
CYPstrate: A Set of Machine Learning Models for the Accurate Classification of Cytochrome P450 Enzyme Substrates and Non-Substrates


Abstract
:1. Introduction
2. Results and Discussion
2.1. Characterization of Data Sets
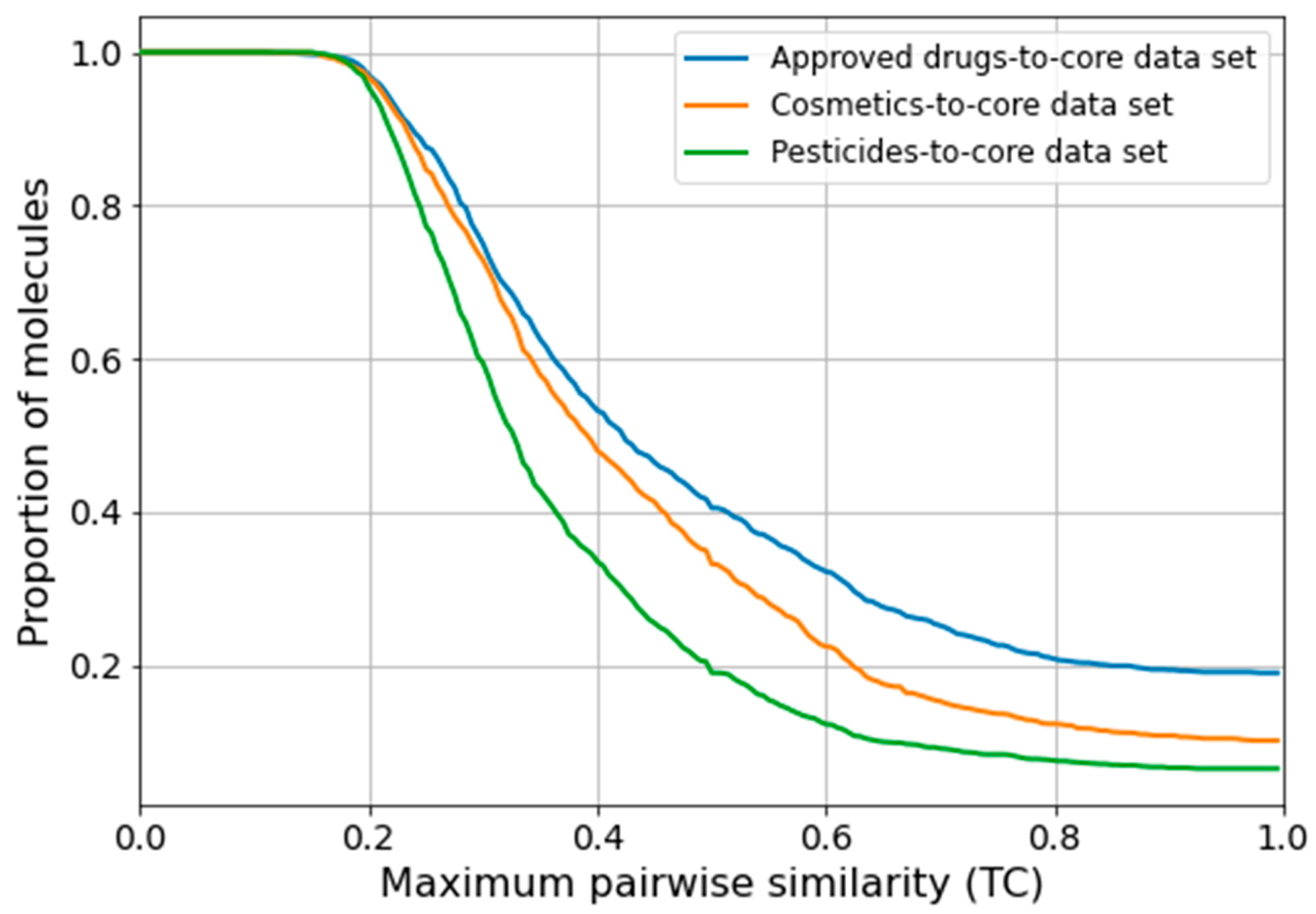
Analysis of the Relevance of the Core Data Set to Small-Molecule Research
