
Modeling Chemical Reactivity at the Interfaces of Emulsions: Effects of Partitioning and Temperature


Abstract
:1. Introduction
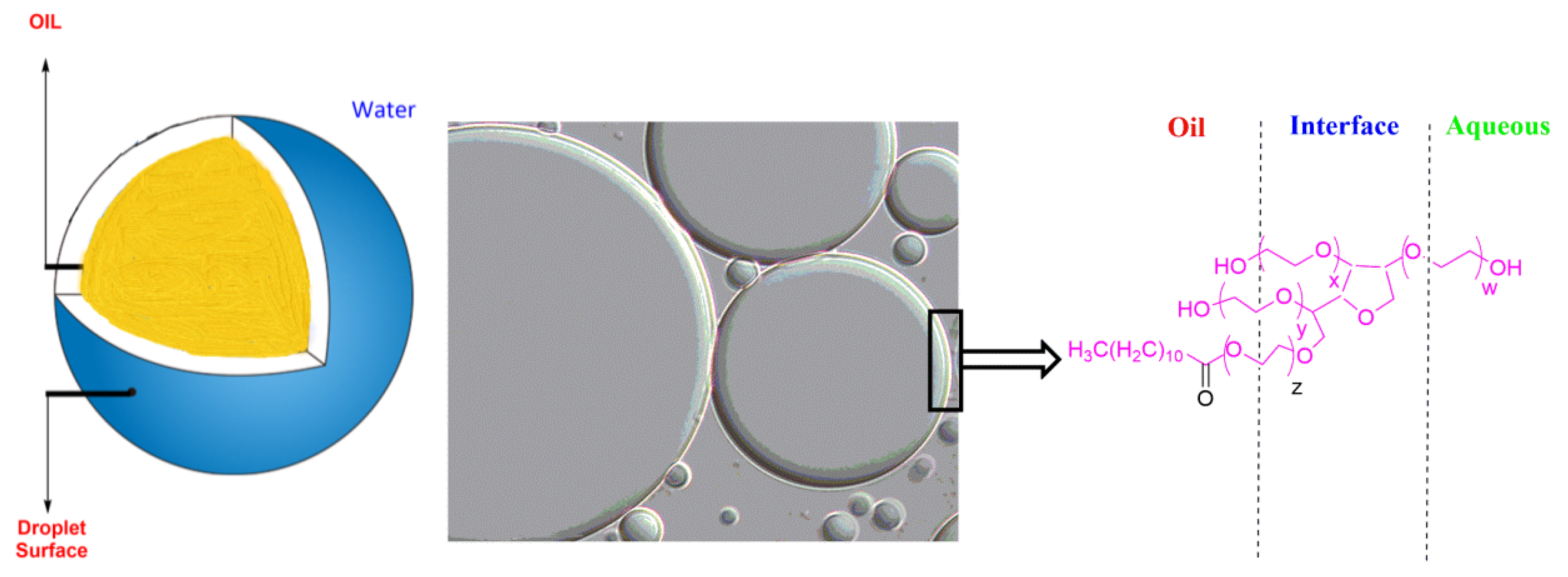
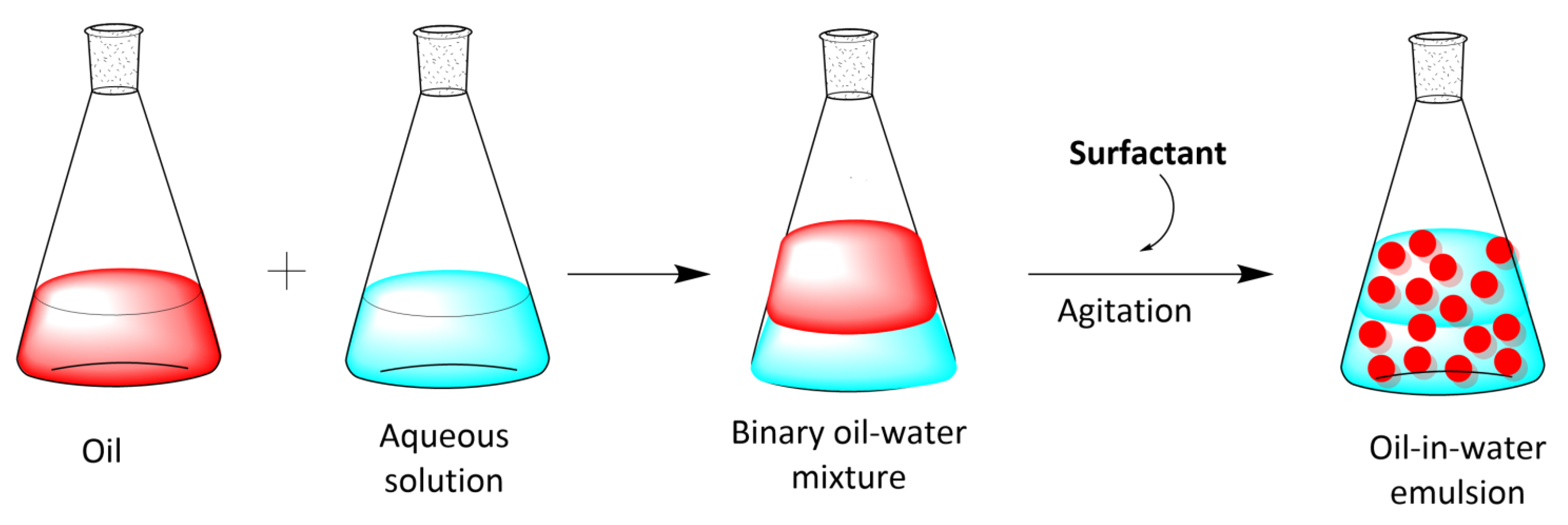
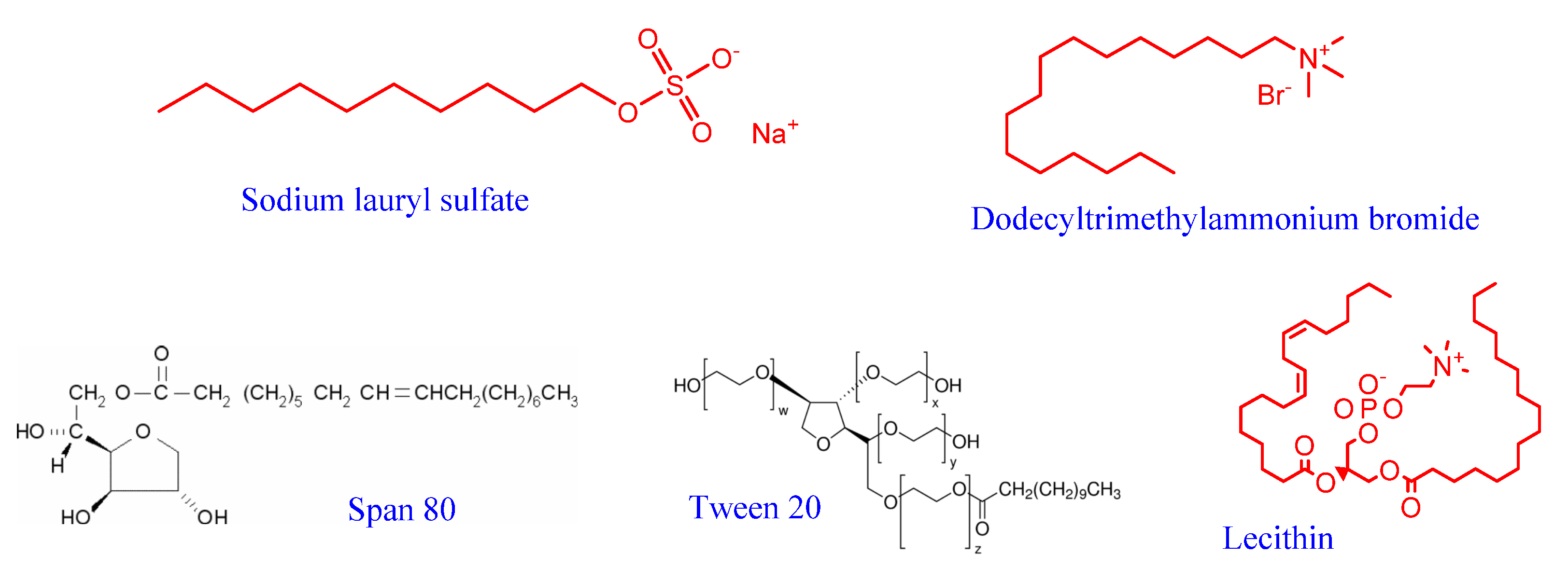
2. Emulsions: Classification, Physical Stabilization, and Main Properties
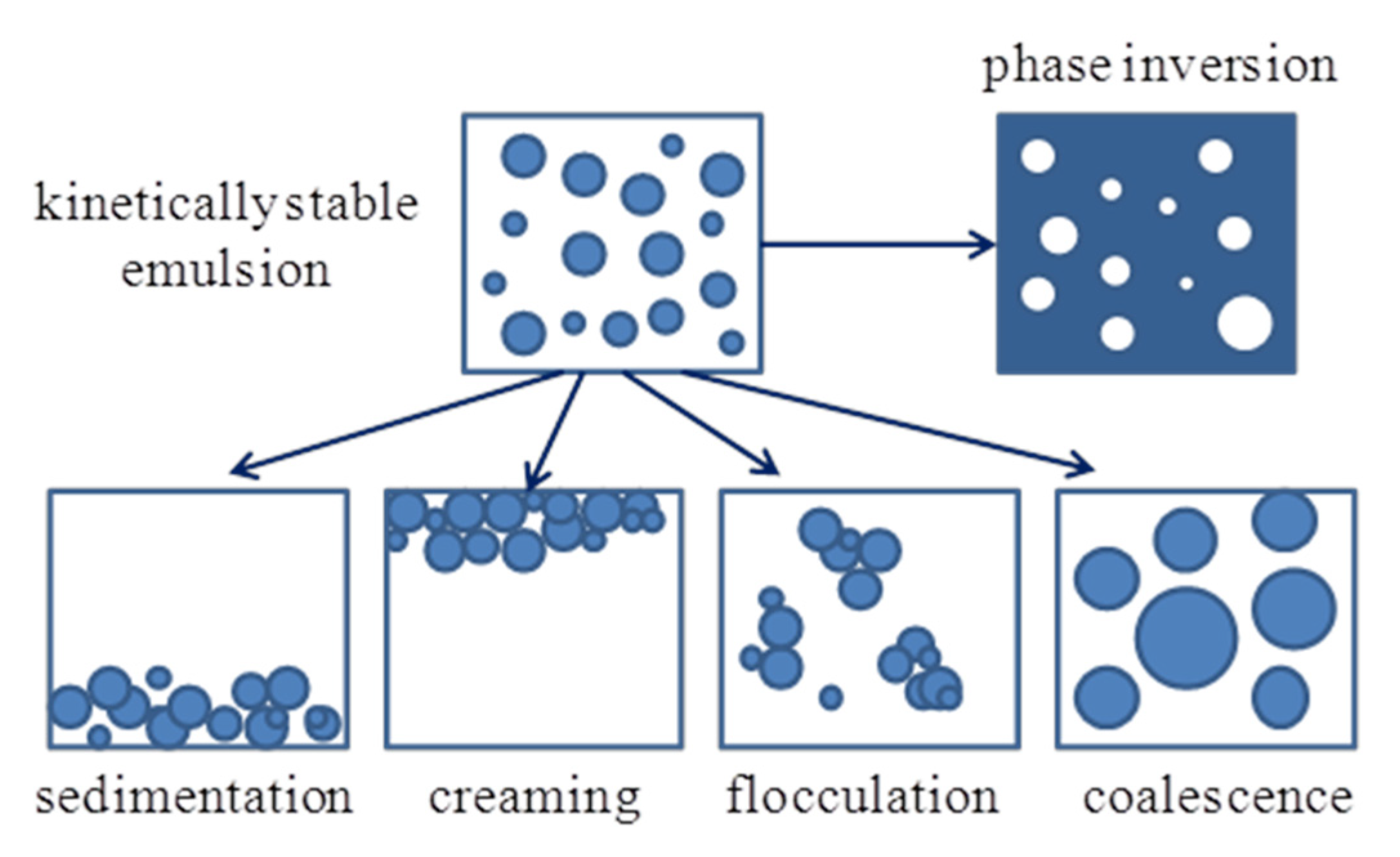
3. Dynamic Aspects of Emulsions and Mass Transfer
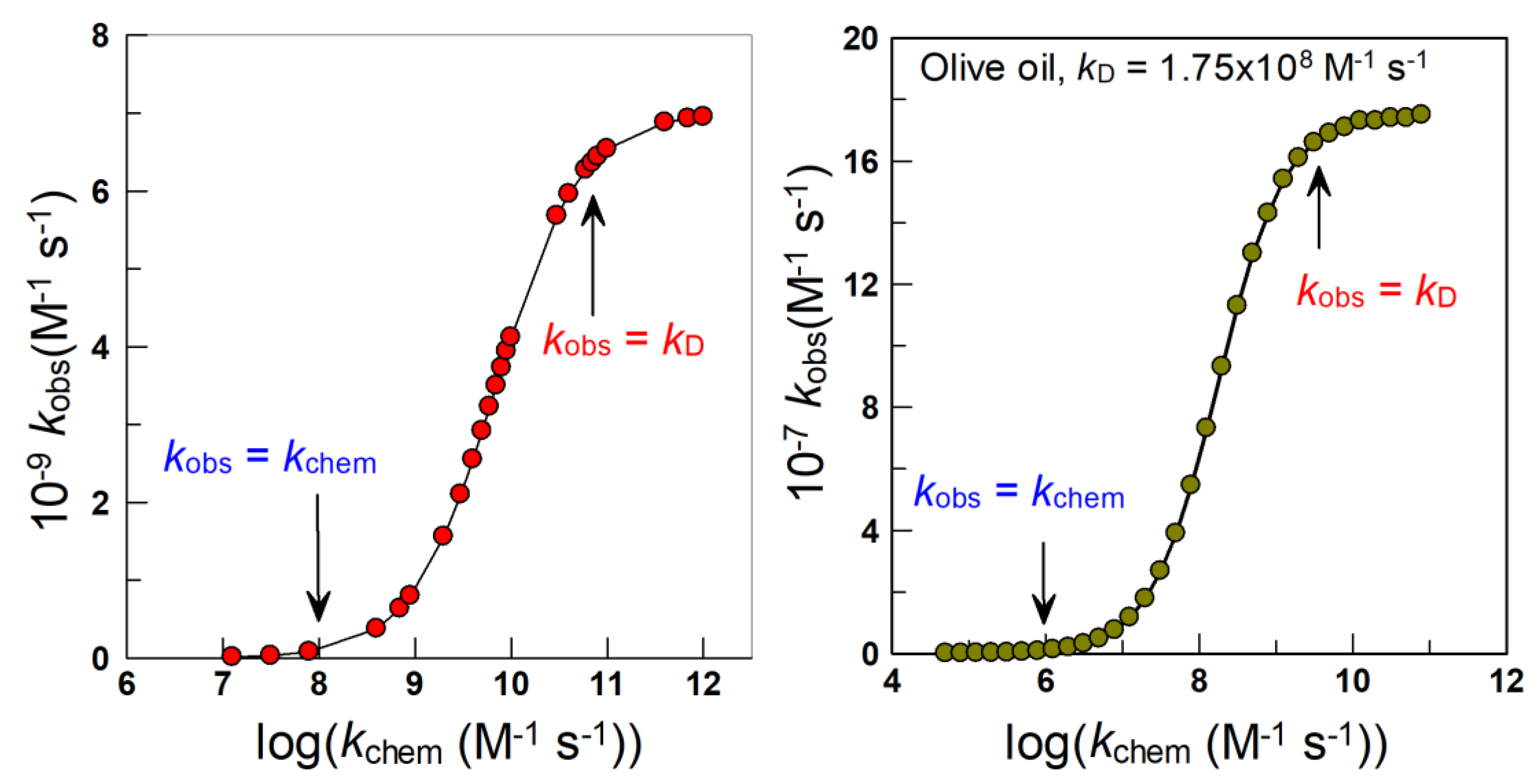
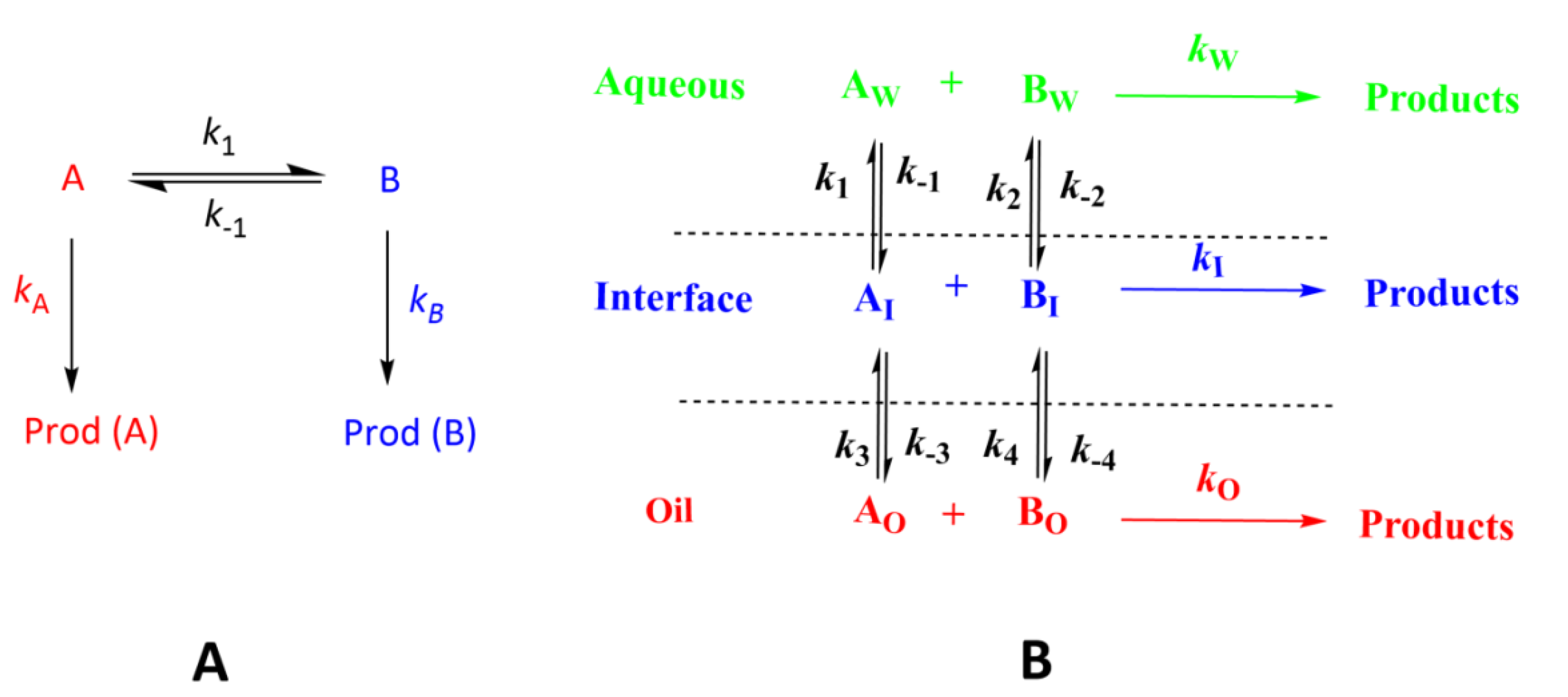
3.1. Kinetics of Chemical Reactions at Interfaces: Diffusive vs. Reactive Systems
3.2. Modeling Chemical Reactivity at the Interfaces of Emulsions
4. Main Equations Derived from the Application of the Pseudophase Model Emulsions
4.1. Reactions in Non-Ionic Emulsions
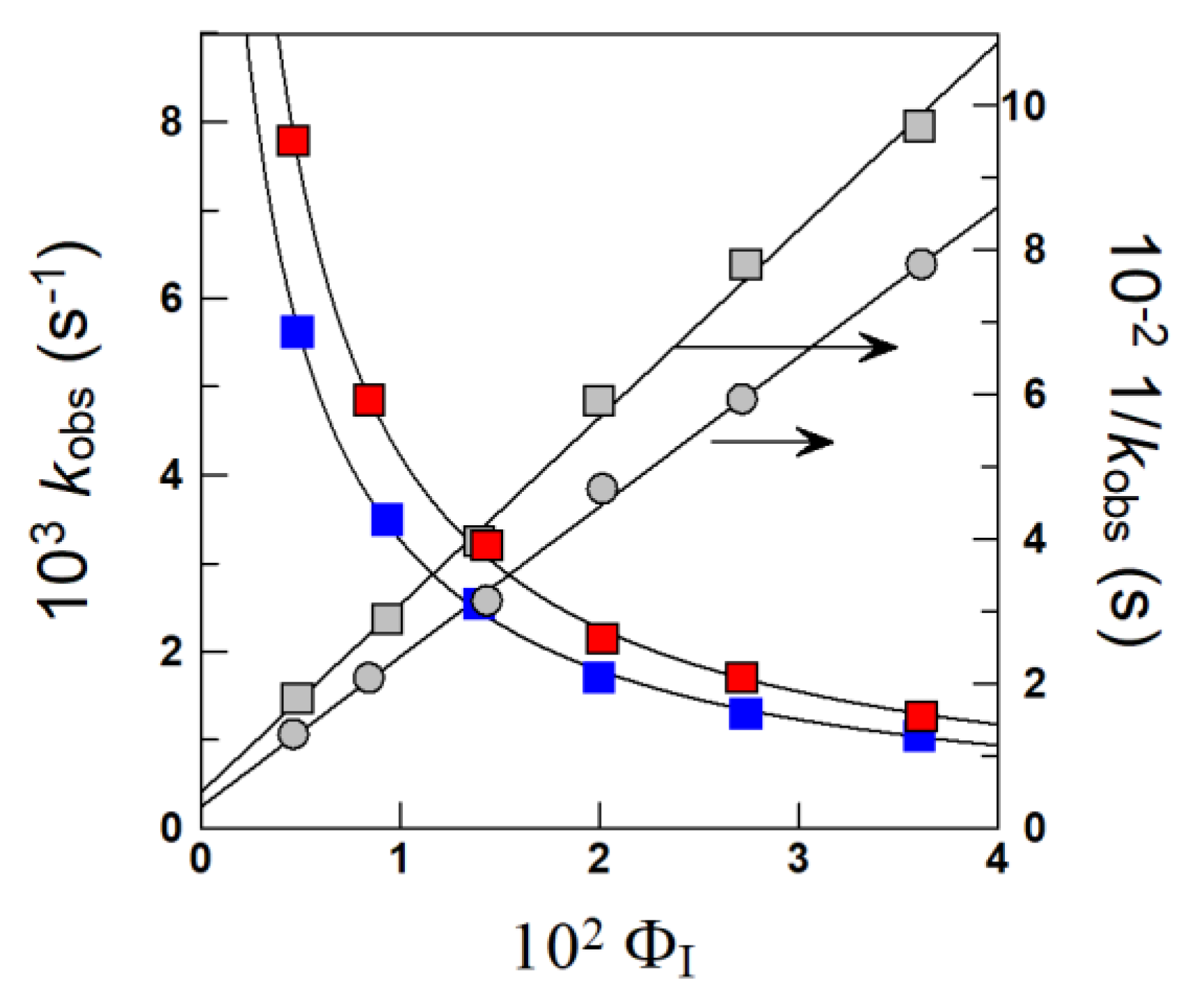
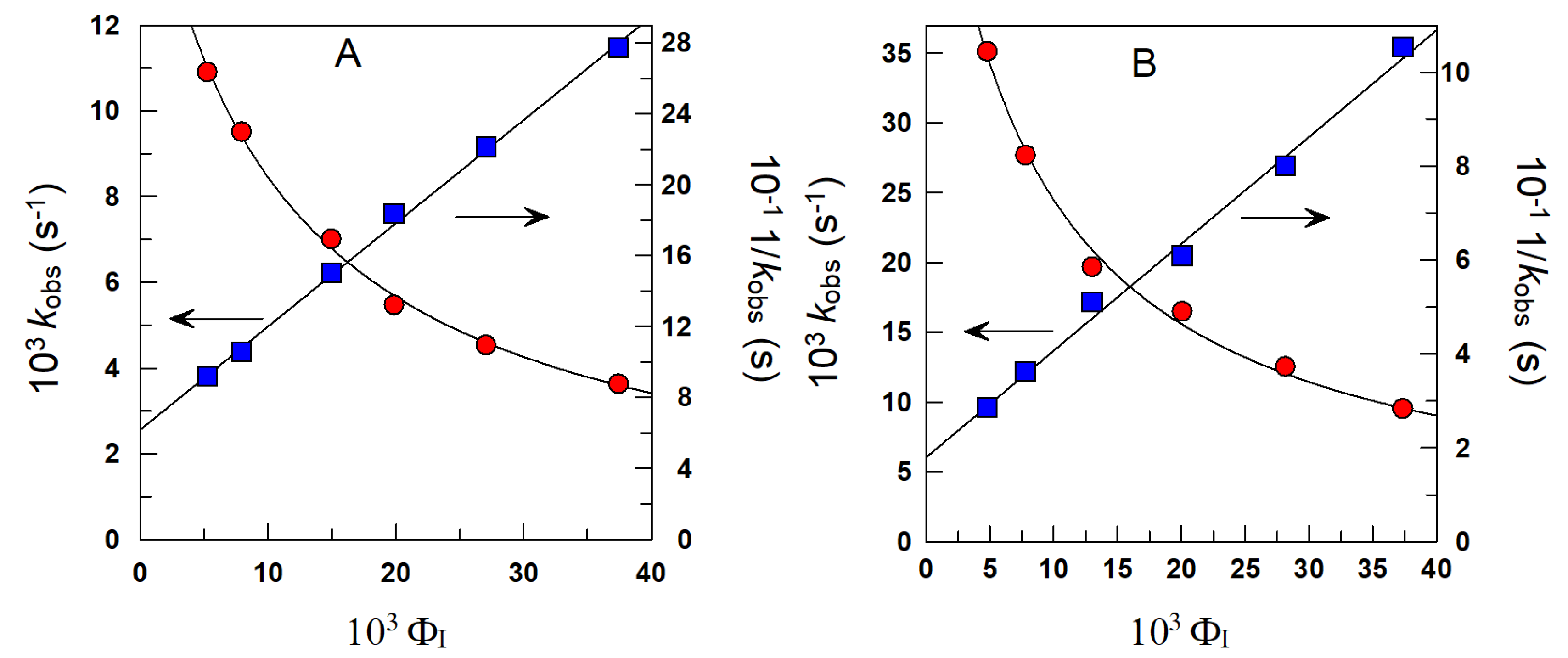
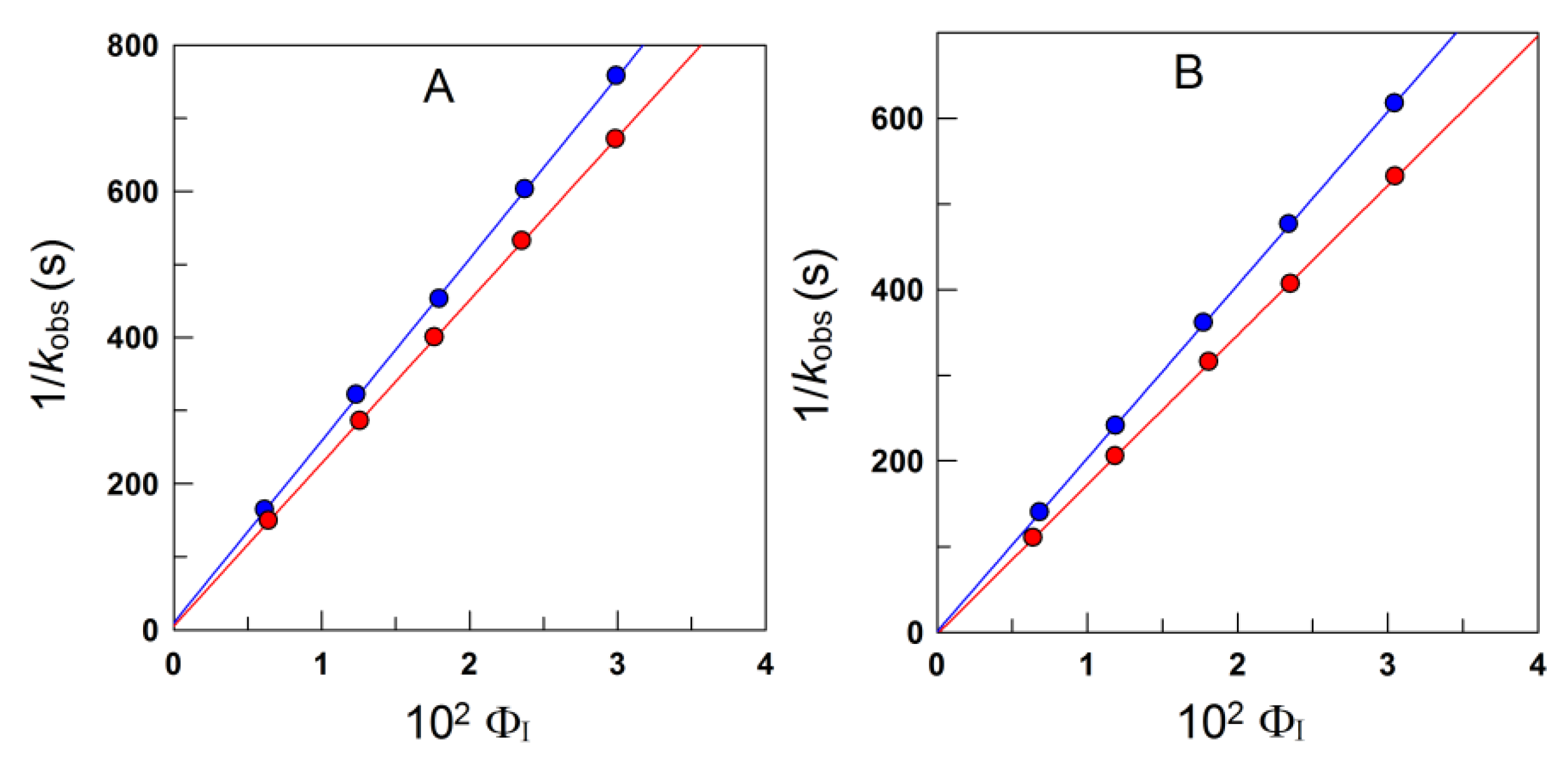
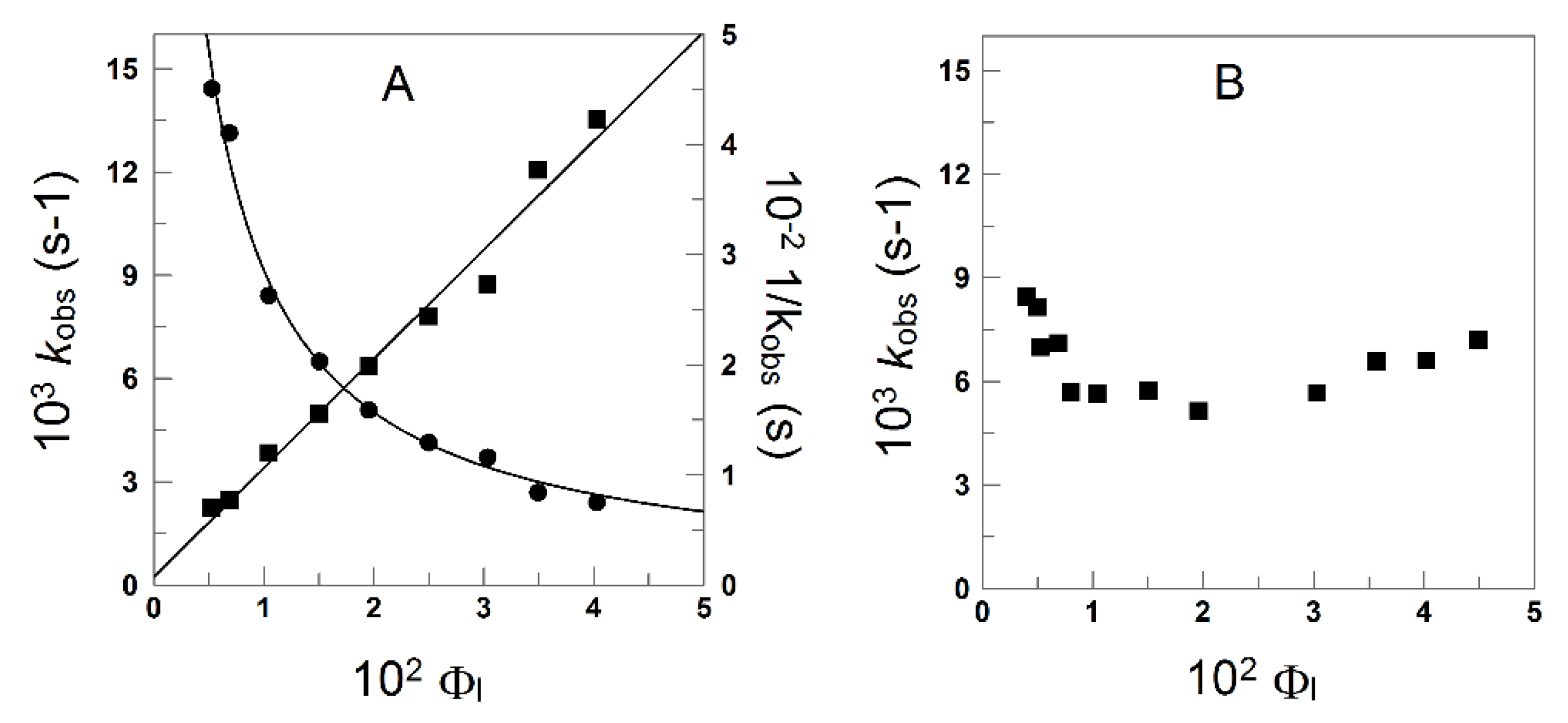
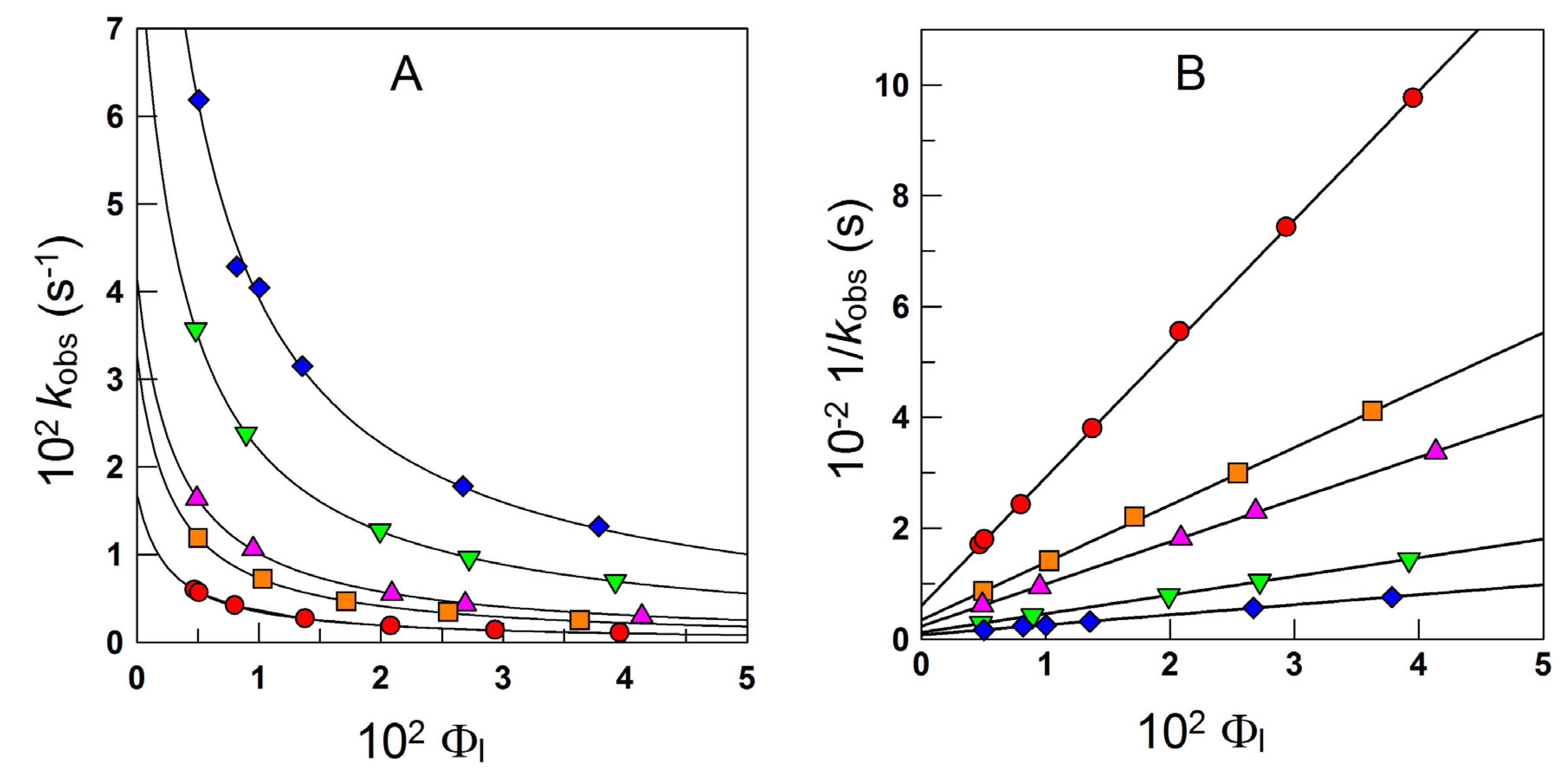
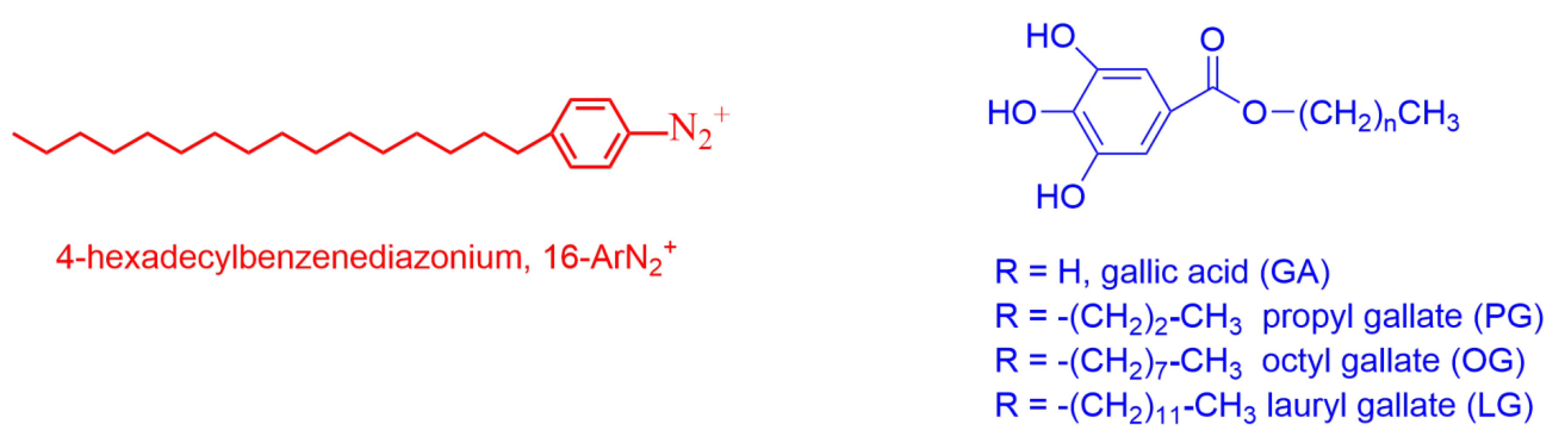
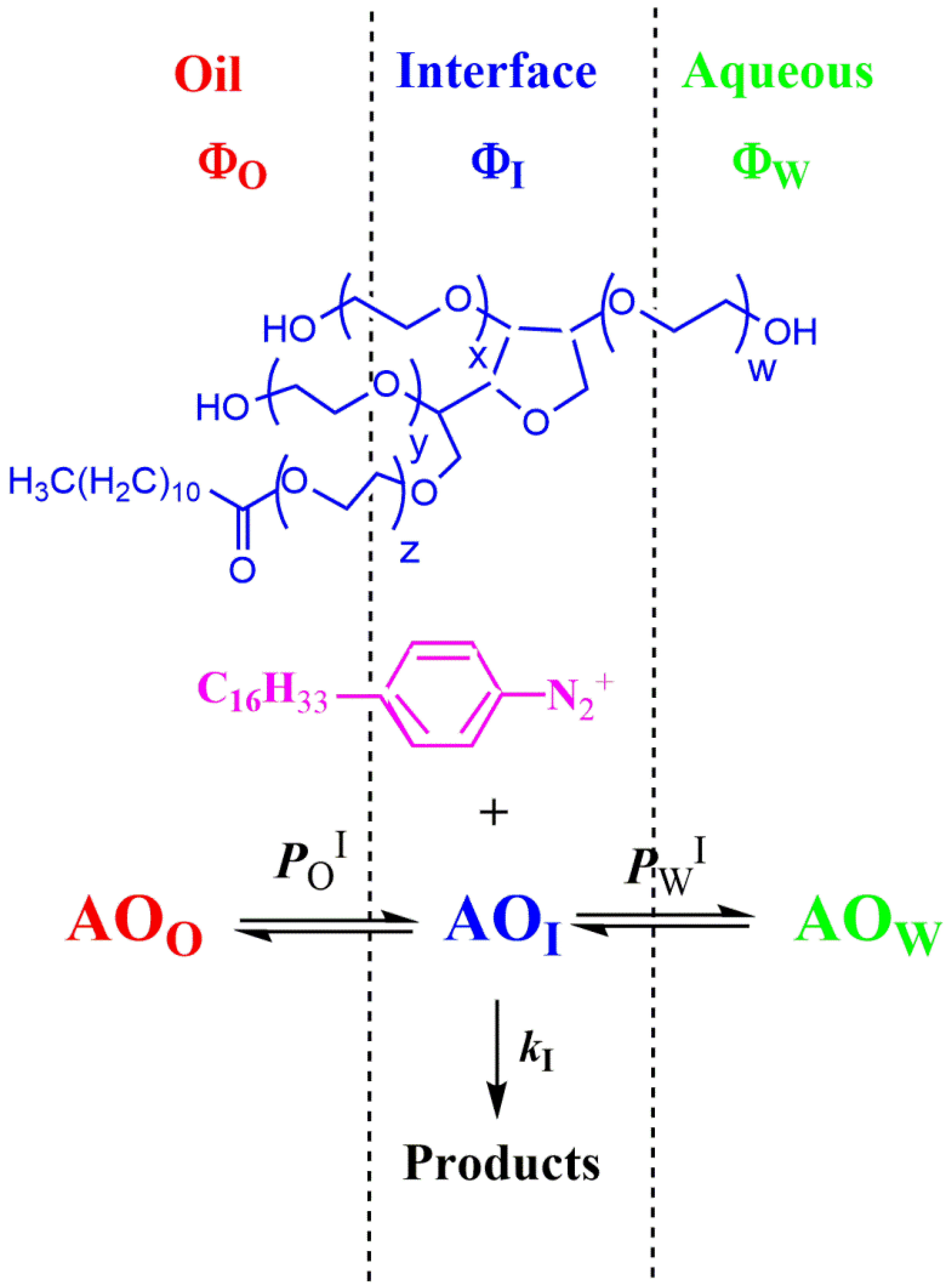
4.1.1. Reactions Where the Antioxidant Partitions between the Three Regions
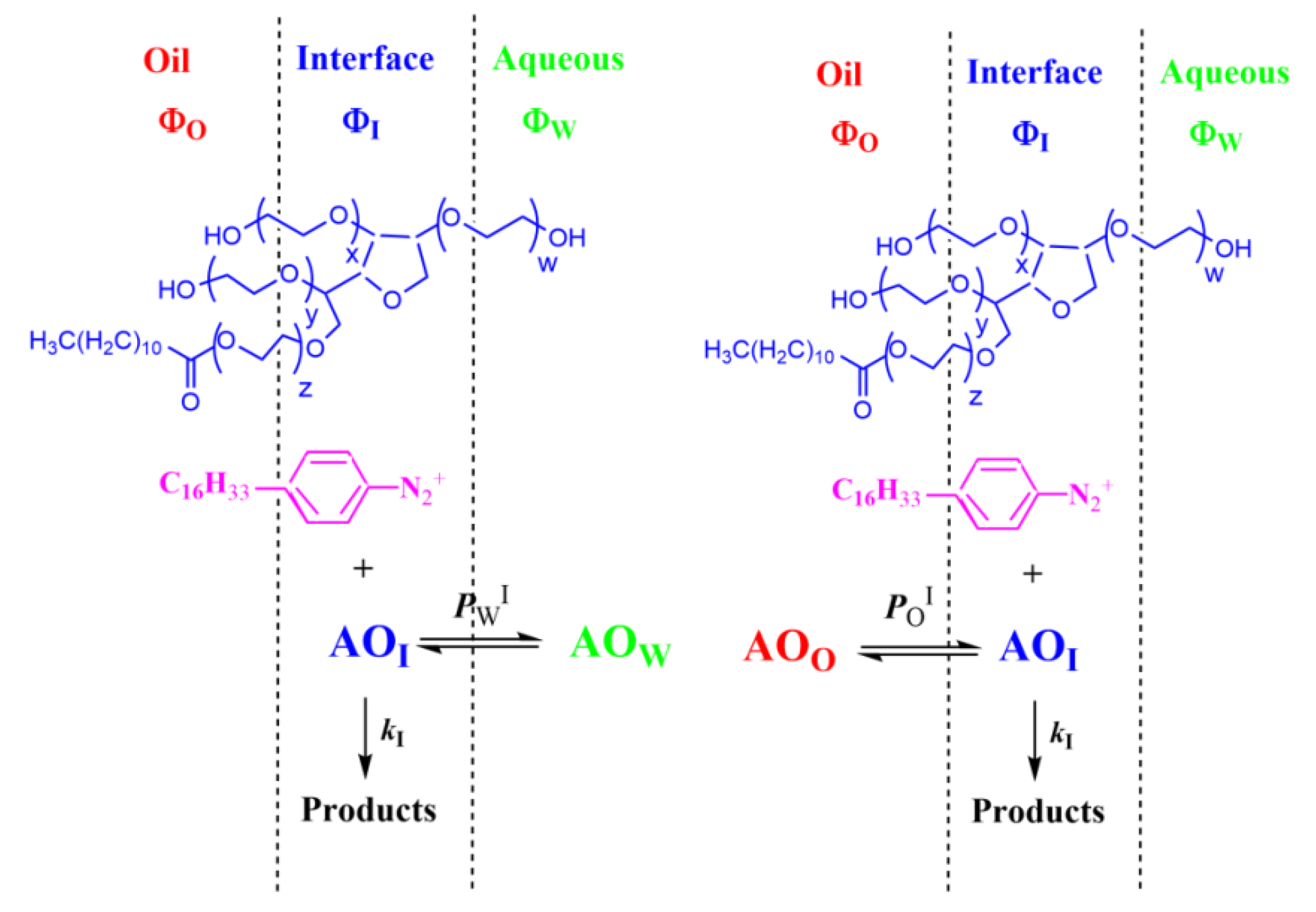
4.1.2. Reactions Where the Antioxidant Partitions between Two Regions
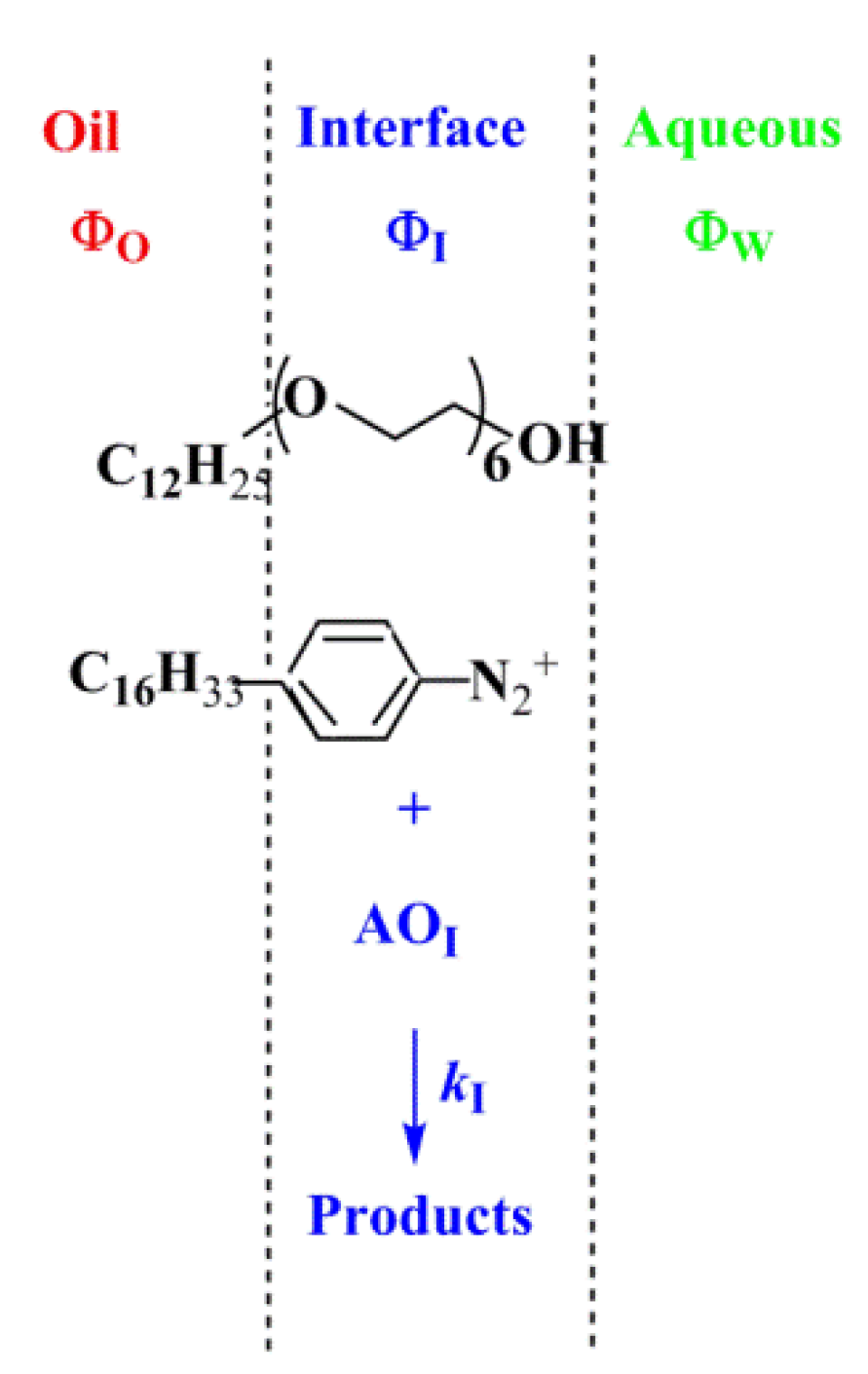
4.1.3. Reactions Taking Place Exclusively at the Interfacial Region
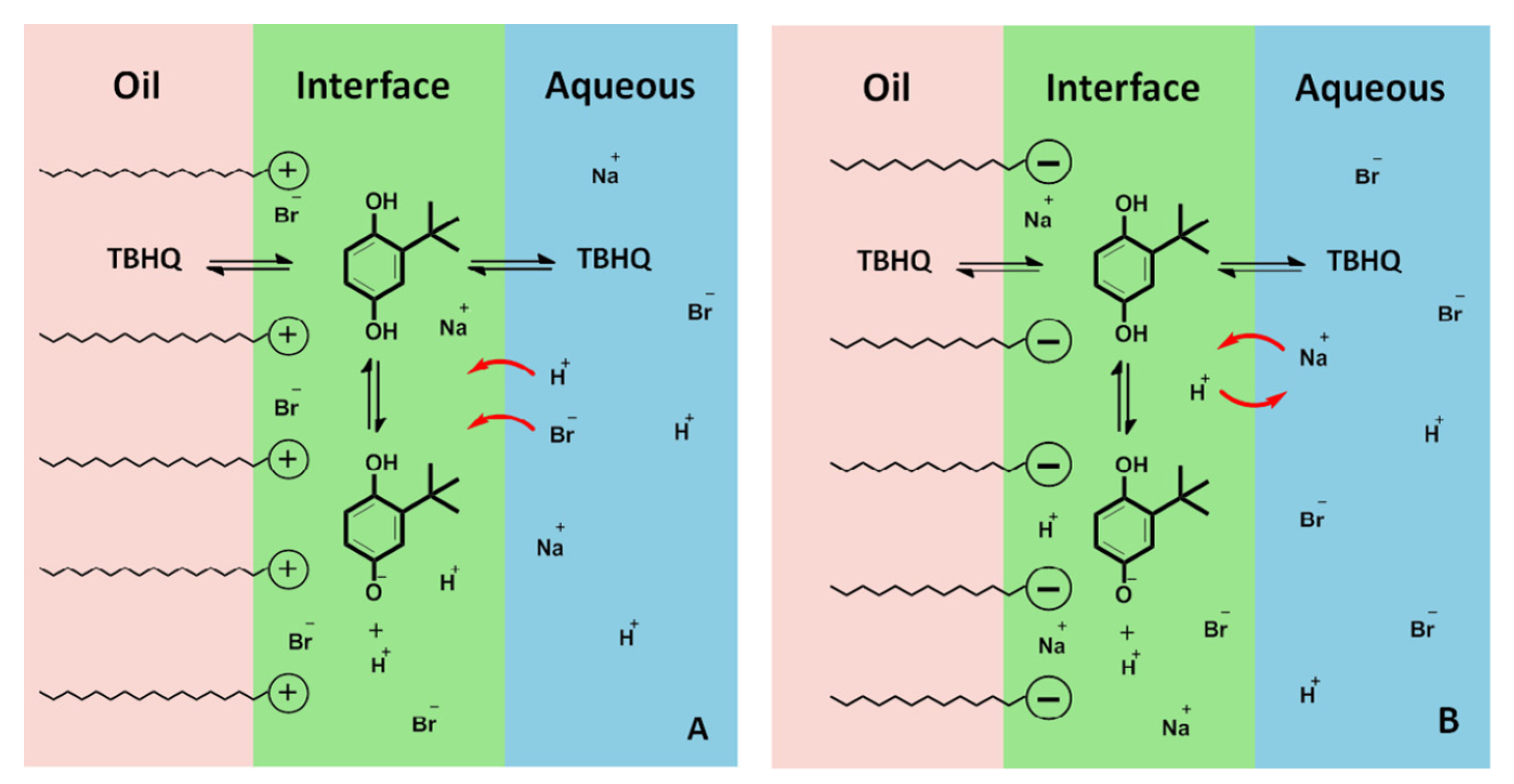
4.2. Reactions in Ionic Emulsions
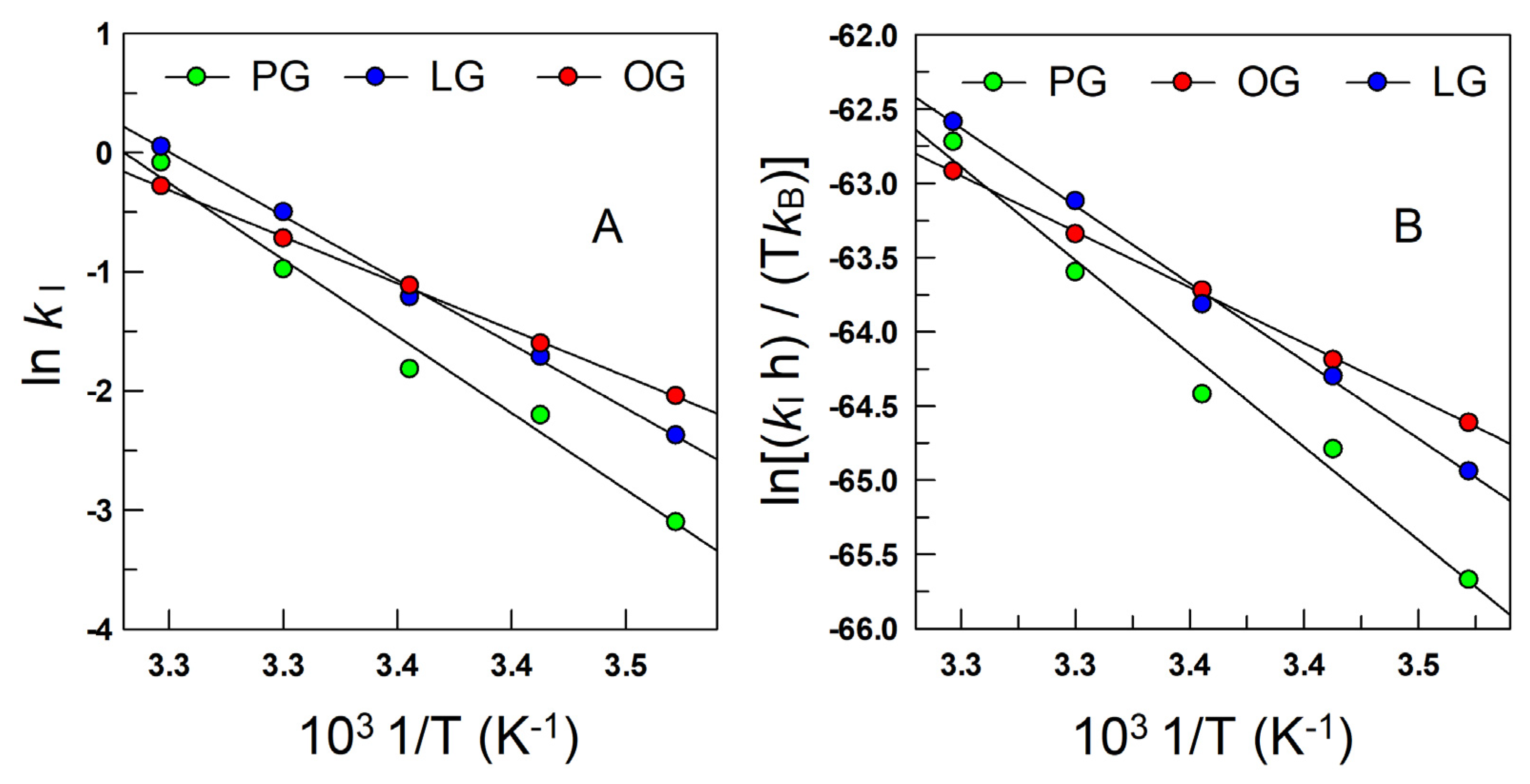
5. Effects of Temperature on the Kinetics in Emulsions: Interfacial Activation Parameters
6. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shahidi, F. Handbook of Antioxidants for Food Preservation, 1st ed.; Woodhead Publishing: Shaxton, UK, 2015. [Google Scholar]
- Frankel, E.N. Lipid Oxidation; The Oily Press: Bridgwater, UK; PJ Barnes & Associates: Bridgwater, UK, 2005. [Google Scholar]
- Costa, M.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Polyphenolic Antioxidants in Lipid Emulsions: Partitioning Effects and Interfacial Phenomena. Foods 2021, 10, 539. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Bravo-Díaz, C.; Paiva-Martins, F. Why encapsulate antioxidants in emulsion-based systems, where they are located, and how location affects their efficiency. In Emulsion-Based Encapsulation of Antioxidants; Aboudzadeh, M.A., Ed.; Springer Nature: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Larsson, K.; Fribera, S.E. Food Emulsions; Marcel Dekker: New York, NY, USA, 1990. [Google Scholar]
- Raman, M.; Almutairdi, A.; Mulesa, L.; Alberda, C.; Beattie, C.; Gramlich, L. Parenteral Nutrition and Lipids. Nutrients 2017, 9, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala-Vila, A.; Barbosa, V.M.; Calder, P.C. Olive oil in parenteral nutrition. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 165–174. [Google Scholar] [CrossRef]
- Gupta, A.; Eral, H.B.; Hatton, T.A.; Doyle, P.S. Nanoemulsions: Formation, properties and applications. Soft Matter 2016, 12, 2826–2841. [Google Scholar] [CrossRef] [Green Version]
- McClements, D.J.; Rao, J. Food-Grade Nanoemulsions: Formulation, Fabrication, Properties, Performance, Biological Fate, and Potential Toxicity. Crit. Rev. Food Sci. Nutr. 2011, 51, 285–330. [Google Scholar] [CrossRef]
- McClements, D.J. Emulsion Design to Improve the Delivery of Functional Lipophilic Components. Annu. Rev. Food Sci. Technol. 2010, 1, 241–269. [Google Scholar] [CrossRef]
- Grumezescu, A.M. Emulsions, Nanotechnology in the Agri-Food Industry; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Friberg, S.E.; Larsson, K. Food Emulsions; Marcel Dekker: New York, NY, USA, 1997. [Google Scholar]
