
Chloroquine Potentiates the Anticancer Effect of Pterostilbene on Pancreatic Cancer by Inhibiting Autophagy and Downregulating the RAGE/STAT3 Pathway

 , , and
, , and
Abstract
:1. Introduction
2. Results
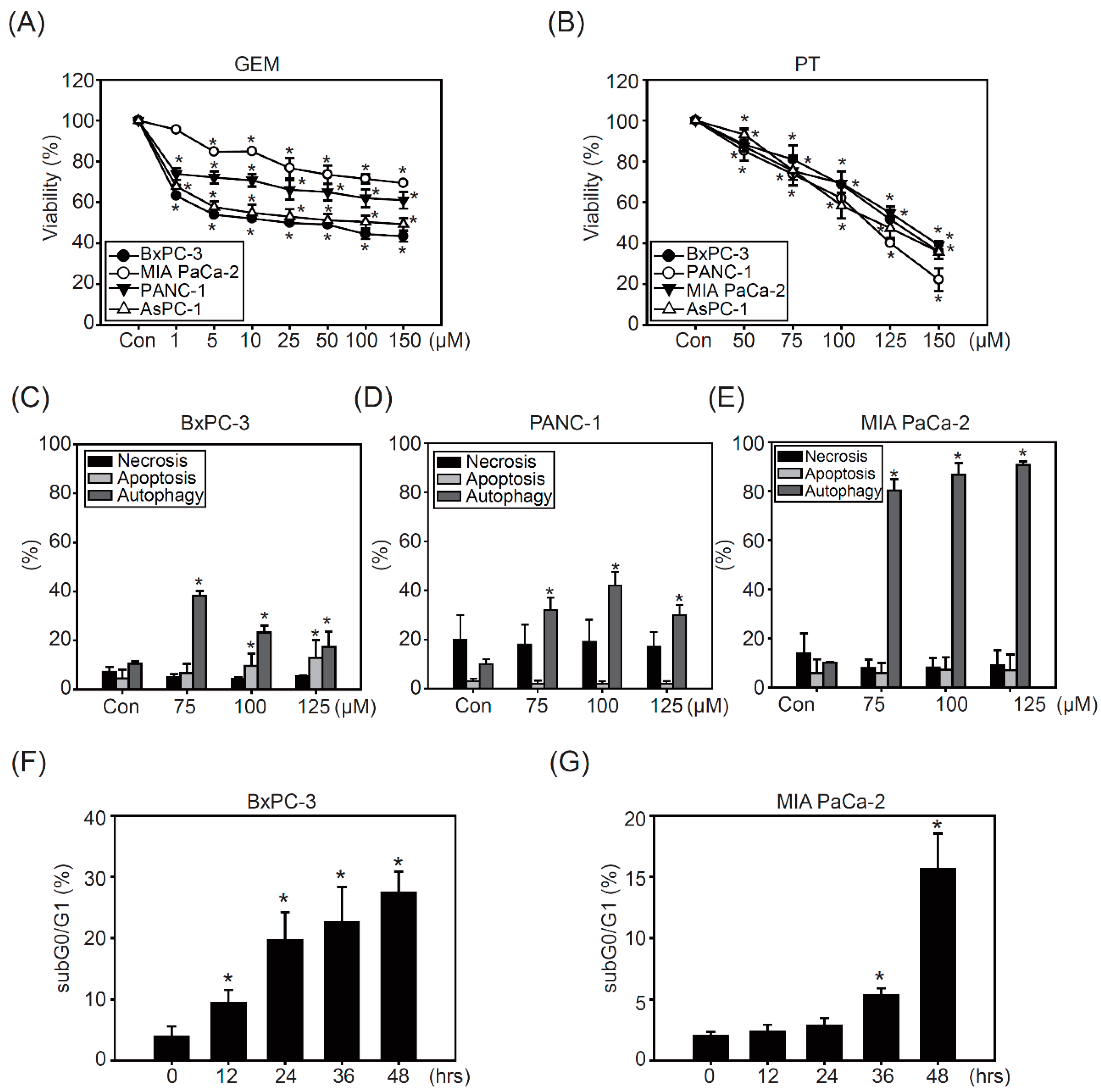
2.1. Pterostilbene