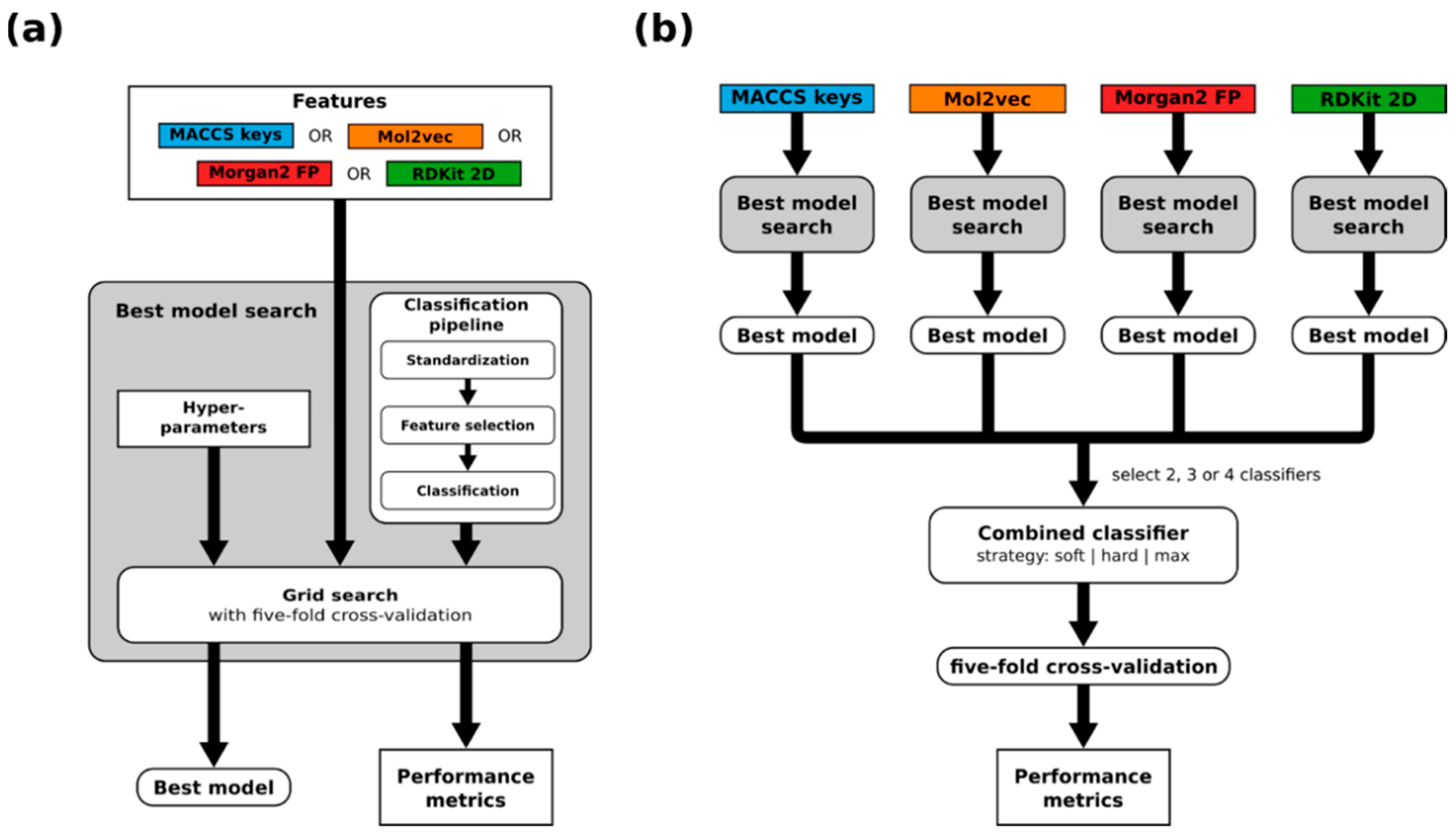
2.2. Model Development
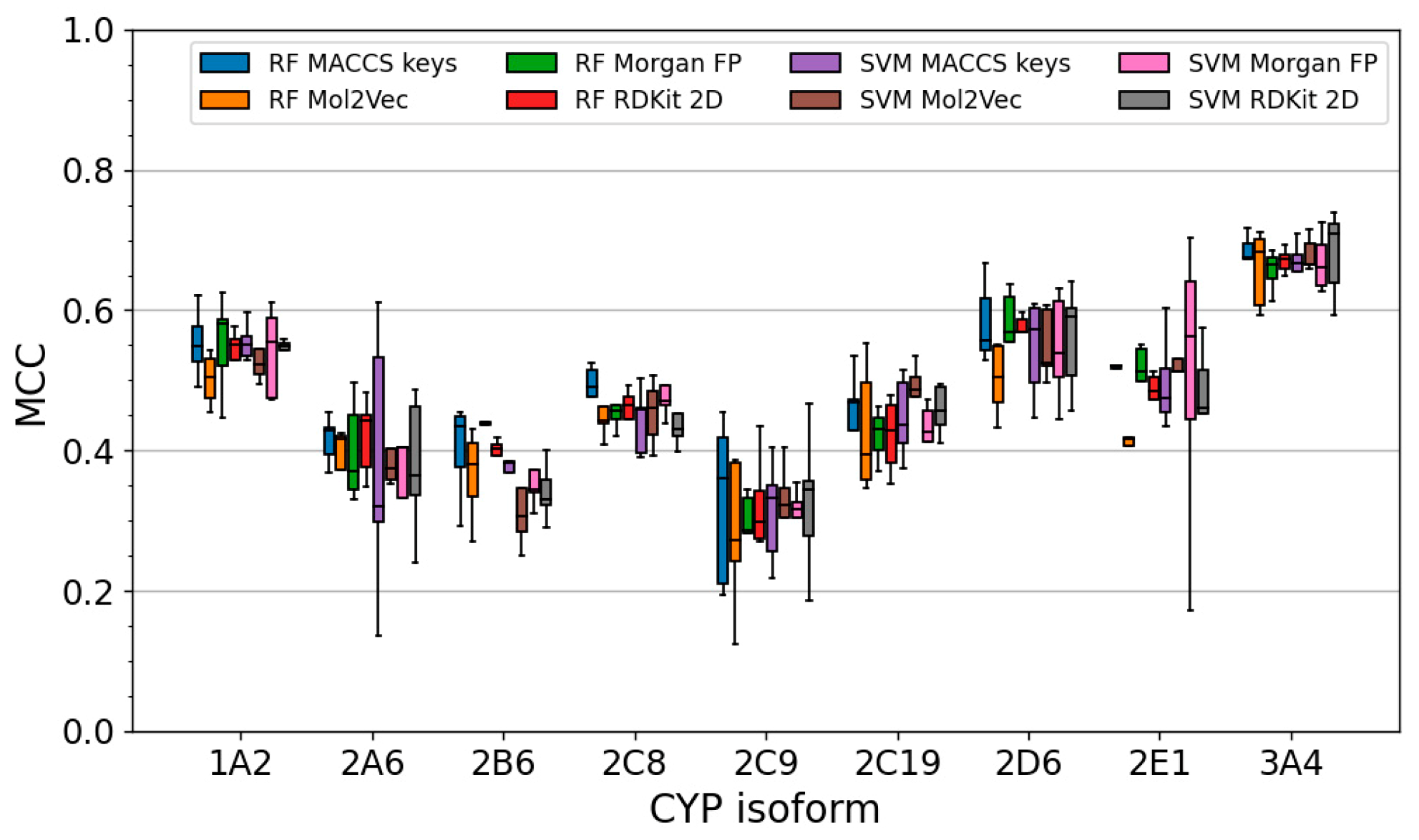
2.3. Performance of Single Classifiers
2.3.1. Performance during Cross-Validation
2.3.2. Performance on the Test Set
2.3.3. Performance Dependence on the Level of Structural Similarity during Cross-Validation
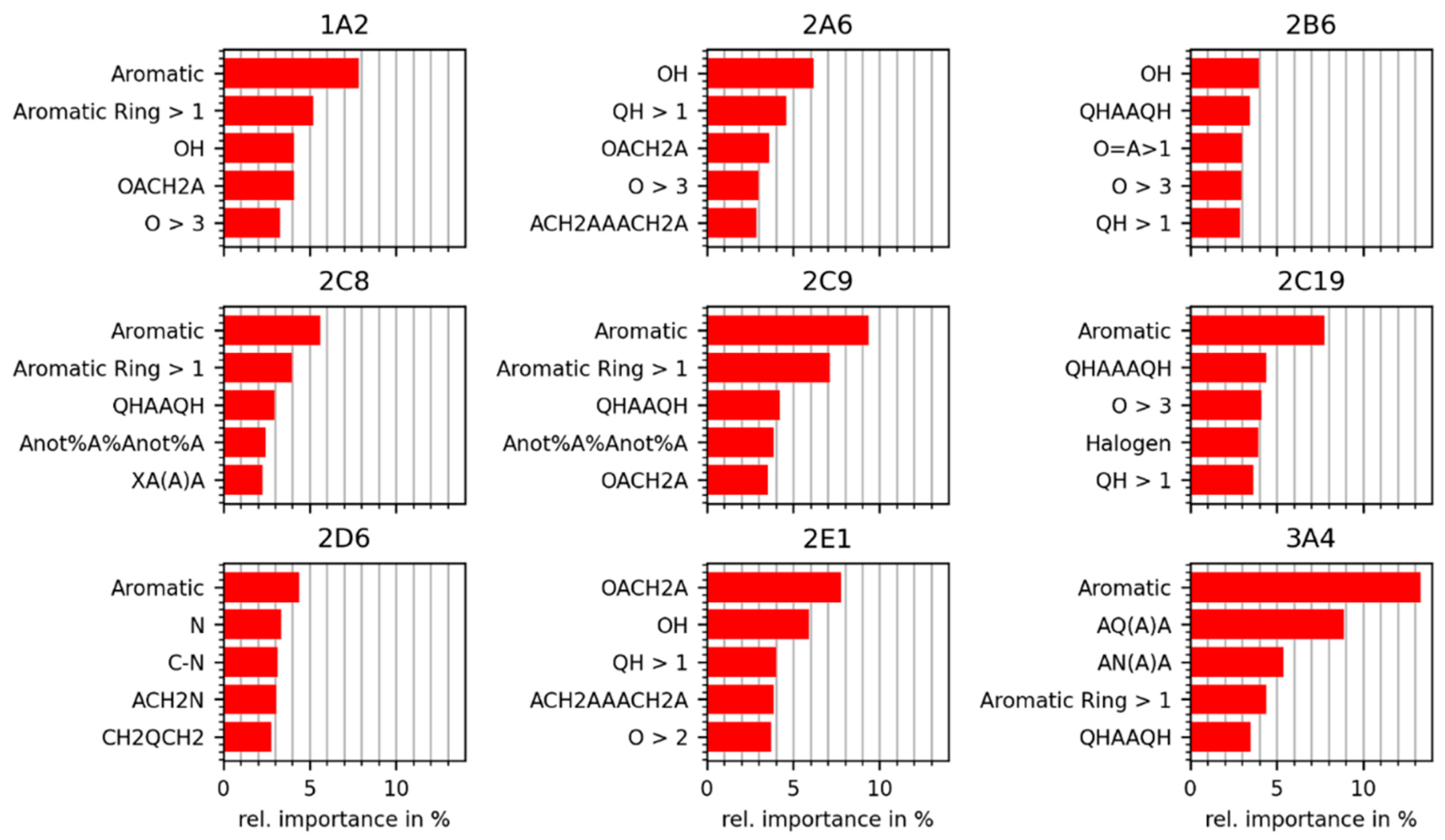
