases. Bisindole alkaloids including semisynthetic derivatives have been found to possess significant bioactivity, including anticancer, antileishmanial, and antimalarial properties, and thus are promising leads for nature-inspired drug discovery and development. Unnatural medicinal compounds formed by combining bioactive mismatched monomeric units can furnish novel medicinal compounds. Incorporating unnatural enantiomers of monomeric alkaloids into bisindoles can provide access to novel and unnatural bioactive compounds that may have better activity and in vivo stability depending on their metabolism.
Author Contributions
Conceptualization: J.M.C., M.T.R. and K.P.P.; Writing—original draft: K.P.P.; Writing—review and editing: K.P.P., M.T.R. and J.M.C.; Supervision: J.M.C. All authors have read and agreed to the published version of the manuscript.
Funding
We wish to acknowledge the NIH-NIDA (R21-AA14617) and NIDA (DA043204) for generous financial support. We also thank the Milwaukee Institute for Drug Discovery and the University of Wisconsin-Milwaukee’s Shimadzu Laboratory for Advanced and Applied Analytical Chemistry for help with spectroscopy and the National Science Foundation, Division of Chemistry [CHE-1625735].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural products for drug discovery in the 21st century: Innovations for novel drug discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, A.T.; George, G.; Yadav, N.; Jeswani, A.; Auti, P.S. Pharmaceutical Application of Bio-actives from Alstonia Genus: Current Findings and Future Directions. In Bioactive Natural Products for Pharmaceutical Applications; Pal, D., Nayak, A.K., Eds.; Springer International Publishing: New York, NY, USA, 2021; pp. 463–533. [Google Scholar]
- Yeap, J.S.; Saad, H.M.; Tan, C.H.; Sim, K.S.; Lim, S.H.; Low, Y.Y.; Kam, T.S. Macroline-sarpagine bisindole alkaloids with antiproliferative activity from Alstonia penangiana. J. Nat. Prod. 2019, 82, 3121–3132. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Tiruveedhula, V.V.; Cook, J.M. Synthesis of bisindole alkaloids from the Apocynaceae which contain a macroline or sarpagine unit: A review. Molecules 2016, 21, 1525. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.T.; Deschamps, J.R.; Imler, G.H.; Cook, J.M. Total Synthesis of Sarpagine-Related Bioactive Indole Alkaloids. Chemistry 2018, 24, 2354–2359. [Google Scholar] [CrossRef]
- Tan, M.C.S.; Carranza, M.S.S.; Linis, V.C.; Malabed, R.S.; Oyong, G.G. Antioxidant, cytotoxicity, and antiophidian potential of Alstonia macrophylla bark. ACS Omega 2019, 4, 9488–9496. [Google Scholar] [CrossRef] [Green Version]
- Fielding, B.C.; da Silva Maia Bezerra Filho, C.; Ismail, N.S.M.; de Sousa, D.P. Alkaloids: Therapeutic potential against human coronaviruses. Molecules 2020, 25, 5496. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.E.; Abdul-Hameed, Z.H.; Alotaibi, M.O.; Bawakid, N.O.; Sobahi, T.R.; Abdel-Lateff, A.; Alarif, W.M. Chemical diversity and bioactivities of monoterpene indole alkaloids (MIAs) from six Apocynaceae genera. Molecules 2021, 26, 488. [Google Scholar] [CrossRef]
- Lu, J.J.; Bao, J.L.; Chen, X.P.; Huang, M.; Wang, Y.T. Alkaloids isolated from natural herbs as the anticancer agents. Evid. Based Complement. Altern. Med. 2012, 2012, 485042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvert, M.B.; Sperry, J. Bioinspired total synthesis and structural revision of yuremamine, an alkaloid from the entheogenic plant Mimosa tenuiflora. Chem. Commun. 2015, 51, 6202–6205. [Google Scholar] [CrossRef]
- Namjoshi, O.A.; Cook, J.M. Chapter Two—Sarpagine and Related Alkaloids. In The Alkaloids: Chemistry and Biology; Knӧlker, H.-J., Ed.; Academic Press: San Diego, CA, USA, 2016; Volume 76, pp. 63–169. [Google Scholar]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, D.-B.; Zhou, P.-P.; Zhang, Q.-L.; Zhao, L.; Chen, J.-J.; Gao, K. Rauvomines A and B, two monoterpenoid indole alkaloids from Rauvolfia vomitoria. Org. Lett. 2017, 19, 3998–4001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Z.; Chen, Q.; Yang, G.-F. A review on recent developments of indole-containing antiviral agents. Eur. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kogure, N.; Kitajima, M.; Takayama, H. Total syntheses of pleiocarpamine, normavacurine, and C-mavacurine. Organic Lett. 2019, 21, 3342–3345. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H. Alkaloids from Alstonia Macrophylla. Ph.D. Thesis, University of Malaya, Kuala Lampur, Malaysia, 2013. [Google Scholar]