Inhibits Growth in PDAC Cell Lines
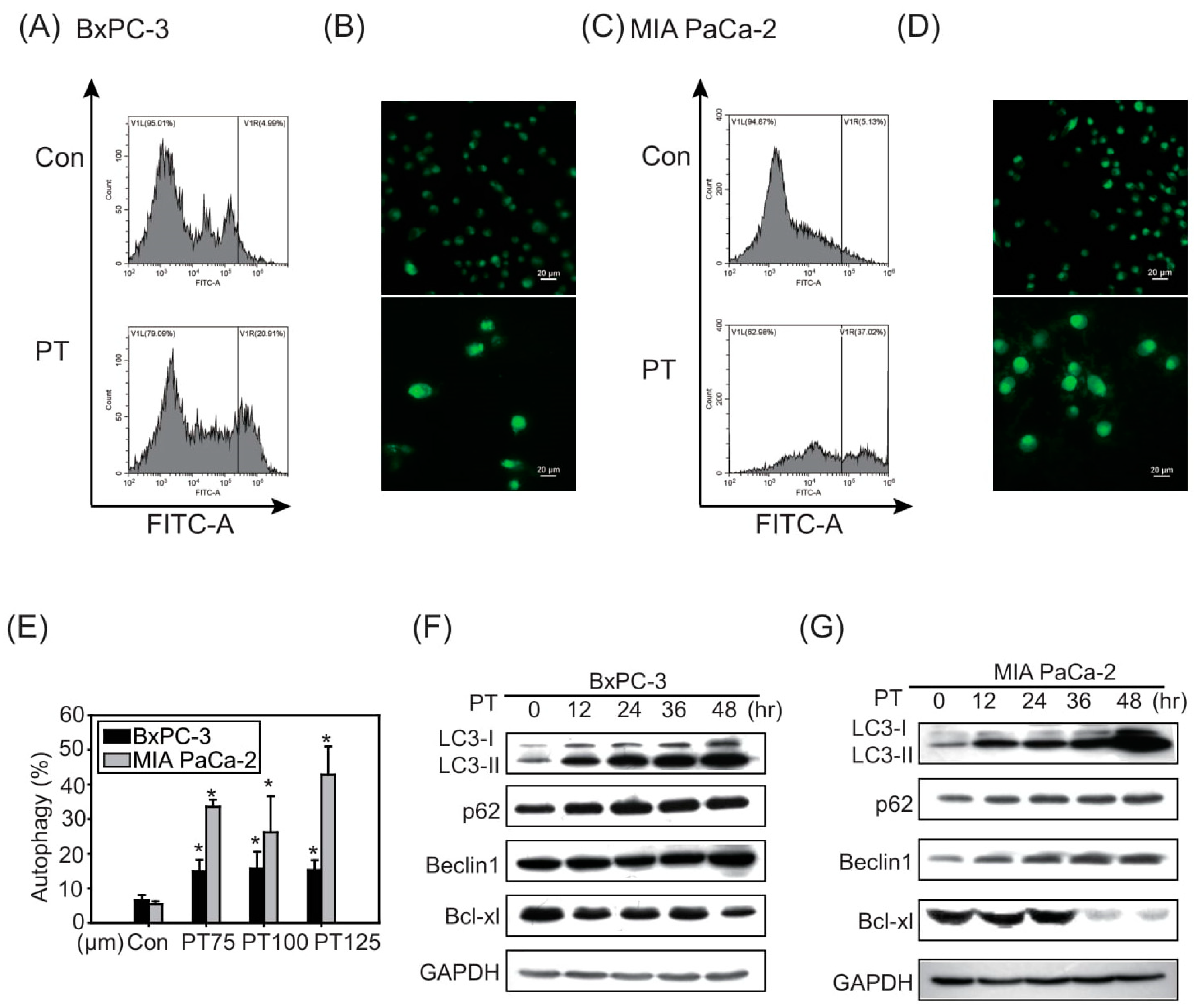
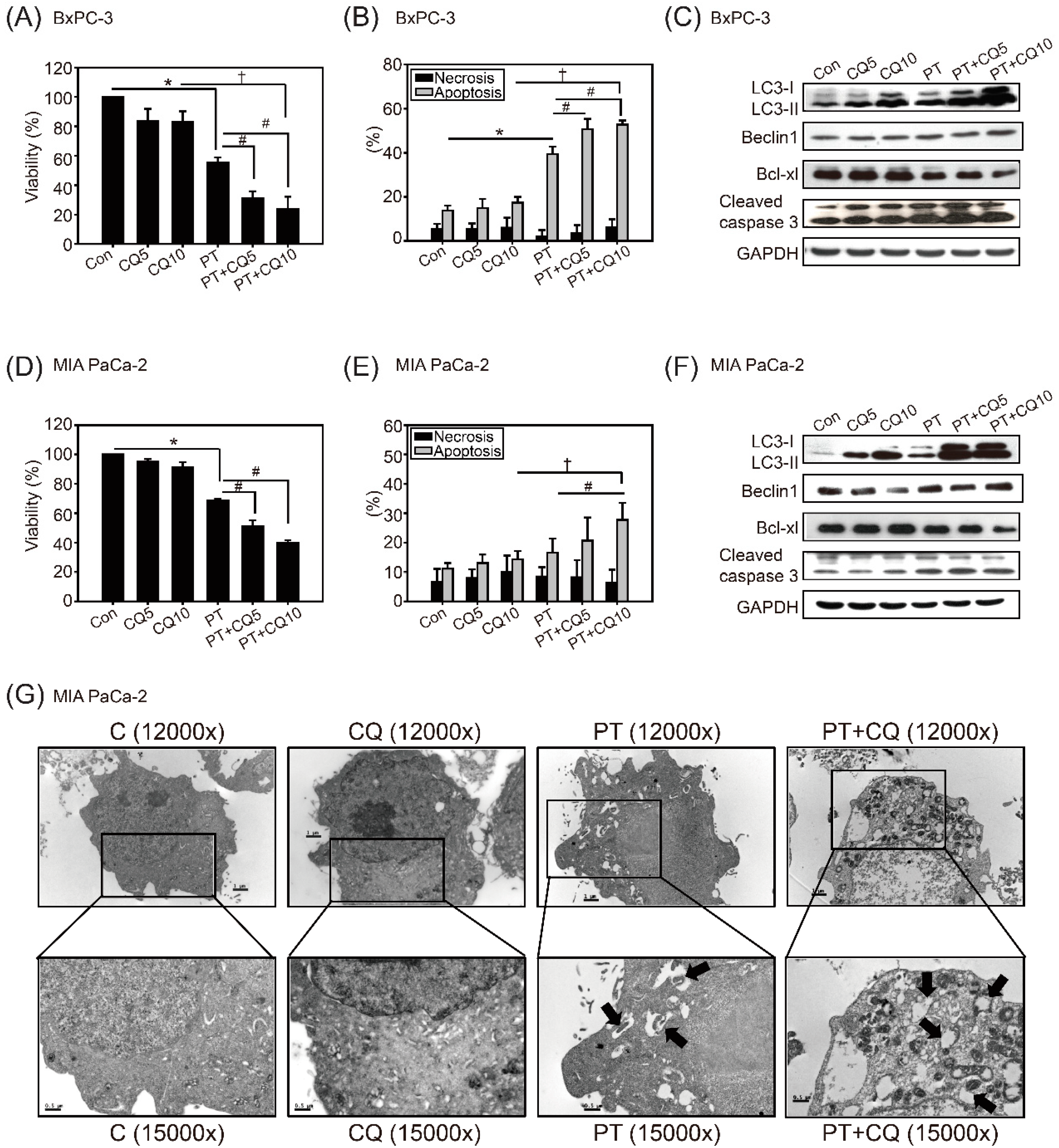
2.2. The Combination