2.4. Analysis of Discriminative Features in Substrates and Non-Substrates
2.4.1. Importance Calculated for Individual MACCS Keys

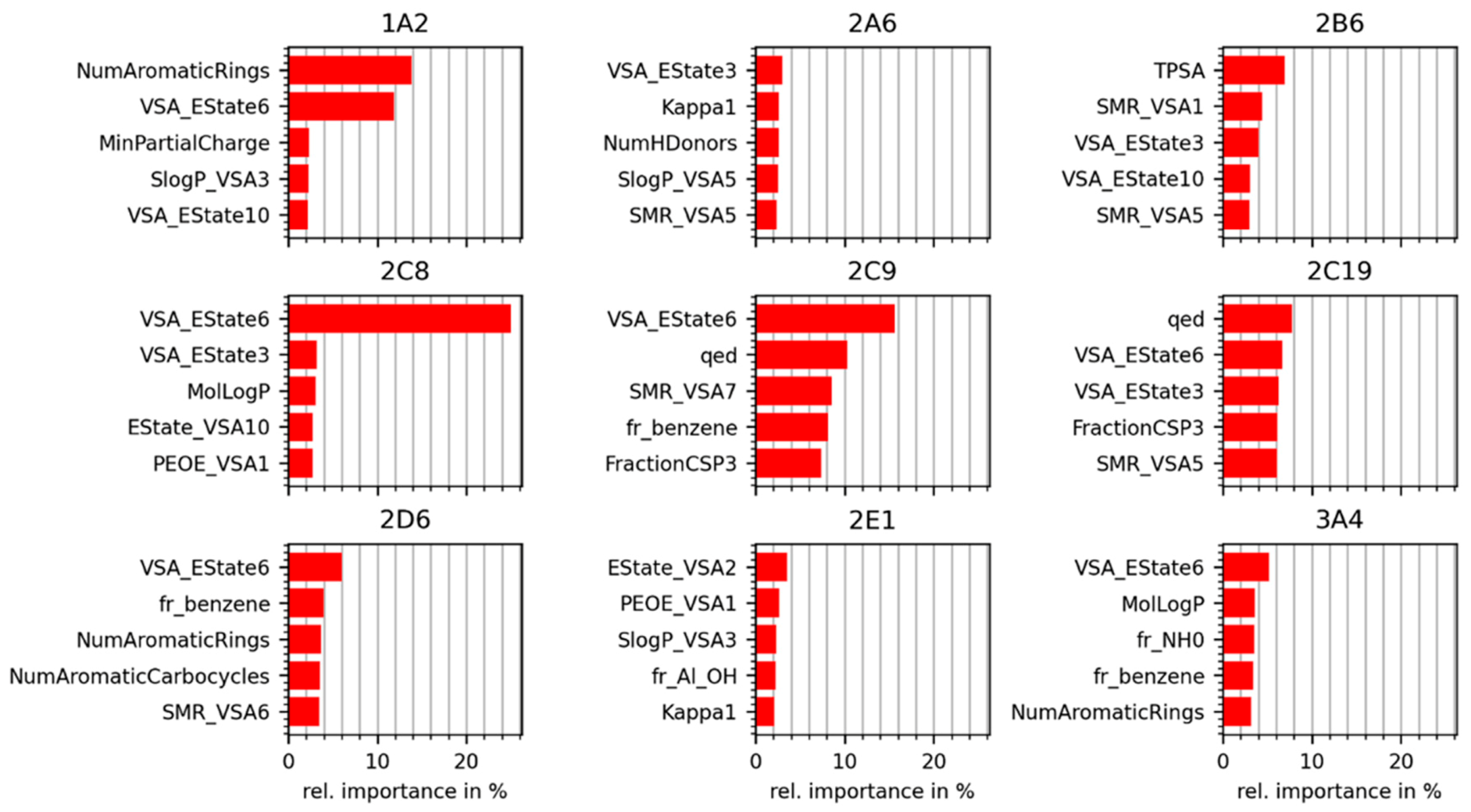
2.4.2. Importances Calculated for Individual RDKit 2D Descriptors

2.5. Performance of Combined Models
- Soft voting, where the class probabilities reported by the individual models are averaged.