- Keawpradub, N.; Houghton, P.J. Indole alkaloids from Alstonia macrophylla. Phytochemistry 1997, 46, 757–762. [Google Scholar] [CrossRef]
- Keawpradub, N.; Kirby, G.C.; Steele, J.C.; Houghton, P.J. Antiplasmodial activity of extracts and alkaloids of three Alstonia species from Thailand. Planta Med. 1999, 65, 690–694. [Google Scholar] [CrossRef]
- Elderfield, R.C.; Gilman, R.E. Alkaloids of Alstonia muelleriana. Phytochemistry 1972, 11, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Burke, D.E.; Cook, G.A.; Cook, J.M.; Haller, K.G.; Lazar, H.A.; Le Quesne, P.W. Further alkaloids of Alstonia muelleriana. Phytochemistry 1973, 12, 1467–1474. [Google Scholar] [CrossRef] [Green Version]
- Hoard, L.G. The Crystal Structures of Altstonisidine, C42H48N4O4, and Anhydrous Cholesterol, C27H46O. Ph.D. Thesis, University of Michigan, Ann Arbor, MI, USA, 1977. [Google Scholar]
- Hamaker, L.K.; Cook, J.M. The Synthesis of Macroline Related Sarpagine Alkaloids. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S.W., Ed.; Elsevier Publications: Amsterdam, The Netherlands, 1995; Volume 9. [Google Scholar]
- Kitajima, M.; Takayama, H. Chapter Four-Monoterpenoid Bisindole Alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: San Diego, CA, USA, 2016; Volume 76, pp. 259–310. [Google Scholar]
- Lim, S.-H.; Tan, S.-J.; Low, Y.-Y.; Kam, T.-S. Lumutinines A–D, linearly fused macroline–macroline and macroline–sarpagine bisindoles from alstonia macrophylla. J. Nat. Prod. 2011, 74, 2556–2562. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-H.; Low, Y.-Y.; Subramaniam, G.; Abdullah, Z.; Thomas, N.F.; Kam, T.-S. Lumusidines A− D and villalstonidine F, macroline–macroline and macroline–pleiocarpamine bisindole alkaloids from Alstonia macrophylla. Phytochemistry 2013, 87, 148–156. [Google Scholar] [CrossRef]
- Tan, S.-J.; Lim, K.-H.; Subramaniam, G.; Kam, T.-S. Macroline–sarpagine and macroline–pleiocarpamine bisindole alkaloids from Alstonia angustifolia. Phytochemistry 2013, 85, 194–202. [Google Scholar] [CrossRef]
- Sharp, T.M. 265. The alkaloids of Alstonia barks. Part II. A. macrophylla, wall., A. somersetensis, FM Bailey, A. verticillosa, F. Muell., A. villosa, blum. J. Chem. Soc. 1934, 1227–1232. [Google Scholar] [CrossRef]
- Kishi, T.; Hesse, M.; Vetter, W.; Gemenden, C.; Taylor, W.; Schmid, H. Macralstonin. Helv. Chim. Acta 1966, 49, 946–964. [Google Scholar] [CrossRef]
- Hart, N.; Johns, S.; Lamberton, J. Tertiary alkaloids of Alstonia spectabilis and Alstonia glabriflora (Apocynaceae). Aust. J. Chem. 1972, 25, 2739–2741. [Google Scholar] [CrossRef] [Green Version]
- Keawprdub, N.; Houghton, P.; Eno-Amooquaye, E.; Burke, P. Activity of extracts and alkaloids of Thai Alstonia species against human lung cancer cell lines. Planta Med. 1997, 63, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-H.; Low, Y.-Y.; Tan, S.-J.; Lim, K.-H.; Thomas, N.F.; Kam, T.-S. Perhentidines A–C: Macroline–macroline bisindoles from Alstonia and the absolute configuration of perhentinine and macralstonine. J. Nat. Prod. 2012, 75, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Changwichit, K.; Khorana, N.; Suwanborirux, K.; Waranuch, N.; Limpeanchob, N.; Wisuitiprot, W.; Suphrom, N.; Ingkaninan, K. Bisindole alkaloids and secoiridoids from Alstonia macrophylla Wall. ex G. Don. Fitoterapia 2011, 82, 798–804. [Google Scholar] [CrossRef]
- Cook, J.; Le Quesne, P. Macralstonine from Alstonia muelleriana. Phytochemistry 1971, 10, 437–439. [Google Scholar] [CrossRef] [Green Version]
- Ghedira, K.; Zeches-Hanrot, M.; Richard, B.; Massiot, G.; Le Men-Olivier, L.; Sevenet, T.; Goh, S. Alkaloids of Alstonia angustifolia. Phytochemistry 1988, 27, 3955–3962. [Google Scholar] [CrossRef]
- Cook, J.M.; Le Quesne, P.; Elderfield, R. Alstonerine, a new indole alkaloid from Alstonia muelleriana. J. Chem. Soc. Chem. Commun. 1969, 1306–1307. [Google Scholar] [CrossRef]