- McClements, D.J. Food Emulsions: Principles, Practices and Techniques, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Coupland, J.N.; McClements, D.J. Lipid oxidation in food emulsions. Trends Food Sci. Technol. 1996, 7, 83–91. [Google Scholar] [CrossRef]
- Laguerre, M.; Bily, A.; Birtić, S. Lipid oxidation in food. In Lipids and Edible Oils; Galanakis, C.M., Ed.; Academic Press: London, UK, 2020; Chapter 7; pp. 243–287. [Google Scholar] [CrossRef]
- Berton-Carabin, C.C.; Ropers, M.-H.; Genot, C. Lipid Oxidation in Oil-in-Water Emulsions: Involvement of the Interfacial Layer. Compr. Rev. Food Sci. Food Saf. 2014, 13, 945–977. [Google Scholar] [CrossRef]
- McClemments, D.J.; Decker, E.A. Lipid Oxidation in Oil-in Water Emulsions: Impact of Molecular Environment on Chemical Reactions in Heterogeneous Food Systems. J. Food Sci. 2000, 65, 1270–1282. [Google Scholar] [CrossRef]
- Berton, C.; Ropers, M.H.; Viau, M.; Genot, C. Contribution of the interfacial layer to the protection of emulsified lipids against oxidation. J. Agric. Food Chem. 2011, 59, 5052–5061. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Díaz, C.; Romsted, L.S.; Liu, C.; Losada-Barreiro, S.; Pastoriza-Gallego, M.J.; Gao, X.; Gu, Q.; Krishnan, G.; Sánchez-Paz, V.; Zhang, Y.; et al. To Model Chemical Reactivity in Heterogeneous Emulsions, Think Homogeneous Microemulsions. Langmuir 2015, 31, 8961–8979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J. Unit D.35: Key Concepts of Interfacial Properties in Food Chemistry. In Current Protocols in Food Analytical Chemistry, Supplement; J. Wiley &Sons: New York, NY, USA, 2002; Supplement 5. [Google Scholar]
- Calder, P.C.; Waitzberg, D.L.; Klek, S.; Martindale, R.G. Lipids in Parenteral Nutrition: Biological Aspects. J. Parenter. Enter. Nutr. 2020, 44, S21–S27. [Google Scholar] [CrossRef]
- de Melo Barbosa, R.; Severino, P.; Finkler, C.L.L.; de Paula, E. Lipid-based colloidal carriers for transdermal administration of bioactives. In Materials for Biomedical Engineering; Holban, A.-M., Grumezescu, A.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Chapter 11; pp. 369–397. [Google Scholar] [CrossRef]
- Ghosh, N.; Das, A.; Chaffee, S.; Roy, S.; Sen, C.K. Reactive Oxygen Species, Oxidative Damage and Cell Death. In Immunity and Inflammation in Health and Disease; Chatterjee, S., Jungraithmayr, W., Bagchi, D., Eds.; Academic Press: London, UK, 2018; Chapter 4; pp. 45–55. [Google Scholar] [CrossRef]
- Romsted, L.S.; Bravo-Díaz, C. Modelling chemical reactivity in emulsions. Curr. Opin. Colloid Interface Sci. 2013, 18, 3–14. [Google Scholar] [CrossRef]
- Gunaseelan, K.; Romsted, L.S.; Pastoriza-Gallego, M.J.; González-Romero, E.; Bravo-Díaz, C. Determining a-tocopherol distributions betweeen the Oil, Water and Interfacial Regions of Macroemulsions: Novel Applications of Electrocanalytical Chemistry and a Pseudophase Kinetic Model. Adv. Colloid Interface Sci. 2006, 123, 303–311. [Google Scholar] [CrossRef]
- Astray, G.; Cid, A.; Garcia Rio, L.; Hervella, P.; Mejuto, J.C.; Pérez-Lorenzo, M. Organic reactivity in AOT-stabilized microemulsions. Prog. React. Kin. Mech. 2008, 33, 81–97. [Google Scholar] [CrossRef]
- Lopez-Quintela, M.A.; Tojo, C.; Blanco, M.C.; Garcia Rio, L.; Leis, J.R. Microemulsion dynamics and reactions in microemulsions. Curr. Opin. Colloid Interface Sci. 2004, 9, 264–278. [Google Scholar] [CrossRef]
- Garti, N. Microemulsions as microreactors for food applications. Curr. Opin. Colloid Interface Sci. 2003, 8, 197–211. [Google Scholar] [CrossRef]