- Hard voting, where the classifications of the individual classifiers are combined by identifying the consensus of the individual binary predictions. Importantly, hard voting does not produce predictions in some cases, because we require consensus models composed of two or three single classifiers to agree in their classifications in order to produce a prediction. For consensus models composed of four models, we tested two scenarios: one in which all models are required to agree and one in which at least three models are required to agree in order to produce a prediction.
- Max voting, where the maximum class probability reported by any of the classifiers (taking into account both classes) is used for making a binary prediction. The maximum class probability is determined considering the class probabilities for both the substrate and non-substrate classes.
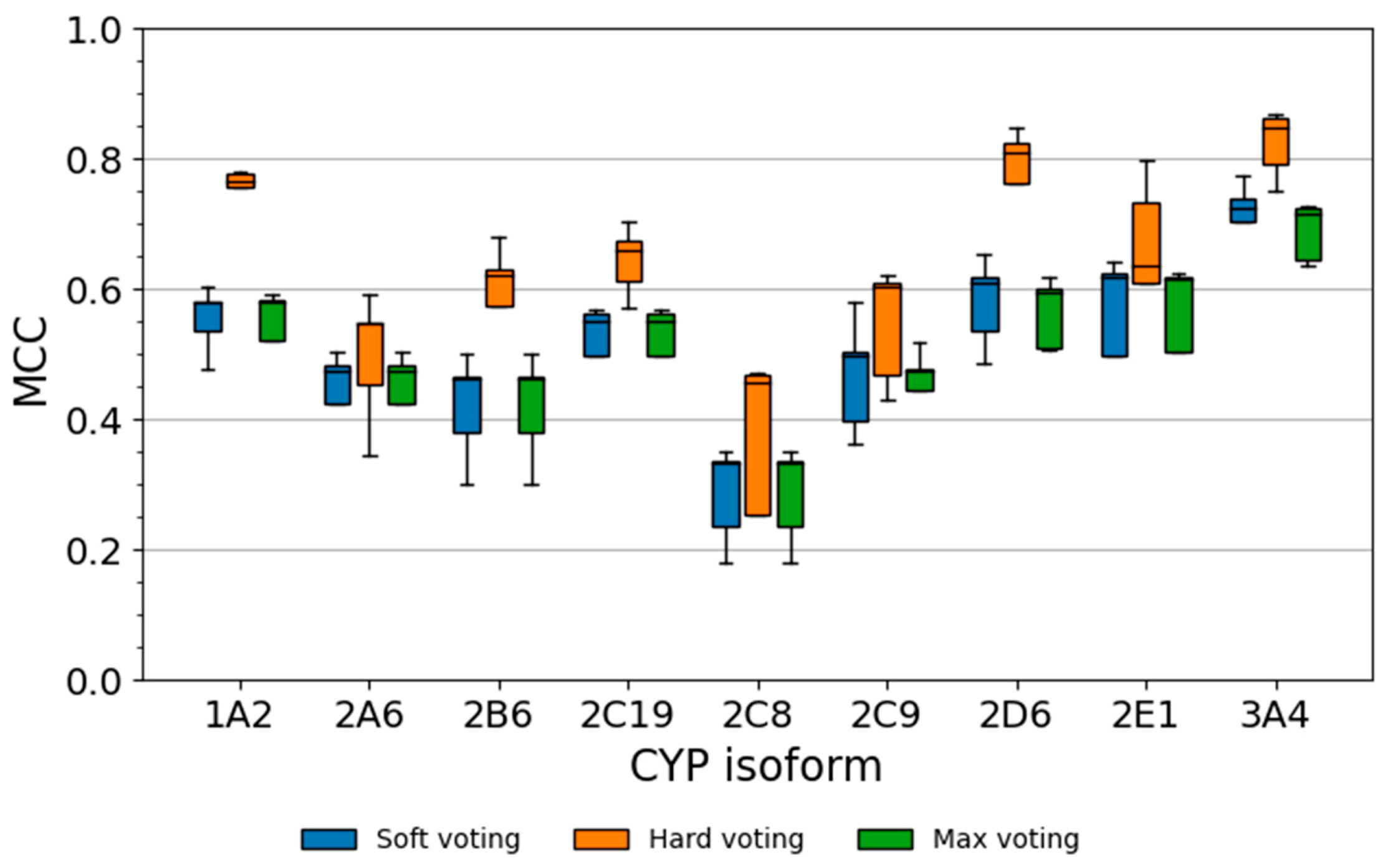
2.5.1. Performance of Soft Voting Classifiers
2.5.2. Performance of Hard Voting Classifiers
2.5.3. Performance of Max Voting Classifiers
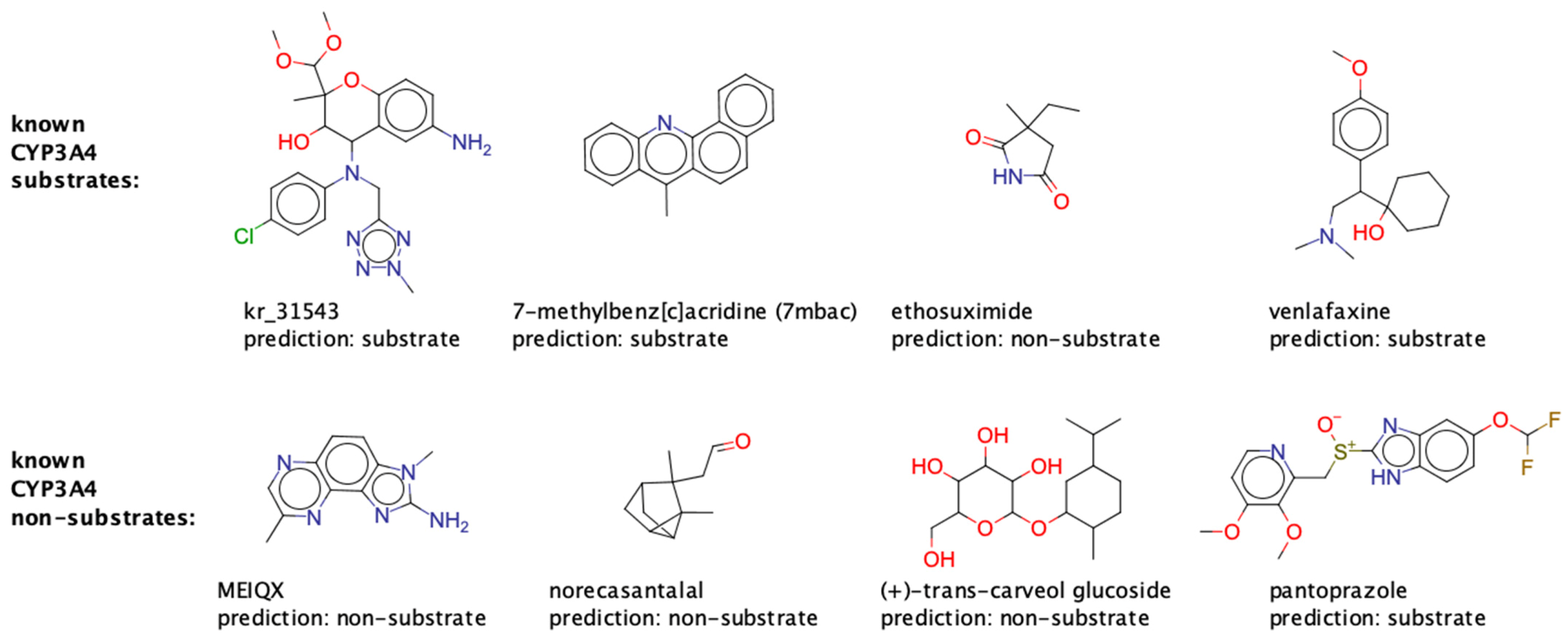
2.6. Comparison of Our Models to CypReact
2.6.1. Comparison of the Data Sets Employed for Model Training and Testing
2.6.2. Comparison of the Cross-Validation Performance of the Models
2.6.3. Comparison of the Applicability Domains
3. Materials and Methods
3.1. Data Sets
3.1.1. Core Data Set
3.1.2. Reference Data Sets for Approved Drugs, Cosmetic Ingredients, and Pesticides
3.2. Chemical Data Preprocessing
- Where not provided by the data source (Tian data set), SMILES representations were generated with RDKit [24] using default parameters. Any entries without chemical information were discarded.
- Molecules for which the SMILES representation could not be parsed by RDKit were discarded.
- The molecular structures were standardized with the standardize_mol method of the ChEMBL structure pipeline [36,37]. This procedure includes the standardization of unknown chemistry, removal of S group annotations, kekulization, removal of non-explicit H atoms, normalization of frequent substructures, and neutralization. For molecule entries consisting of several connected components or molecules with specified isotopes, the parent molecule was identified with the get_parent_mol method. By this method, all isotope information is removed, known salts and solvents are stripped, duplicate fragments are removed, and the molecule is neutralized.