- Hesse, M.; Hürzeler, H.; Gemenden, C.; Joshi, B.; Taylor, W.; Schmid, H. Die Struktur des Alstonia-Alkaloides Villalstonin Vorläufige Mitteilung. Helv. Chim. Acta 1965, 48, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Terrazas, C.; Muñoz Acuña, U.; Ninh, T.N.; Chai, H.; Carcache de Blanco, E.J.; Soejarto, D.D.; Satoskar, A.R.; Kinghorn, A.D. Bioactive indole alkaloids isolated from Alstonia angustifolia. Phytochem. Lett. 2014, 10, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordman, C.; Kumra, S. The structure of villalstonine1. J. Am. Chem. Soc. 1965, 87, 2059–2060. [Google Scholar] [CrossRef]
- Buckingham, J.; Baggaley, K.H.; Roberts, A.D.; Szabo, L.F. Dictionary of Alkaloids with CD-ROM; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Keawpradub, N.; Eno-Amooquaye, E.; Burke, P.; Houghton, P. Cytotoxic activity of indole alkaloids from Alstonia macrophylla. Planta Med. 1999, 65, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Allen, D.; Cai, Y.; Phillipson, J.; Said, I.; Kirby, G.; Warhurst, D. In vitro antiamoebic and antiplasmodial activities of alkaloids isolated from Alstonia angustifolia roots. Phytother. Res. 1992, 6, 121–124. [Google Scholar] [CrossRef]
- Gan, T.; Cook, J.M. General approach for the synthesis of macroline/sarpagine related indole alkaloids via the asymmetric Pictet-Spengler reaction: The enantiospecific synthesis of (−)-anhydromacrosalhine-methine. Tetrahedron Lett. 1996, 37, 5033–5036. [Google Scholar] [CrossRef]
- Bi, Y.; Zhang, L.-H.; Hamaker, L.K.; Cook, J.M. Enantiospecific synthesis of (-)-alstonerine and (+)-macroline as well as a partial synthesis of (+)-villalstonine. J. Am. Chem. Soc. 1994, 116, 9027–9041. [Google Scholar] [CrossRef]
- Mayerl, F.; Hesse, M. Macrocarpamin, ein neues Bisindolalkaloid aus Alstonia macrophylla WALL. 167. Mittelung über organische Naturstoffe. Helv. Chim. Acta 1978, 61, 337–351. [Google Scholar] [CrossRef]
- Khan, Z.M.; Hesse, M.; Schmid, H. Die Struktur des quartären Alkaloides Macrosalhin. Helv. Chim. Acta 1967, 50, 1002–1010. [Google Scholar] [CrossRef]
- Kump, W.; Schmid, H. Über die alkaloide von Pleiocarpa mutica BENTH. Helv. Chim. Acta 1961, 44, 1503–1516. [Google Scholar] [CrossRef]
- Kam, T.-S.; Subramaniam, G.; Chen, W. Alkaloids from Kopsia dasyrachis. Phytochemistry 1999, 51, 159–169. [Google Scholar] [CrossRef]
- Bartlett, M.; Sklar, R.; Smith, A.; Taylor, W. The alkaloids of Hunteria eburnea Pichon. III. 1 The tertiary bases. J. Org. Chem. 1963, 28, 2197–2199. [Google Scholar] [CrossRef]
- Burke, D.E.; Cook, J.M.; Le Quesne, P. Biomimetic synthesis of the bisindole alkaloids villalstonine and alstonisidine. J. Am. Chem. Soc. 1973, 95, 546–552. [Google Scholar] [CrossRef]
- Kam, T.-S.; Tan, S.-J.; Ng, S.-W.; Komiyama, K. Bipleiophylline, an unprecedented cytotoxic bisindole alkaloid constituted from the bridging of two indole moieties by an aromatic spacer unit. Org. Lett. 2008, 10, 3749–3752. [Google Scholar] [CrossRef]
- Jiménez, J.-M.; Zulaica, E.; Bennasar, M.-L.; Bosch, J. A new synthetic entry to the alkaloids of the mavacurine group. First total synthesis of (±)-2, 7-dihydropleiocarpamine. J. Chem. Soc. Chem. Commun. 1993, 732–733. [Google Scholar] [CrossRef]
- Jarret, M.; Tap, A.; Kouklovsky, C.; Poupon, E.; Evanno, L.; Vincent, G. Bioinspired oxidative cyclization of the geissoschizine skeleton for the total synthesis of (-)-17-nor-excelsinidine. Angew. Chem. Int. Ed. 2018, 57, 12294–12298. [Google Scholar] [CrossRef] [PubMed]
- Le Men, J.; Taylor, W.I. A uniform numbering system for indole alkaloids. Experientia 1965, 21, 508–510. [Google Scholar] [CrossRef]
- Rahman, M.T.; Cook, J.M. The C-19 Methyl Substituted Sarpagine-Macroline-Ajmaline Alkaloids: Diversity, Occurrence, Bioactivity, and Synthesis. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Ruppert, M.; Woll, J.; Giritch, A.; Genady, E.; Ma, X.; Stöckigt, J. Functional expression of an ajmaline pathway-specific esterase from Rauvolfia in a novel plant-virus expression system. Planta 2005, 222, 888–898. [Google Scholar] [CrossRef]