- Bunton, C.; Yao, J.; Romsted, L.S. Micellar Catalysis: A useful misnomer. Curr. Opin. Colloid Interface Sci. 1997, 2, 622–628. [Google Scholar]
- Khan, M.N. Micellar Catalysis; Science, S., Ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Laguerre, M.; Bily, A.; Roller, M.; Birtić, S. Mass Transport Phenomena in Lipid Oxidation and Antioxidation. Annu. Rev. Food Sci. Technol. 2017, 8, 391–411. [Google Scholar] [CrossRef]
- McClements, D.J. Food Emulsions; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Tadros, T.F. Surfactants; Academic Press: London, UK, 1984. [Google Scholar]
- Costa, M.; Freiría-Gándara, J.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Effects of droplet size on the interfacial concentrations of antioxidants in fish and olive oil-in-water emulsions and nanoemulsions and on their oxidative stability. J. Colloid Interface Sci. 2020, 562, 352–362. [Google Scholar] [CrossRef]
- McClements, D.J. Enhanced delivery of lipophilic bioactives using emulsions: A review of major factors affecting vitamin, nutraceutical and lipid bioaccessibility. Food Funct. 2018, 9, 22–41. [Google Scholar] [CrossRef]
- García-Pérez, P.; Lozano-Milo, E.; Gallego, P.P.; Tojo, C.; Losada-Barreiro, S.; Bravo-Díaz, C. Plant Antioxidants in Food Emulsions. In Some New Aspects of Colloidal Systems in Foods; Milani, J.M., Ed.; BoD–Books on Demand: Hamburg, Germany, 2018; Volume Plant Antioxidants in Food Emulsions. [Google Scholar]
- Quina, F.H. Dynamics and prototropic reactivity of electronically excited states in simple surfactant aggregates. Curr. Opin. Colloid Interface Sci. 2013, 18, 35–39. [Google Scholar] [CrossRef]
- Malassagne-Bulgarelli, N.; McGrath, K.M. Emulsion ageing: Effect on the dynamics of oil exchange in oil-in-water emulsions. Soft Matter 2013, 9, 48–59. [Google Scholar] [CrossRef]
- Skhiri, Y.; Gruner, P.; Semin, B.; Brosseau, Q.; Pekin, D.; Mazutis, L.; Goust, V.; Kleinschmidt, F.; El Harrak, A.; Hutchison, J.B.; et al. Dynamics of molecular transport by surfactants in emulsions. Soft Matter 2012, 8, 10618–10627. [Google Scholar] [CrossRef]
- Malassagne-Bulgarelli, N.; McGrath, K.M. Dynamics of oil transfer in oil-in-water emulsions. Soft Matter 2009, 5, 4804–4813. [Google Scholar] [CrossRef]
- Wooster, T.J.; Golding, M.; Sanguansri, P. Impact of Oil Type on Nanoemulsion Formation and Ostwald Ripening Stability. Langmuir 2008, 24, 12758–12765. [Google Scholar] [CrossRef] [PubMed]
- Colafemmina, G.; Palazzo, G.; Ceglie, A.; Ambrosone, L.; Cinelli, G.; Di Lorenzo, V. Restricted Diffusion: An Effective Tool to Investigate Food Emulsions. Prog. Colloid Polym. Sci. 2002, 120, 23–27. [Google Scholar]
- Soderman, O.; Nyden, M. NMR translational diffusion studies of a model microemulsion. Colloid Surf. A: Physicochem. Eng. Aspects 1999, 158, 273–280. [Google Scholar] [CrossRef]
- Levinger, N.E. Ultrafast dynamics in reverse micelles, microemulsions, and vesicles. Curr. Opin. Colloid Interface Sci. 2000, 5, 118–124. [Google Scholar] [CrossRef]
- Kabalnov, A.S. Can Micelles Mediate a Mass Transfer between Oil Droplets? Langmuir 1994, 10, 680–684. [Google Scholar] [CrossRef]
- Smoluchowski, M.V. Versuch Einer Mathematischen Theorie der Koagulationskinetik kolloider Losungen. Z. Phys. Chem. 1918, 92, 129–168. [Google Scholar] [CrossRef] [Green Version]
- Andraos, J. The Contributions of Solomon F. Acree (1875–1957) and the Centennial Anniversary of the Discovery of the Acree–Curtin–Hammett Principle. Chem. Educator 2008, 13, 170–178. [Google Scholar]