- Molecules containing any elements other than H, B, C, N, O, F, Si, P, S, Cl, Se, Br, and I were discarded.
- Tautomers were canonicalized using the TautomerEnumerator class of RDKit. All stereochemical information was removed during tautomer canonicalization.
Processing of Duplicate Molecule Entries
3.3. Compilation of the Core Data Set
3.4. Splitting of the Core Data Set into a Training and a Test Set
3.5. Principal Component Analysis
3.6. Descriptors
3.7. Model Development
3.7.1. General Model Development
3.7.2. Feature Selection
3.7.3. Cross-Validation and Hyperparameter Optimization
3.8. Analysis of Feature Importance
3.9. Performance Metrics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Guengerich, F.P. Cytochromes P450, Drugs, and Diseases. Mol. Interv. 2003, 3, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.S.; Dalvie, D.K. Cytochrome P450 and Non-Cytochrome P450 Oxidative Metabolism: Contributions to the Pharmacokinetics, Safety, and Efficacy of Xenobiotics. Drug Metab. Dispos. 2016, 44, 1229–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, B.; Pedretti, A.; Vistoli, G. Reactions and Enzymes in the Metabolism of Drugs and Other Xenobiotics. Drug Discov. Today 2012, 17, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Common and Uncommon Cytochrome P450 Reactions Related to Metabolism and Chemical Toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting Drug Metabolism: Experiment And/or Computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Kirchmair, J.; Williamson, M.J.; Tyzack, J.D.; Tan, L.; Bond, P.J.; Bender, A.; Glen, R.C. Computational Prediction of Metabolism: Sites, Products, SAR, P450 Enzyme Dynamics, and Mechanisms. J. Chem. Inf. Model. 2012, 52, 617–648. [Google Scholar] [CrossRef]
- Dixit, V.A.; Deshpande, S. Advances in Computational Prediction of Regioselective and Isoform-Specific Drug Metabolism Catalyzed by CYP450s. ChemistrySelect 2016, 1, 6571–6597. [Google Scholar] [CrossRef]
- Kato, H. Computational Prediction of Cytochrome P450 Inhibition and Induction. Drug Metab. Pharmacokinet. 2020, 35, 30–44. [Google Scholar] [CrossRef]
- Tyzack, J.D.; Kirchmair, J. Computational Methods and Tools to Predict Cytochrome P450 Metabolism for Drug Discovery. Chem. Biol. Drug Des. 2019, 93, 377–386. [Google Scholar] [CrossRef]
- Simulations Plus. ADMET Predictor Metabolism Module. Available online: https://www.simulations-plus.com/software/admetpredictor/ (accessed on 24 June 2021).