- Mattern-Dogru, E.; Ma, X.; Hartmann, J.; Decker, H.; Stöckigt, J. Potential activ-site residues in polyneuridine aldehyde esterase, a central enzyme of indole alkaloid biosynthesis, by modelling and site-directed mutagenesis. Eur. J. Biochem. 2002, 269, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Pfitzner, A.; Stöckigt, J. Biogenetic link between sarpagine and ajmaline type alkaloids. Tetrahedron Lett. 1983, 24, 5197–5200. [Google Scholar] [CrossRef]
- Wu, F.; Kerčmar, P.; Zhang, C.; Stöckigt, J. Sarpagan-Ajmalan-Type Indoles: Biosynthesis, structural biology, and chemo-enzymatic significance. In The Alkaloids: Chemistry and Biology; Knӧlker, H.-J., Ed.; Academic Press: San Diego, CA, USA, 2016; Volume 76, pp. 1–61. [Google Scholar]
- Esmond, R.W.; Le Quesne, P.W. Biomimetic synthesis of macroline. J. Am. Chem. Soc. 1980, 102, 7116–7117. [Google Scholar] [CrossRef]
- Rahman, M.T.; Deschamps, J.R.; Imler, G.H.; Schwabacher, A.W.; Cook, J.M. Total synthesis of macrocarpines D and E via an enolate-driven copper-mediated cross-coupling process: Replacement of catalytic palladium with copper iodide. Org. Lett. 2016, 18, 4174–4177. [Google Scholar] [CrossRef] [Green Version]
- Edwankar, C.R.; Edwankar, R.V.; Namjoshi, O.A.; Rallapalli, S.K.; Yang, J.; Cook, J.M. Recent progress in the total synthesis of indole alkaloids. Curr. Opin. Drug Discov. Dev. 2009, 12, 752–771. [Google Scholar]
- Edwankar, C.R. Part. I: The First Regio-and Atropdiastereoselective Total Synthesis of the Dimeric Indole Alkaloid (+)-Dispegatrine, as Well as The First Total Synthesis of the Sarpagine Alkaloids (+)-Spegatrine, Lochvinerine,(+)-Lochneram and an Improved Total Synthesis of (+)-10-Methoxyvellosimine,(+)-Lochnerine and (+)-Sarpagine. Part. II: Studies Directed toward the Total Synthesis of The Carbon-19 Methyl Substituted Sarpagine-Macroline Alkaloids (+)-Macro-salhine Chloride as Well as Macrocarpine A, B and C. Ph.D. Thesis, The University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2011. [Google Scholar]
- Gan, T.; Cook, J.M. Partial synthesis of the antiamoebic bisindole alkaloid (−)-macrocarpamine. Tetrahedron Lett. 1996, 37, 5037–5038. [Google Scholar] [CrossRef]
- Liao, X.; Zhou, H.; Wearing, X.Z.; Ma, J.; Cook, J.M. The first regiospecific, enantiospecific total synthesis of 6-oxoalstophylline and an improved total synthesis of alstonerine and alstophylline as well as the bisindole alkaloid macralstonine. Org. Lett. 2005, 7, 3501–3504. [Google Scholar] [CrossRef]
- Kam, T.-S.; Choo, Y.-M.; Komiyama, K. Unusual spirocyclic macroline alkaloids, nitrogenous derivatives, and a cytotoxic bisindole from Alstonia. Tetrahedron 2004, 60, 3957–3966. [Google Scholar] [CrossRef]
- Kishi, T.; Hesse, M.; Gemenden, C.; Taylor, W.; Schmid, H. Alstophyllin, ein neues indolalkaloid aus Alstonia macrophylla WALL. Helv. Chim. Acta 1965, 48, 1349–1362. [Google Scholar] [CrossRef]
- Liu, X.; Deschamp, J.R.; Cook, J.M. Regiospecific, enantiospecific total synthesis of the alkoxy-substituted indole bases, 16-e pi-N a-methylgardneral, 11-methoxyaffinisine, and 11-methoxymacroline as well as the indole alkaloids alstophylline and macralstonine. Org. Lett. 2002, 4, 3339–3342. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.S.; Hamaker, L.K.; La Loggia, A.J.; Cook, J.M. Entry into 6-methoxy-D (+)-tryptophans. Stereospecific synthesis of 1-benzenesulfonyl-6-methoxy-D (+)-tryptophan ethyl ester. Synth. Commun. 1992, 22, 2077–2102. [Google Scholar] [CrossRef]
- Ma, C.; Liu, X.; Li, X.; Flippen-Anderson, J.; Yu, S.; Cook, J.M. Efficient asymmetric synthesis of biologically important tryptophan analogues via a palladium-mediated heteroannulation reaction. J. Org. Chem. 2001, 66, 4525–4542. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J.; Nagashima, H.; Hori, K. A new preparative method for 1, 3-dicarbonyl compounds by the regioselective oxidation of alpha, beta-unsaturated carbonyl compounds, catalyzed by PdCl2 using hydroperoxides as the reoxidant of Pd0. Chem. Lett. 1980, 9, 257–260. [Google Scholar] [CrossRef]