- Bravo-Díaz, C.; Romsted, L.S.; Losada-Barreiro, S.; Paiva-Martins, F. Using a pseudophase model to determine AO distributions in emulsions: Why dynamic equilibrium matters. Eur. J. Lipid Sci. Technol. 2017, 119, 1600277. [Google Scholar] [CrossRef]
- Anslyn, E.V.; Dougherty, D.A. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, USA, 2006. [Google Scholar]
- Laidler, K.J. Chemical Kinetics, 3rd ed.; Harper Cpllins Pub.: New York, NY, USA, 1987. [Google Scholar]
- Van Boekel, M.A.J.S. Kinetic Modeling of Reactions in Foods; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- van Boekel, M.A.J.S. Kinetic Modeling of Food Quality: A Critical Review. Compr. Rev. Food Sci. Food Saf. 2008, 7, 144–158. [Google Scholar] [CrossRef]
- Espenson, J.H. Chemical Kinetics and Reaction Mechanisms, 2nd ed.; McGraw-Hill: New York, NY, USA, 1995. [Google Scholar]
- Atkins, P.; de Paula, J. Physical Chemistry; W. H. Freeman: New York, NY, USA, 2010. [Google Scholar]
- Pace, T.C.S.; Bohne, C. Dynamics of guest binding to supramolecular systems: Techniques and selected examples. In Advances in Physical Organic Chemistry; Richard, J.P., Ed.; Academic Press: London, UK, 2007; Volume 42, pp. 167–223. [Google Scholar]
- Quina, F.H.; Lissi, E.A. Photoprocesses in Microaggregates. Acc. Chem. Res. 2004, 37, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Meireles, M.; Losada-Barreiro, S.; Costa, M.; Paiva-Martins, F.; Bravo-Díaz, C.; Monteiro, L.S. Control of antioxidant efficiency of chlorogenates in emulsions: Modulation of antioxidant interfacial concentrations. J. Sci. Food Agric. 2020, 99, 3917–3925. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Losada-Barreiro, S.; Bravo-Díaz, C.; Vicente, A.A.; Monteiro, L.S.; Paiva-Martins, F. Influence of AO chain length, droplet size and oil to water ratio on the distribution and on the activity of gallates in fish oil-in-water emulsified systems: Emulsion and nanoemulsion comparison. Food Chem. 2020, 310, 125716. [Google Scholar] [CrossRef] [Green Version]
- Raimúndez-Rodríguez, E.A.; Losada-Barreiro, S.; Bravo-Díaz, C. Enhancing the fraction of antioxidants at the interfaces of oil-in-water emulsions: A kinetic and thermodynamic analysis of their partitioning. J. Colloid Interface Sci. 2019, 555, 224–233. [Google Scholar] [CrossRef]
- Mitrus, O.; Żuraw, M.; Losada-Barreiro, S.; Bravo-Díaz, C.; Paiva-Martins, F. Targeting Antioxidants to Interfaces: Control of the Oxidative Stability of Lipid-Based Emulsions. J. Agric. Food Chem. 2019, 67, 3266–3274. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Bravo-Díaz, C. Free radicals and polyphenols: The redox chemistry of neurodegenerative diseases. Eur. J. Med. Chem 2017, 133, 379–402. [Google Scholar] [CrossRef]
- Dar, A.A.; Bravo-Diaz, C.; Nazir, N.; Romsted, L.S. Chemical kinetic and chemical trapping methods: Unique approaches for determining respectively the antioxidant distributions and interfacial molarities of water, counter-anions, and other weakly basic nucleophiles in association colloids. Curr. Opin. Colloid Interface Sci. 2017, 32, 84–93. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Sánchez-Paz, V.; Bravo-Díaz, C. Transfer of antioxidants at the interfaces of model food emulsions: Distributions and thermodynamic parameters. Org. Biomol. Chem. 2015, 13, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Menger, F.M.; Portnoy, C.E. Chemistry of reactions proceeding inside molecular aggregates. J. Am. Chem. Soc. 1967, 89, 4698–4703. [Google Scholar] [CrossRef]