- Fujitsu ADMEWORKS Properties Prediction System. Available online: https://www.fujitsu.com/global/solutions/business-technology/tc/sol/admeworks/index.html (accessed on 24 June 2021).
- ACD/Labs. Percepta Platform. Available online: https://www.acdlabs.com (accessed on 24 June 2021).
- Hunt, P.A.; Segall, M.D.; Tyzack, J.D. WhichP450: A Multi-Class Categorical Model to Predict the Major Metabolising CYP450 Isoform for a Compound. J. Comput. Aided Mol. Des. 2018, 32, 537–546. [Google Scholar] [CrossRef]
- Optibrium. Stardrop. Available online: https://www.optibrium.com/stardrop/ (accessed on 28 June 2021).
- Tian, S.; Djoumbou-Feunang, Y.; Greiner, R.; Wishart, D.S. CypReact: A Software Tool for in Silico Reactant Prediction for Human Cytochrome P450 Enzymes. J. Chem. Inf. Model. 2018, 58, 1282–1291. [Google Scholar] [CrossRef]
- Zaretzki, J.; Rydberg, P.; Bergeron, C.; Bennett, K.P.; Olsen, L.; Breneman, C.M. RS-Predictor Models Augmented with SMARTCyp Reactivities: Robust Metabolic Regioselectivity Predictions for Nine CYP Isozymes. J. Chem. Inf. Model. 2012, 52, 1637–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The Human Metabolome Database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- United States Environmental Protection Agency. CompTox Chemical List COSMOS DB. Available online: https://comptox.epa.gov/dashboard/chemical_lists/COSMOSDB (accessed on 26 October 2020).
- United States Environmental Protection Agency. CompTox Chemical List EPAPCS. Available online: https://comptox.epa.gov/dashboard/chemical_lists/EPAPCS (accessed on 26 October 2020).
- Stork, C.; Wagner, J.; Friedrich, N.-O.; de Bruyn Kops, C.; Šícho, M.; Kirchmair, J. Hit Dexter: A Machine-Learning Model for the Prediction of Frequent Hitters. ChemMedChem 2018, 13, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit (Version 2019.09.3). Available online: http://www.rdkit.org (accessed on 22 March 2021).
- Matthews, B.W. Comparison of the Predicted and Observed Secondary Structure of T4 Phage Lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- De Montellano, P.R.O. (Ed.) Cytochrome P450: Structure, Mechanism, and Biochemistry; Springer: Berlin/Heidelberg, Germany, 2015; ISBN 9783319374376. [Google Scholar]
- Beck, T.C.; Beck, K.R.; Morningstar, J.; Benjamin, M.M.; Norris, R.A. Descriptors of Cytochrome Inhibitors and Useful Machine Learning Based Methods for the Design of Safer Drugs. Pharmaceuticals 2021, 14, 472. [Google Scholar] [CrossRef]
- Brown, C.M.; Reisfeld, B.; Mayeno, A.N. Cytochromes P450: A Structure-Based Summary of Biotransformations Using Representative Substrates. Drug Metab. Rev. 2008, 40, 1–100. [Google Scholar] [CrossRef]
- Landrum, G. RDKit-MACCS Keys Module. Available online: https://github.com/rdkit/rdkit/blob/master/rdkit/Chem/MACCSkeys.py (accessed on 17 June 2021).
- Molecular Operating Environment (MOE). Chemical Computing Group: Montreal, QC, USA, 2021. Available online: www.chemcomp.com/MOE-Molecular_Operating_Environment.htm/ (accessed on 1 August 2021).
- Kier, L.B.; Hall, L.H. An Electrotopological-State Index for Atoms in Molecules. Pharm. Res. 1990, 7, 801–807. [Google Scholar] [CrossRef]
- Tyzack, J.D.; Hunt, P.A.; Segall, M.D. Predicting Regioselectivity and Lability of Cytochrome P450 Metabolism Using Quantum Mechanical Simulations. J. Chem. Inf. Model. 2016, 56, 2180–2193. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn Kops, C.; Friedrich, N.-O.; Kirchmair, J. Alignment-Based Prediction of Sites of Metabolism. J. Chem. Inf. Model. 2017, 57, 1258–1264. [Google Scholar] [CrossRef]
- DrugBank (Version 5.1.7). Available online: https://go.drugbank.com/releases/5-1-7 (accessed on 26 October 2020).
- Lowe, C.N.; Williams, A.J. Enabling High-Throughput Searches for Multiple Chemical Data Using the U.S.-EPA CompTox Chemicals Dashboard. J. Chem. Inf. Model. 2021, 61, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Hersey, A.; Félix, E.; Landrum, G.; Gaulton, A.; Atkinson, F.; Bellis, L.J.; De Veij, M.; Leach, A.R. An Open Source Chemical Structure Curation Pipeline Using RDKit. J. Cheminform. 2020, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- ChEMBL Structure Pipeline (Version 1.0.0). Available online: https://github.com/chembl/ChEMBL_Structure_Pipeline (accessed on 4 April 2021).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Jaeger, S.; Fulle, S.; Turk, S. Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition. J. Chem. Inf. Model. 2018, 58, 27–35. [Google Scholar] [CrossRef]
- Turk, S. Mol2vec-Example Models. Available online: https://github.com/samoturk/mol2vec/tree/master/examples/models (accessed on 30 April 2021).