- Takayama, H.; Phisalaphong, C.; Kitajima, M.; Aimi, N.; Sakai, S.-I. An efficient synthetic pathway to the macroline-type indole alkaloids, talcarpine and alstonerine from ajmaline. Tetrahedron 1991, 47, 1383–1392. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Bi, Y.-Z.; Yu, F.-X.; Menzia, G.; Cook, J.M. Stereospecificity in the Pictet-Spengler reaction. Enantiospecific synthesis of (6S, 10S)-5# 75)-5-methyl-9-oxo-12-benzyl-6, 7, 8, 9, 10, 11-hexahydro-6, 10-imino-5H-cyclooct [b] indole, a template for preparation of macroline/sarpagine alkaloids. Heterocycles 1992, 34, 517–547. [Google Scholar]
- Zhang, L.; Cook, J. General approach to the synthesis of macroline-related alkaloids. Stereospecific total synthesis of (-)-alstonerine. J. Am. Chem. Soc. 1990, 112, 4088–4090. [Google Scholar] [CrossRef]
- Liao, X.; Zhou, H.; Yu, J.; Cook, J.M. An improved total synthesis of (+)-macroline and alstonerine as well as the formal total synthesis of (−)-talcarpine and (−)-anhydromacrosalhine− methine. J. Org. Chem. 2006, 71, 8884–8890. [Google Scholar] [CrossRef]
- Gorman, M.; Sweeny, J. Perivine. Tetrahedron Lett. 1964, 5, 3105–3111. [Google Scholar] [CrossRef]
- Neukomm, G.; Kletzhäundler, E.; Hesse, M. Die absolute konfiguration von macrolin, einem abbauprodukt des alkaloides villalstonin 179. Mitteilung über organische naturstoffe. Helv. Chim. Acta 1981, 64, 90–96. [Google Scholar] [CrossRef]
- Bi, Y.; Cook, J.M. General approach for the synthesis of macroline/sarpagine alkaloids. The total synthesis of (+)-macroline. Tetrahedron Lett. 1993, 34, 4501–4504. [Google Scholar] [CrossRef]
- Hesse, M.; Bodmer, F.; Gemenden, C.; Joshi, B.; Taylor, W.; Schmid, H. Die struktur des Alstonia-alkaloides villalstonin. Helv. Chim. Acta 1966, 49, 1173–1182. [Google Scholar] [CrossRef]
- Tran, Y.S.; Kwon, O. An application of the phosphine-catalyzed [4 + 2] annulation in indole alkaloid synthesis: Formal syntheses of (±)-alstonerine and (±)-macroline. Org. Lett. 2005, 7, 4289–4291. [Google Scholar] [CrossRef]
- Kadam, V.D.; Rao, B.S.S.; Mahesh, S.; Chakraborty, M.; Vemulapalli, S.P.B.; Dayaka, S.N.; Sudhakar, G. Stereoselective access to the core structure of macroline-type indole alkaloids: Total synthesis of macroline and alstomicine. Org. Lett. 2018, 20, 4782–4786. [Google Scholar] [CrossRef]
- Zhao, S.; Liao, X.; Wang, T.; Flippen-Anderson, J.; Cook, J.M. The enantiospecific, stereospecific total synthesis of the ring-A oxygenated sarpagine indole alkaloids (+)-majvinine,(+)-10-methoxyaffinisine, and (+)-N a-methylsarpagine, as well as the total synthesis of the Alstonia bisindole alkaloid macralstonidine. J. Org. Chem. 2003, 68, 6279–6295. [Google Scholar] [CrossRef]
- Zhao, S.; Liao, X.; Cook, J.M. Enantiospecific, stereospecific total synthesis of (+)-majvinine,(+)-10-methoxyaffinisine, and (+)-N a-methylsarpagine as well as the total synthesis of the alstonia bisindole macralstonidine. Org. Lett. 2002, 4, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Burke, D.E.; Cook, J.M.; Le Quesne, P. Biomimetic synthesis and structure of the bisindole alkaloid alstonisidine. J. Chem. Soc. Chem. Commun. 1972, 697. [Google Scholar] [CrossRef]
- Lin, M.; Yang, B.-Q.; Yu, D.-Q. Studies on the quaternary alkaloids of Rauvolfia verticillata (lour.) Baill var. Hainanensis Tsiang. Acta Pharmacol. Sin. 1986, 21, 114–118. [Google Scholar]
- Edwankar, C.R.; Edwankar, R.V.; Deschamps, J.R.; Cook, J.M. Nature-inspired stereospecific total synthesis of p-(+)-dispegatrine and four other monomeric sarpagine indole alkaloids. Angew. Chem. Int. Ed. 2012, 51, 11762–11765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwankar, C.R.; Edwankar, R.V.; Namjoshi, O.A.; Liao, X.; Cook, J.M. Stereospecific approach to the synthesis of ring-A oxygenated sarpagine indole alkaloids. Total synthesis of the dimeric indole alkaloid P-(+)-dispegatrine and six other monomeric indole alkaloids. J. Org. Chem. 2013, 78, 6471–6487. [Google Scholar] [CrossRef] [Green Version]
- Burke, D.E.; DeMarkey, C.A.; Le Quesne, P.; Cook, J.M. Biomimetic synthesis of the bis-indole alkaloid macralstonine. J. Chem. Soc. Chem. Commun. 1972, 1346–1347. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Cook, J.M. Pictet-Spengler reactions in aprotic media: Nb-benzyl promoted retention of optical activity in the synthesis of an indolo substituted azabicyclo[3.3.1]nonane, a key template for the synthesis of macroline alkaloids. Heterocycles 1988, 27, 2795–2802. [Google Scholar] [CrossRef]