- Romsted, L.S. Micellar Effects on Reaction Rates and Equilibria. In Surfactants in Solution; Mittal, K.L., Lindman, J., Eds.; Plenum Press: New York, NY, USA, 1984. [Google Scholar]
- Romsted, L.S.; Zanette, D. Quantitative Treatment of Indicator Equilibria in Micellar Solutions of Sodium Decyl Phosphate amd Sodium Dodecyl Sulfate. J. Phys. Chem. 1988, 92, 4690. [Google Scholar] [CrossRef]
- Bunton, C.A.; Nome, F.; Quina, F.H.; Romsted, L.S. Ion Binding and Reactivity at Charged Aqueous Interfaces. Acc. Chem. Res. 1991, 24, 357–364. [Google Scholar] [CrossRef]
- Garcia-Rio, L.; Leis, J.R.; Mejuto, J.C.; Perez-Lorenzo, M. Microemulsions as microreactors in physical organic chemistry. Pure Appl. Chem. 2007, 79, 1111–1123. [Google Scholar] [CrossRef] [Green Version]
- Muriel-Delgado, F.; Jiménez, R.; Gómez-Herrera, C.; Sánchez, F. Use of the Brönsted Equation in the Interpretation of Micellar Effects in Kinetics (II). Study of the Reaction [Fe(CN)5(4-CNpy)]3− + CN− ⇄ Fe(CN)64− + 4-CNpy in CTACl Micellar Solutions. Langmuir 1999, 15, 4344–4350. [Google Scholar] [CrossRef]
- Blasko, A.; Bunton, C.A.; Hong, Y.S.; Mhala, M.M.; Moffat, J.R.; Wright, S. Micellar Rate Effects on Reactions of Hydroxide Ion with Phosphinate and Thiophosphinate Esters. J. Phys. Org. Chem. 1991, 4, 618–628. [Google Scholar] [CrossRef]
- Bunton, C.A. The dependence of micellar rate effects upon reaction mechanism. Adv. Colloid Interface Sci. 2006, 123, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Bunton, C.A.; Hong, Y.-S.; Romsted, L.S. A Quantitative Treatment of the Deprotonation Equilibria of Benzimidazole in Basic Solutions of Cetyltrimethylammonium Ion (CTAX) Surfactants. In Solution Behaviour of Surfactants, Theoretical and Applied Aspects; Mittal, K.L., Fendler, E.J., Eds.; Springer: New York, NY, USA, 1982; Volume 2, pp. 1137–1155. [Google Scholar]
- Behera, G.; Mishra, B.; Behera, P.; Panda, M. Fluorescent probes for structural and distance effect studies in micelles, reversed micelles and microemulsions. Adv. Colloid Interface Sci. 1999, 82, 1–42. [Google Scholar] [CrossRef]
- Shoemaker, R.; Holmberg, K. Reactions in organized surfactant systems. In Microemulsions: Background, New Concepts, Applications, Perspectives; Stubenrauch, S., Ed.; Blackwell Publishing: Oxford, UK, 2009; p. 148. [Google Scholar]
- Williams, D.; Lyn, H. Denitrosation of N-Methyl-N-nitrosotoluene-p-sulphonamide in Acid Solution. J. Chem. Soc. Perkin Trans. II 1976, 15, 1838–1841. [Google Scholar] [CrossRef]
- Bravo, C.; Herves, P.; Leis, J.R.; Peña, M.E. Micellar Effects in the Acid Denitrosation of N-Nitroso-N-methyl-p-toluensulfonamide. J. Phys. Chem. 1990, 94, 8816–8820. [Google Scholar] [CrossRef]
- Bravo, C.; Leis, J.R.; Peña, M.E. Effect of Alcohols on Catalysis by Dodecyl Sulfate Micelles. J. Phys. Chem. 1992, 96, 1957–1961. [Google Scholar] [CrossRef]
- Iglesias, E.; Garcia-Rio, L.; Leis, J.R.; Casado, J. Nitrosation reactions and nitrosocompounds in microheterogeneous media. Recent Res. Devel. Phys. Chem. 1997, 1, 403. [Google Scholar]
- Iglesias, E. Cyclodextrins as Enzyme Models in Nitrosation and in Acid−Base-Catalyzed Reactions of Alkyl Nitrites. J. Am. Chem. Soc. 1998, 120, 13057–13069. [Google Scholar] [CrossRef]
- Aliaga, C.; Bravo-Moraga, F.; Gonzalez-Nilo, D.; Márquez, S.; Lürh, S.; Mena, G.; Rezende, M.C. Location of TEMPO derivatives in micelles: Subtle effect of the probe orientation. Food Chem. 2016, 192, 395–401. [Google Scholar] [CrossRef]
- Aliaga, C.; Celis, F.; Lürh, S.; Oñate, R. TEMPO-Attached Pre-fluorescent Probes Based on Pyridinium Fluorophores. J. Fluoresc. 2015, 25, 979–983. [Google Scholar] [CrossRef]