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Stork, C.; Embruch, G.; Šícho, M.; de Bruyn Kops, C.; Chen, Y.; Svozil, D.; Kirchmair, J. NERDD: A Web Portal Providing Access to in Silico Tools for Drug Discovery. Bioinformatics 2020, 36, 1291–1292. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Substrates | Number of Non-Substrates | |||||
|---|---|---|---|---|---|---|
| CYP | Without Hunt et al. | With Hunt et al. (This Is the Core Data Set) | Difference in % | Without Hunt et al. | With Hunt et al. (This Is the Core Data Set) | Difference in % 1 |
| 1A2 | 280 | 296 | +5.4 | 1441 | 1428 | −0.9 |
| 2A6 | 107 | 107 | 0.0 | 1607 | 1607 | 0.0 |
| 2B6 | 150 | 150 | 0.0 | 1561 | 1561 | 0.0 |
| 2C8 | 140 | 149 | +6.0 | 1572 | 1565 | −0.5 |
| 2C9 | 237 | 253 | +6.3 | 1484 | 1469 | −1.0 |
| 2C19 | 225 | 242 | +7.0 | 1493 | 1481 | −0.8 |
| 2D6 | 276 | 304 | +9.2 | 1441 | 1425 | −1.1 |
| 2E1 | 148 | 156 | +5.1 | 1566 | 1556 | −0.6 |
| 3A4 | 478 | 520 | +8.1 | 1241 | 1239 | −0.2 |
| CYP | 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A4 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Algorithm 1 | RF | RF | RF | RF | SVM | RF | SVM | SVM | SVM | |
| Descriptors 2 | MFP | R2D | MFP | MK | M2V | MK | R2D | MFP | R2D | |
| Performance during CV 3 | MCC | 0.58 | 0.44 | 0.44 | 0.36 | 0.49 | 0.49 | 0.59 | 0.56 | 0.71 |
| Jaccard | 0.48 | 0.31 | 0.32 | 0.26 | 0.38 | 0.39 | 0.49 | 0.42 | 0.66 | |
| AUC | 0.88 | 0.89 | 0.86 | 0.80 | 0.87 | 0.86 | 0.92 | 0.88 | 0.92 | |
| Performance on the test set | MCC | 0.50 | 0.32 | 0.33 | 0.40 | 0.48 | 0.59 | 0.61 | 0.40 | 0.70 |
| Jaccard | 0.41 | 0.22 | 0.24 | 0.29 | 0.37 | 0.48 | 0.50 | 0.28 | 0.65 | |
| AUC | 0.88 | 0.85 | 0.85 | 0.90 | 0.89 | 0.91 | 0.92 | 0.80 | 0.90 | |
| No. Classifiers Underlying a Combined Model | Feature Sets | Classification Algorithms | Voting Strategy | No. Combinations (i.e., Number of Consensus Models) |
|---|---|---|---|---|
| 2 | MACCS keys Morgan2 fingerprints RDKit 2D descriptors Mol2vec descriptors | RF, SVM | soft max hard (min consensus = 2) | 72 1 |
| 3 | soft max hard (min consensus = 3) | 96 1 | ||
| 4 | soft max hard (min consensus = 3) hard (min consensus = 4) | 64 1 |
| CYP | 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A4 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Model Setup 1 | RF MFP + RF MK | RF R2D + RF MK | RF R2D + RF MFP | RF R2D + RF M2V | RF R2D + RF MFP + SVM MK + RF M2V | RF R2D + SVM MFP | RF R2D + RF MFP + RF MK + SVM M2V | RF R2D + SVM MFP + RF MK + SVM M2V | SVM R2D + RF MK + SVM M2V | |
| Performance during CV (Median over the 5 Folds of CV) | MCC | 0.58 | 0.47 | 0.46 | 0.33 | 0.50 | 0.55 | 0.61 | 0.62 | 0.72 |
| Jaccard | 0.48 | 0.32 | 0.33 | 0.24 | 0.40 | 0.40 | 0.51 | 0.44 | 0.67 | |
| AUC | 0.90 | 0.89 | 0.88 | 0.79 | 0.87 | 0.88 | 0.93 | 0.89 | 0.94 | |
| Performance on the Test Set | MCC | 0.58 | 0.39 | 0.36 | 0.38 | 0.50 | 0.54 | 0.58 | 0.46 | 0.70 |
| Jaccard | 0.49 | 0.26 | 0.26 | 0.28 | 0.41 | 0.41 | 0.47 | 0.32 | 0.65 | |
| AUC | 0.91 | 0.84 | 0.86 | 0.87 | 0.90 | 0.90 | 0.92 | 0.89 | 0.91 | |
| CYP | 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A4 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Model Setup 1 | RF R2D + RF MFP + RF MK + RF M2V | RF R2D + RF MK + RF M2V | RF R2D + RF MFP + RF MK + RF M2V | RF MFP + RF MK | RF R2D + SVM MFP + RF MK + RF M2V | RF R2D + SVM MFP + RF MK + RF M2V | RF R2D + RF MFP + SVM MK + RF M2V | RF R2D + RF MK + SVM M2V | RF R2D + SVM MFP + RF MK + SVM M2V | |
| Minimum Consensus 2 | 4 | 3 | 4 | 2 | 4 | 4 | 4 | 3 | 4 | |
| Performance during CV (Median over the 5 Folds of CV) | MCC | 0.76 | 0.55 | 0.62 | 0.45 | 0.6 | 0.66 | 0.81 | 0.63 | 0.85 |
| Jaccard | 0.66 | 0.39 | 0.48 | 0.33 | 0.44 | 0.52 | 0.72 | 0.45 | 0.79 | |
| Coverage of Substrate | 0.52 | 0.71 | 0.62 | 0.67 | 0.42 | 0.54 | 0.54 | 0.56 | 0.66 | |
| Coverage of Non-Substrate | 0.82 | 0.91 | 0.79 | 0.88 | 0.79 | 0.81 | 0.84 | 0.93 | 0.85 | |
| Performance on the Test Set | MCC | 0.81 | 0.37 | 0.54 | 0.49 | 0.64 | 0.72 | 0.72 | 0.58 | 0.81 |
| Jaccard | 0.72 | 0.25 | 0.38 | 0.35 | 0.5 | 0.58 | 0.61 | 0.42 | 0.75 | |