- Liao, X. The First Total Synthesis of The Indole Alkaloids, Macralstonidine, 6-Oxoalstophylline, 10-Methoxyvellosimine, Lochnerine, Sarpagine and an Improved Total Synthesis of Macralstonine and Macroline, as Well as The Formal Total Synthesis of Dispegatrine. Ph.D. Thesis, University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2007. [Google Scholar]
- Rahman, M.T.; Cook, J.M. The ambidextrous Pictet–Spengler reaction: Access to the (+)- or (–)-enantiomers of the bioactive C-19 methyl-substituted sarpagine/macroline/ajmaline alkaloids from either d- or l-tryptophan. Synthesis 2019, 51, 1980–1988. [Google Scholar] [CrossRef]
- Edwankar, R.V.; Edwankar, C.R.; Deschamps, J.R.; Cook, J.M. General strategy for synthesis of C-19 methyl-substituted sarpagine/macroline/ajmaline indole alkaloids including total synthesis of 19 (S), 20 (R)-dihydroperaksine, 19 (S), 20 (R)-dihydroperaksine-17-al, and peraksine. J. Org. Chem. 2014, 79, 10030–10048. [Google Scholar] [CrossRef] [PubMed]
- Edwankar, R.V.; Edwankar, C.R.; Deschamps, J.; Cook, J.M. Regiospecific, enantiospecific total synthesis of C-19 methyl substituted sarpagine alkaloids dihydroperaksine-17-al and dihydroperaksine. Org. Lett. 2011, 13, 5216–5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.T.; Cook, J.M. Unprecedented stereocontrol in the synthesis of 1, 2, 3-trisubstituted tetrahydro-β-carbolines via a new asymmetric Pictet—Spengler reaction towards sarpagine-type indole alkaloids. Eur. J. Org. Chem. 2018, 2018, 3224–3229. [Google Scholar] [CrossRef]
- Sheludko, Y.; Gerasimenko, I.; Kolshorn, H.; Stöckigt, J. New alkaloids of the sarpagine group from Rauvolfia serpentina hairy root culture. J. Nat. Prod. 2002, 65, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T. Shorter and Improved Access to the Key Tetracyclic Core of Sarpagine-Macroline-Ajmaline Indole Alkaloids: The Total Synthesis of Alkaloids Macrocarpines Ag, Talcarpine, N (4)-methyl-n (4), 21-secotalpinine, Deoxyperaksine, Dihydroperaksine, Talpinine, O-acetyltalpinine, and N (4)-methyltalpinine. Ph.D. Thesis, University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2018. [Google Scholar]
- Brossi, A.; Pei, X.-F. Chapter 3 Biological Activity of Unnatural Alkaloid Enantiomers. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: San Diego CA, USA, 1998; Volume 50, pp. 109–139. [Google Scholar]
- Liotta, D.C.; Painter, G.R. Discovery and development of the anti-human immunodeficiency virus drug, emtricitabine (Emtriva, FTC). Acc. Chem. Res. 2016, 49, 2091–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.L.; Rower, J.E. Zidovudine and Lamivudine for HIV Infection. Clin. Med. Rev. Ther. 2010, 2, a2004. [Google Scholar]
- Barker, T.J.; Duncan, K.K.; Otrubova, K.; Boger, D.L. Potent vinblastine C20′ ureas displaying additionally improved activity against a vinblastine-resistant cancer cell cine. ACS Med. Chem. Lett. 2013, 4, 985–988. [Google Scholar] [CrossRef]
- Wender, P.A.; DeChristopher, B.A.; Schrier, A.J. Efficient synthetic access to a new family of highly potent bryostatin analogues via a Prins-driven macrocyclization strategy. J. Am. Chem. Soc. 2008, 130, 6658–6659. [Google Scholar] [CrossRef] [Green Version]
- Carney, D.W.; Lukesh, J.C.; Brody, D.M.; Brütsch, M.M.; Boger, D.L. Ultrapotent vinblastines in which added molecular complexity further disrupts the target tubulin dimer–dimer interface. Proc. Natl. Acad. Sci. USA 2016, 113, 9691–9698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Colby, D.A.; Seto, S.; Va, P.; Tam, A.; Kakei, H.; Rayl, T.J.; Hwang, I.; Boger, D.L. Total synthesis of vinblastine, vincristine, related natural products, and key structural analogues. J. Am. Chem. Soc. 2009, 131, 4904–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, H.; Duncan, K.K.; Robertson, W.M.; Boger, D.L. 10′-fluorovinblastine and 10′-fluorovincristine: Synthesis of a key series of modified Vinca alkaloids. ACS Med. Chem. Lett. 2011, 2, 948–952. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Proposed biogenetic pathway to (+)-angustilongine G 8 and (+)-angustilongine H 9.