- Lopez de Arbina, A.; Losada-Barreiro, S.; Rezende, M.C.; Vidal, M.; Aliaga, C. The location of amphiphobic antioxidants in micellar systems: The diving-swan analogy. Food Chem. 2019, 279, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Romsted, L.S. Interfacial Compositions of Surfactant Assemblies by Chemical Trapping with Arenediazonium Ions: Method and Applications. In Reactions and Synthesis in Surfactant Systems; Texter, J., Ed.; Marcel Dekker: New York, NY, USA, 2001. [Google Scholar]
- Losada-Barreiro, S.; Bravo Díaz, C.; Paiva Martins, F.; Romsted, L.S. Maxima in antioxidant distributions and efficiencies with increasing hydrophobicity of gallic acid and its alkyl esters. The pseudophase model interpretation of the ‘‘cut-off’’ effect. J. Agric. Food Chem. 2013, 61, 6533–6543. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Bravo-Díaz, C.; Romsted, L.S. Interpreting ion-specific effects on the reduction of an arenediazonium Ion by t-butylhydroquinone (TBHQ) using the pseudophase kinetic model in emulsions prepared with a zwitterionic sulfobetaine surfactant. Langmuir 2013, 29, 4928–4933. [Google Scholar] [CrossRef] [PubMed]
- Lisete-Torres, P.; Losada-Barreiro, S.; Albuquerque, H.; Sánchez-Paz, V.; Paiva-Martins, F.; Bravo-Díaz, C. Distribution of hydroxytyrosol and hydroxytyrosol acetate in olive oil emulsions and their antioxidant efficiency. J. Agric. Food Chem. 2012, 60, 7318–7325. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Bravo-Díaz, C.; Romsted, L.S. Using the pseudophase kinetic model to interpret chemical reactivity in ionic emulsions: Determining antioxidant partition constants and interfacial rate constants. J. Colloid Interface Sci. 2012, 400, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Chauduri, A.; Loughlin, J.A.; Romsted, L.S.; Yao, J. Arenediazonium Salts: New Probes of Interfacial Compositions of Association Colloids. 1. Basic Approach, Methods and Ilustrative Applications. J. Am. Chem. Soc. 1993, 115, 8351. [Google Scholar] [CrossRef]
- Chauduri, A.; Romsted, L.S.; Yao, J. Arenediazonium Salts: New Probes of Interfacial Compositions of Association Colloids. 2. Binding Constants of Butanol and Hexanol in Aqueous Three-Component Cetyltrimehylamonium Bromide Microemulsions. J. Am. Chem. Soc. 1993, 115, 8362. [Google Scholar] [CrossRef]
- Freiría-Gándara, J.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Differential Partitioning of Bioantioxidants in Edible Oil–Water and Octanol–Water Systems: Linear Free Energy Relationships. J. Chem. Eng. Data 2018, 63, 2999–3007. [Google Scholar] [CrossRef]
- Figueroa-Espinoza, M.C.; Villeneuve, P. Phenolic acids enzymatic lipophilization. J. Agric. Food Chem. 2005, 53, 2779–2787. [Google Scholar] [CrossRef] [PubMed]
- Kahveci, D.; Laguerre, M.; Villeneuve, P. Phenolipids as New Antioxidants: Production, Activity, and Potential Applications. In Polar Lipids; Elsevier: Amsterdam, The Netherlands, 2015; pp. 185–214. [Google Scholar] [CrossRef]
- Romsted, L.S.; Zhang, J. Kinetic method for Determining Antioxidant Distributions in Model Food Emulsions: Distribution constants of TBHQ in Mixtures of Octane, Water and a Nonionic Emulsifier. J. Agric. Food. Chem. 2002, 50, 3328–3336. [Google Scholar] [CrossRef]
- Gunaseelan, K.; Romsted, L.S.; González-Romero, E.; Bravo-Díaz, C. Determining Partition Constants of Polar Organic Molecules between the Oil/Interfacial and Water/ Interfacial Regions in Emulsions: A Combined Electrochemical and Spectrometric Method. Langmuir 2004, 20, 3047–3055. [Google Scholar] [CrossRef]