| Coverage of substrate | 0.56 | 0.62 | 0.5 | 0.7 | 0.39 | 0.4 | 0.52 | 0.68 | 0.72 | |
| Coverage of Non-Substrate | 0.82 | 0.9 | 0.83 | 0.9 | 0.8 | 0.81 | 0.85 | 0.91 | 0.92 | |
| CYP | 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A4 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Model Setup 1 | RF R2D + SVM MFP + RF MK + SVM M2V | RF R2D + RF MK | RF R2D + RF MFP | RF R2D + RF M2V | RF R2D + SVM MFP + SVM MK + RF M2V | RF R2D + SVM MFP | RF R2D + SVM MK + RF M2V | SVM R2D + RF MFP + RF MK + SVM M2V | RF R2D + SVM MK + RF M2V | |
| Performance during CV (Median over the 5 Folds of CV) | MCC | 0.58 | 0.47 | 0.46 | 0.33 | 0.47 | 0.55 | 0.60 | 0.61 | 0.71 |
| Jaccard | 0.45 | 0.32 | 0.33 | 0.24 | 0.37 | 0.40 | 0.49 | 0.42 | 0.66 | |
| AUC | 0.90 | 0.88 | 0.88 | 0.79 | 0.86 | 0.88 | 0.91 | 0.88 | 0.92 | |
| Performance on the Test Set | MCC | 0.70 | 0.39 | 0.36 | 0.38 | 0.54 | 0.54 | 0.56 | 0.46 | 0.71 |
| Jaccard | 0.58 | 0.26 | 0.26 | 0.28 | 0.43 | 0.41 | 0.45 | 0.32 | 0.66 | |
| AUC | 0.92 | 0.83 | 0.86 | 0.87 | 0.90 | 0.90 | 0.90 | 0.88 | 0.92 | |
| CYP | Single Classifier | Hard Voting Classifier | CypReact | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Algorithm 1 | Descriptors 2 | Jaccard Score | AUC | Model Setup | Minimum Consensus 3 | Jaccard Score | Jaccard Score | AUC | |
| 1A2 | RF | Morgan FP | 0.48 | 0.88 | RF RDKit 2D + RF Morgan FP + RF MACCS keys + RF Mol2Vec | 4 | 0.66 | 0.39 | 0.86 |
| 2A6 | RF | RDKit 2D | 0.31 | 0.89 | RF RDKit 2D + RF MACCS keys + RF Mol2Vec | 3 | 0.39 | 0.28 | 0.84 |
| 2B6 | RF | Morgan FP | 0.32 | 0.86 | RF RDKit 2D + RF Morgan FP + RF MACCS keys + RF Mol2Vec | 4 | 0.48 | 0.28 | 0.86 |
| 2C8 | RF | MACCS keys | 0.26 | 0.80 | RF Morgan FP + RF MACCS keys | 2 | 0.33 | 0.25 | 0.84 |
| 2C9 | SVM | Mol2Vec | 0.38 | 0.87 | RF RDKit 2D + SVM Morgan FP + RF MACCS keys + RF Mol2Vec | 4 | 0.44 | 0.30 | 0.83 |
| 2C19 | RF | MACCS keys | 0.39 | 0.86 | RF RDKit 2D + SVM Morgan FP + RF MACCS keys + RF Mol2Vec | 4 | 0.52 | 0.30 | 0.83 |
| 2D6 | SVM | RDKit 2D | 0.49 | 0.92 | RF RDKit 2D + RF Morgan FP + SVM MACCS keys + RF Mol2Vec | 4 | 0.72 | 0.40 | 0.87 |
| 2E1 | SVM | Morgan FP | 0.42 | 0.88 | RF RDKit 2D + RF MACCS keys + SVM Mol2Vec | 3 | 0.45 | 0.30 | 0.87 |
| 3A4 | SVM | RDKit 2D | 0.66 | 0.92 | RF RDKit 2D + SVM Morgan FP + RF MACCS keys + SVM Mol2Vec | 4 | 0.79 | 0.55 | 0.92 |
| Number of Compounds | Core Data Set | COSMOS DB | EPAPCS | DrugBank |
|---|---|---|---|---|
| Forming the unprocessed data set | 2284 1 | 7035 | 4035 | 2506 |
| Removed due to missing SMILES | 0 | 1618 | 751 | 0 |
| Removed due to unparsable SMILES | 0 | 23 | 43 | 6 |
| Removed by the element filter | 0 | 392 | 344 | 208 |
| Removed by the duplicate filter | 453 | 522 | 369 | 74 |
| Forming the processed data set | 1831 | 4480 | 2528 | 2218 |
| Algorithm | Parameter | Explored Values |
|---|---|---|
| RF | min_samples_split | 2, 4, 8, 16, 32, 64, 128 |
| max_features | 0.05, 0.1, 0.2, 0.4, 0.8, sqrt | |
| SVM | C | 1 × 10−2, 1 × 10−1, 1 × 100, 1 × 101, 1 × 102, 1 × 103 |
| gamma | 1, 1 × 10−1, 1 × 10−2, 1 × 10−3, 1 × 10−4, 1 × 10−5 | |
| SelectPercentile | percentile | 10, 40, 70, 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holmer, M.; de Bruyn Kops, C.; Stork, C.; Kirchmair, J. CYPstrate: A Set of Machine Learning Models for the Accurate Classification of Cytochrome P450 Enzyme Substrates and Non-Substrates. Molecules 2021, 26, 4678. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154678
Holmer M, de Bruyn Kops C, Stork C, Kirchmair J. CYPstrate: A Set of Machine Learning Models for the Accurate Classification of Cytochrome P450 Enzyme Substrates and Non-Substrates. Molecules. 2021; 26(15):4678. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154678
Chicago/Turabian StyleHolmer, Malte, Christina de Bruyn Kops, Conrad Stork, and Johannes Kirchmair. 2021. "CYPstrate: A Set of Machine Learning Models for the Accurate Classification of Cytochrome P450 Enzyme Substrates and Non-Substrates" Molecules 26, no. 15: 4678. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154678