Figure 2.
Proposed biogenetic pathway to (−)-angustilongine E 6, (−)-angustilongine F 7, and (+)-angustilongine J 10.
Figure 2.
Proposed biogenetic pathway to (−)-angustilongine E 6, (−)-angustilongine F 7, and (+)-angustilongine J 10.

Figure 3.
Putative biogenetic pathway to (-)-lumusidines A–D (14–17).

Figure 4.
Putative biogenetic pathway to lumutinines A–D (16–19).

Figure 5.
Possible alternative mechanism of bisindole formation via a Friedel–Crafts alkylation process for (+)-macralstonine 24 and (+)-lumutinine A 18, respectively, as representative examples.
Figure 5.
Possible alternative mechanism of bisindole formation via a Friedel–Crafts alkylation process for (+)-macralstonine 24 and (+)-lumutinine A 18, respectively, as representative examples.

Figure 6.
Pyrolysis of (-)-macrocarpamine 31 and macrosalhine 75 to generate key olefin 33.

Scheme 1.
Synthesis of rac-pleiocarpamine 32 and 16-epi-pleiocarpamine 85 by Sato et al.

Figure 7.
Abbreviated chemical and biosynthetic relations among the sarpagine/macroline/ajmaline family of alkaloids.
Figure 7.
Abbreviated chemical and biosynthetic relations among the sarpagine/macroline/ajmaline family of alkaloids.

Scheme 2.
An improved total synthesis of (-)-alstophylline 28 using modified Wacker (Pd II) conditions.
Scheme 2.
An improved total synthesis of (-)-alstophylline 28 using modified Wacker (Pd II) conditions.

Scheme 3.
Enantiospecific total synthesis of (-)-anhydromacrosalhine-methine 33 via the asymmetric Pictet–Spengler reaction.
Scheme 3.
Enantiospecific total synthesis of (-)-anhydromacrosalhine-methine 33 via the asymmetric Pictet–Spengler reaction.

Scheme 4.
Improved synthesis of (+)-macroline 5.

Scheme 5.
Stereoselective total synthesis of macroline 5 and alstomicine 123.

Scheme 6.
Enantiomeric total synthesis of (+)-majvinine 128 and (+)-Na-methylsarpagine 124.

Scheme 7.
Biomimetic partial synthesis of (-)-alstonisidine 3.

Scheme 8.
Biomimetic partial synthesis of (+)-dispegatrine 131 by oxidative phenolic coupling by Yu et al. [86].
Scheme 8.
Biomimetic partial synthesis of (+)-dispegatrine 131 by oxidative phenolic coupling by Yu et al. [86].

Scheme 9.
The biomimetic total synthesis of the natural P(S)-atropodiastereomer of (+)-dispegatrine 131.
Scheme 9.
The biomimetic total synthesis of the natural P(S)-atropodiastereomer of (+)-dispegatrine 131.

Scheme 10.
Biomimetic total synthesis of macralstonidine 23 by the condensation of macroline 5 with Na-methyl sarpagine 124 via two possible mechanisms.
Scheme 10.
Biomimetic total synthesis of macralstonidine 23 by the condensation of macroline 5 with Na-methyl sarpagine 124 via two possible mechanisms.

Scheme 11.
The biomimetic total synthesis of macralstonine 24, and O-acetylmacralstonine 25.

Scheme 12.
Partial synthesis of (-)-macrocarpamine 31.

Scheme 13.
Biomimetic partial synthesis of (+)-villalstonine 43.

Scheme 14.
Intramolecular cyclization of macroline 5 into dihydroalstonerine 105.

Scheme 15.
Improved partial synthesis of (+)-villalstonine 43.

Figure 8.
Representative examples of C-19 methyl-substituted monomeric sarpagine/macroline-related alkaloids from Alstonia species and their bioactivity.
Figure 8.
Representative examples of C-19 methyl-substituted monomeric sarpagine/macroline-related alkaloids from Alstonia species and their bioactivity.

Scheme 16.
Improved general strategy toward the tetracyclic ketone intermediates of C-19 methyl-substituted alkaloids.
Scheme 16.
Improved general strategy toward the tetracyclic ketone intermediates of C-19 methyl-substituted alkaloids.

Scheme 17.
Access to both natural and unnatural enantiomers of C-19 methyl-substituted sarpagine/macroline indole alkaloids from the same chiral tryptophan.
Scheme 17.
Access to both natural and unnatural enantiomers of C-19 methyl-substituted sarpagine/macroline indole alkaloids from the same chiral tryptophan.

Figure 9.
Examples of C-19 methylated alkaloids synthesized employing the general strategy developed herein.
Figure 9.
Examples of C-19 methylated alkaloids synthesized employing the general strategy developed herein.