- Sánchez-Paz, V.; Pastoriza-Gallego, M.J.; Losada-Barreiro, S.; Bravo-Diaz, C.; Gunaseelan, K.; Romsted, L.S. Quantitative determination of a-tocopherol distribution in a tributyrin/Brij 30/water model food emulsion. J. Colloid Interface Sci. 2008, 320, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C.; Romsted, L.S. A direct correlation between the antioxidant efficiencies of caffeic acid and its alkyl esters and their concentrations in the interfacial region of olive oil emulsions. The pseudophase model interpretation of the ‘‘cut-off’’ effect. Food Chem. 2015, 175, 233–242. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Sánchez Paz, V.; Bravo Díaz, C. Effects of emulsifier hydrophile–lipophile balance and emulsifier concentration the distributions of gallic acid, propyl gallate, and a-tocopherol in corn oil emulsions. J. Colloid Interface Sci. 2012, in press. [Google Scholar] [CrossRef]
- Freiría-Gándara, J.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Enhancement of the antioxidant efficiency of gallic acid derivatives in intact fish oil-in-water emulsions through optimization of their interfacial concentrations. Food Funct. 2018, 9, 4429–4442. [Google Scholar] [CrossRef]
- Pastoriza-Gallego, M.J.; Losada-Barreiro, S.; Bravo-Díaz, C. Interfacial kinetics in octane based emulsions. Effects of surfactant concentration on the reaction between 16-ArN2+ and octyl and lauryl gallates. Colloids Surf. A Physicochem. Eng. Asp. 2015, 480, 171–177. [Google Scholar] [CrossRef]
- Savelli, G.; Germani, R.; Brinchi, L. Reactivity control by Aqueous self-Assembling Systems. In Reactions and Synthesis in Surfactant Systems; Texter, J., Ed.; Marcel Dekker: New York, NY, USA, 2001. [Google Scholar]
- Oehlke, K.; Heins, A.; Stöckmann, H.; Schwarz, K. Impact of emulsifier microenvironments on acid—Base equilibrium and activity of antioxidants. Food Chem. 2010, 118, 48–55. [Google Scholar] [CrossRef]
- Fendler, J.H.; Fendler, E.F. Catalysis in Micellar and Macromolecular Systems; Academic Press: New York, NY, USA, 1975. [Google Scholar]
- Romsted, L.S. Quantitative Treatment of Benzimidazole Deprotonation Equilibria in Aqueous Micellar Solutions of Cetyltrimetylammonium Ions (CTAX, X = Cl−, Br− and NO3−) Surfactants. J. Phys. Chem. 1985, 89, 5113. [Google Scholar] [CrossRef]
- Romsted, L.S. Do Amphiphile Aggregate Morphologies and Interfacial Compositions Depend Primarily on Interfacial Hydration and Ion-Specific Interactions? The Evidence from Chemical Trapping. Langmuir 2007, 23, 414–424. [Google Scholar] [CrossRef]
- Quina, F.H.; Politi, M.J.; Cuccovia, I.M.; Martins-Franchetti, S.M.; Chaimovich, H. Alkaline hydrolysis in micellar sodium dodecyl sulfate: The binding of hydroxide ion to anionic micelles. In Solution Behavior of Surfactants: Theoretical and Applied Aspects; Mittal, K.L., Fendler, E.J., Eds.; Plenum Press: New York, NY, USA, 1982; Volume 2, p. 1125. [Google Scholar]
- Bravo Díaz, C. Diazohydroxides, diazoethers and related species. In The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids; Rappoport, Z., Liebman, J.F., Eds.; J. Wiley & Sons: Chichester, UK, 2011; Volume 2, p. 853. [Google Scholar]
- Maskill, H. The Physical Basis of Organic Chemistry; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Martinez-Aranda, N.; Losada-Barreiro, N.; Bravo-Díaz, C.; Romsted, L.S. Influence of Temperature on the Distribution of Catechin in Corn oil-in-water Emulsions and some Relevant Thermodynamic Parameters. Food Biophys. 2014, 9, 380–388. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Sánchez Paz, V.; Bravo Díaz, C.; Paiva Martins, F.; Romsted, L.S. Temperature and emulsifier concentration effects on gallic acid distribution in a model food emulsion. J. Colloid Interface Sci. 2012, 370, 73–79. [Google Scholar] [CrossRef]
- Turro, N.J.; Yekta, A. Luminescent probes for detergent solutions. A simple procedure for determination of the mean aggregation number of micelles. J. Am. Chem. Soc. 1978, 100, 5951–5952. [Google Scholar] [CrossRef]
- Bunton, C.A.; Savelli, G. Organic Reactivity in Aqueous Micelles and Similar Assemblies. Adv. Phys. Org. Chem. 1986, 22, 213–309. [Google Scholar]
- Biasutti, M.A.; Abuin, E.; Silber, J.; Correa, M.; Lissi, E.A. Kinetics of reactions catalyzed by enzymes in solutions of surfactants. Adv. Colloid Interface Sci. 2008, 136, 1–24. [Google Scholar] [CrossRef]
- López-Cornejo, P.; Pérez, P.; García, F.; de la Vega, R.; Sánchez, F. Use of the Pseudophase Model in the Interpretation of Reactivity under Restricted Geometry Conditions. An Application to the Study of the [Ru(NH3)5pz]2+ + S2O82- Electron-Transfer Reaction in Different Microheterogeneous Systems. J. Am. Chem. Soc. 2002, 124, 5154–5164. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, I. Chemical Reactions and Solvation at Liquid Interfaces: A Microscopic Perspective. Chem. Rev. 1996, 96, 1449–1476. [Google Scholar] [CrossRef] [PubMed]
- Bondar, A.-N.; Lemieux, M.J. Reactions at Biomembrane Interfaces. Chem. Rev. 2019, 119, 6162–6183. [Google Scholar] [CrossRef] [PubMed]
- Zarbin, A.J.G. Liquid–liquid interfaces: A unique and advantageous environment to prepare and process thin films of complex materials. Mater. Horizons 2021, 8, 1409–1432. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HLB Range | Application |
|---|---|
| <3 | Surface films |
| 3–6 | Water-in-oil emulsions |
| 7–9 | Wetting agents |
| 8–15 | Oil-in-water emulsions |
| 13–15 | Detergents |
| 15–18 | Solubilizers |
| Ea (kJ mol−1) | kchem (dm3 mol−1 s−1) |
|---|---|
| ~0 | ~1010 |
| <25 | 109–1010 |
| 50 | 105 |
| 100 | 10−4 |
| >200 | <10−22 |
| AO | Emulsion | Intercept | 102 kI (M−1 s−1) |
|---|---|---|---|
| OG | 1:1 | 5 ± 5 | 1.0 ± 0.1 |
| OG | 1:4 | 2 ± 2 | 1.1 ± 0.2 |
| LG | 1:1 | 1 ± 1 | 1.2 ± 0.1 |
| LG | 1:4 | −2 ± 2 | 1.4 ± 0.3 |
| CA | TOC | PG | OG | LG | |
|---|---|---|---|---|---|
| T (°C) | 102 kI (M−1 s−1) | 102 kI (M−1 s−1) | 102 kI (M−1 s−1) | 102 kI (M−1 s−1) | 102 kI (M−1 s−1) |
| 15 | 0.97 ± 0.01 | --- | 4.5 ± 0.1 | 12.9 ± 0.1 | 9.3 ± 0.2 |
| 20 | 2.15 ± 0.04 | 9.2 ± 0.5 | 11 ±0.1 | 20 ± 1 | 18 ± 1 |
| 25 | 3.03 ± 0.02 | 16.8 ± 0.5 | 16 ± 0.5 | 33 ± 1 | 29 ± 1 |
| 30 | 6.64 ± 0.03 | 21 ± 0.4 | 37 ± 4 | 48 ± 3 | 61 ± 4 |
| 35 | 12.1 ± 0.05 | 37.6 ±0.6 | 91 ± 2 | 75 ± 5 | 105 ± 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, M.; Paiva-Martins, F.; Losada-Barreiro, S.; Bravo-Díaz, C. Modeling Chemical Reactivity at the Interfaces of Emulsions: Effects of Partitioning and Temperature. Molecules 2021, 26, 4703. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154703
Costa M, Paiva-Martins F, Losada-Barreiro S, Bravo-Díaz C. Modeling Chemical Reactivity at the Interfaces of Emulsions: Effects of Partitioning and Temperature. Molecules. 2021; 26(15):4703. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154703
Chicago/Turabian StyleCosta, Marlene, Fátima Paiva-Martins, Sonia Losada-Barreiro, and Carlos Bravo-Díaz. 2021. "Modeling Chemical Reactivity at the Interfaces of Emulsions: Effects of Partitioning and Temperature" Molecules 26, no. 15: 4703. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154703