Table 1.
Indole alkaloids (monomeric units) in bisindoles from Alstonia species that are reviewed herein.
Table 1.
Indole alkaloids (monomeric units) in bisindoles from Alstonia species that are reviewed herein.
| Types | Indole Alkaloids | |
|---|---|---|
 |  |  |
 |  | |
 |  | |
 |  | |
 |  | |
Table 2.
Bisindoles from Alstonia species that are reviewed herein.
| Bisindole Alkaloids Reviewed Herein | ||
|---|---|---|
| Macroline-macroline | ||
 |  |  |
 |  |  |
 |  |  |
 |  |  |
 |  |  |
| Macroline-sarpagine | ||
 |  |  |
 |  |  |
 |  |  |
 |  | |
| Macroline-ajmaline | ||
 |  | |
| Macroline-pleiocarpamine | ||
 |  |  |
 |  |  |
 | ||
Table 4.
Bioactivity of bisindole alkaloids (including semisynthetic derivatives) from Alstonia species.
Table 4.
Bioactivity of bisindole alkaloids (including semisynthetic derivatives) from Alstonia species.
| Bisindoles | Bioactivity | References |
|---|---|---|
| (+)-Alstomacroline 1 | Antimalarial, with IC50 values of 1.12 ± 0.35 and 10.0 ± 0.4 μM against the K1 strain and T9-96 strain of P. falciparum, respectively. | [20] |
| (+)-Alstomacrophylline 2 | Antimalarial, with an IC50 value of 1.10 ± 0.30 μM against the K1 strain of P. falciparum. | [20] |
| Angustilongines E, F, G, H, J, and K (6–11) | Anticancer, cytotoxic against various human cancer cell lines including KB, vincristine-resistant KB, HCCT 116, PC-3, MDA-MB-231, LNCaP, MCF7, HT-29, and A549 cells with IC50 values ranging from 0.02 to 9.0 μM. | [5] |
| (−)-Lumusidine A, B, and C (14–16) | Anticancer, moderately cytotoxic in KB/VJ300 cells with IC50 values of 0.16, 0.70, and 1.19 μg/mL (μM), respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (−)-Lumusidine D 17 | Anticancer, cytotoxic in KB/VJ300 cells with an IC50 value of 5.03 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| Lumutinine A, B, C, D, and E (18–22) | Anticancer, moderately cytotoxic in KB/VJ300 cells with IC50 0.21, 0.10, 4.61, 3.93, and 2.74 μg/mL (μM) values, respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-Macralstonine 24 | Anticancer, strongly cytotoxic in KB/VJ300 cells with an IC50 1.71 μg/mL (μM) value. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| Antimalarial, active against the K1 strain of P. falciparum with an IC50 8.92 ± 2.95 μM value. | [20] | |
| (−)-Anhydromacralstonine 27 | Anticancer, moderately cytotoxic in KB/VJ300 cells with an IC50 value of 0.44 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-O-Acetyl macralstonine 25 | Antimalarial, with IC50 values 0.53 ± 0.09 and 12.4 ± 1.6 (μM) against the K1 strain and T9-96 strain of P. falciparum, respectively. | [20] |
| (+)-O-Methyl macralstonine 26 | Antimalarial, active against the K1 strain of P. falciparum with an IC50 0.85 ± 0.20 μM value. | [20] |
| O-Acetyl-E-seco-macralstonine 53 | Anticancer, strongly cytotoxic in KB/VJ300 cells with an IC50 value of 0.27 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (−)-Perhentidine A 36 and (-)-perhentidine B 37 | Anticancer, strongly cytotoxic in KB/VJ300 cells with IC50 values of 2.29 and 0.84 μg/mL (μM), respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (−)-O-Acetylperhentidine A 54 and (-)-O-Acetylperhentidine B 55 | Anticancer, strongly cytotoxic in KB/VJ300 cells with IC50 0.36 and 0.28 μg/mL (μM) values, respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (−)-Perhentinine 39 and O-Acetylperhentinine 56 | Anticancer, cytotoxic in KB/VJ300 cells with IC50 values of 0.52 and 0.30 μg/mL (μM), respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-Macralstonidine 23 | Anticancer, moderately cytotoxic in KB/VJ300 cells with an IC50 value of 0.13 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-Macrocarpamine 21 | Anticancer, strongly cytotoxic in KB/VJ300 cells with an IC50 value of 0.53 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| Strong antimalarial activity against the K1 strain of P. falciparum with an IC50 value of 0.36 ± 0.06 μM. Active against the T9-96 strain of P. falciparum with an IC50 >39 μM value. | [20] | |
| Strong antiprotozoal activity in vitro against E. histolytica and P. falciparum with ED50 8.12 (95% C.I.) μM and ED50 9.36 (95% C.I.) μM values, respectively. | [43] | |
| (+)-Villalstonine 43 | Anticancer, cytotoxic in KB/VJ300 cells with an IC50 value of 0.42 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| Anticancer, cytotoxic against the HT-29 cell line with an ED50 8.0 μM value (paclitaxel was used as the positive control). | [39] | |
| Antimalarial, with IC50 values of 0.27 ± 0.06 and 0.94 ± 0.07 μM against the K1 strain and T9-96 strain of P. falciparum, respectively. | [20] | |
| Antiamoebic activity against E. histolytica with an ED50 of 2.04 μM. | [43] | |
| Villalstonine N(4)-oxide 44 | Antileishmanial activity against promastigotes of L. mexicana with an IC50 value of 80.3 μM (amphotericin B was used as the positive control). | [39] |
| Antimalarial, active against the K1 strain of P. falciparum with an IC50 10.7 ± 1.9 (μM) value. | [20] | |
| (+)-Villalstonidine B 48 and (+)-villalstonidine F 52 | Anticancer, strongly cytotoxic in KB/VJ300 cells with IC50 values of 0.35 and 5.64 μg/mL (μM), respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-Villalstodinine D 50 | Antileishmanial, active against promastigotes of L. mexicana with an IC50 value of 120.4 μM (amphotericin B was used as the positive control). | [39] |
| (+)-Villalstonidine E 51 | Anticancer, cytotoxic against HT-29 cell lines with an ED50 6.5 μM value (paclitaxel was used as the positive control). | [39] |
| Antileishmanial against promastigotes of L. mexicana with an IC50 78 μM value (amphotericin B was used as the positive control). | [39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pandey, K.P.; Rahman, M.T.; Cook, J.M. Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis. Molecules 2021, 26, 3459. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113459
AMA Style
Pandey KP, Rahman MT, Cook JM. Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis. Molecules. 2021; 26(11):3459. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113459
Chicago/Turabian StylePandey, Kamal P., Md Toufiqur Rahman, and James M. Cook. 2021. "Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis" Molecules 26, no. 11: 3459. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113459