of Pterostilbene and Chloroquine Exerts Enhanced Cytotoxicity against Pancreatic Cancer Cells
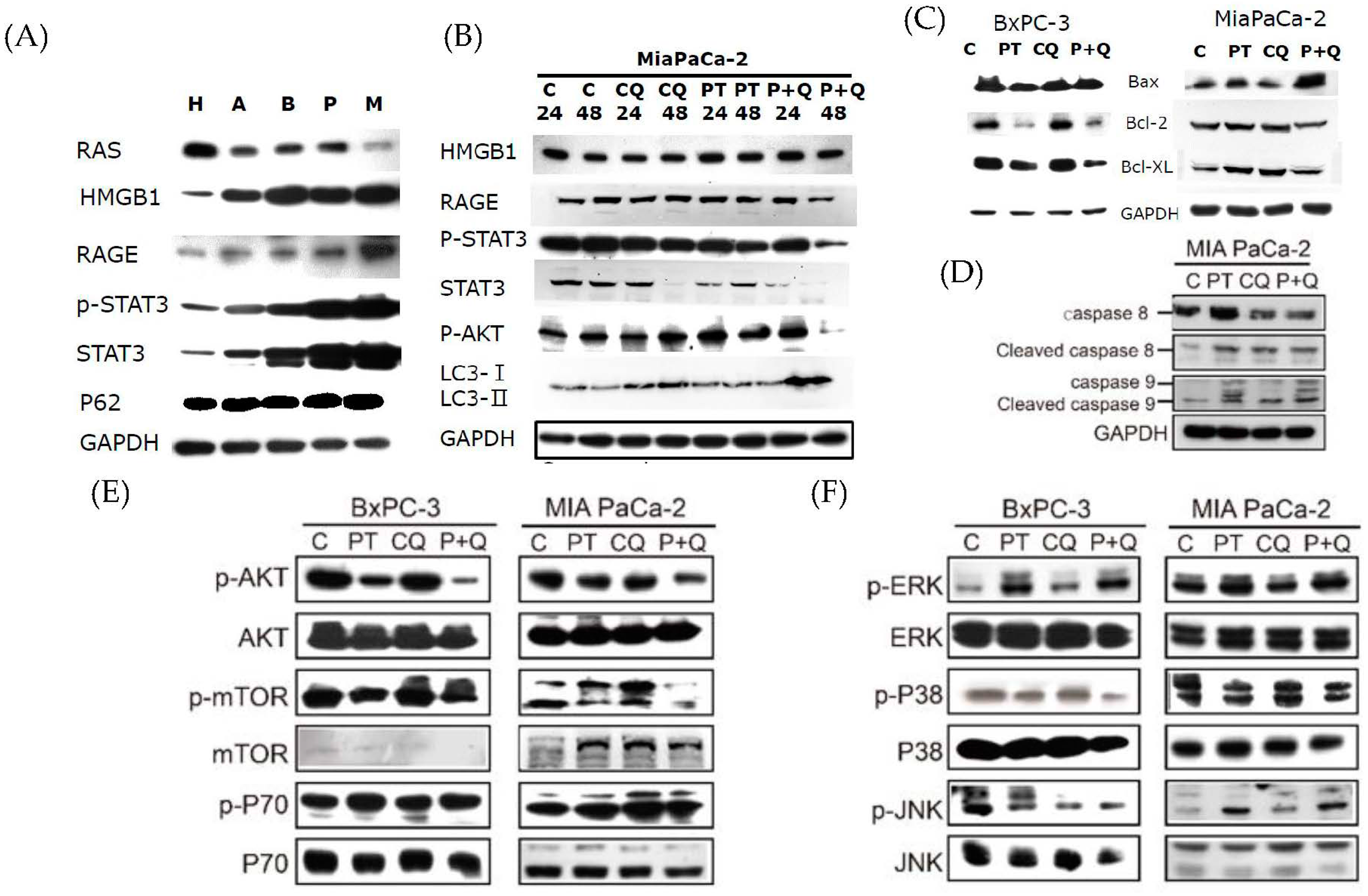
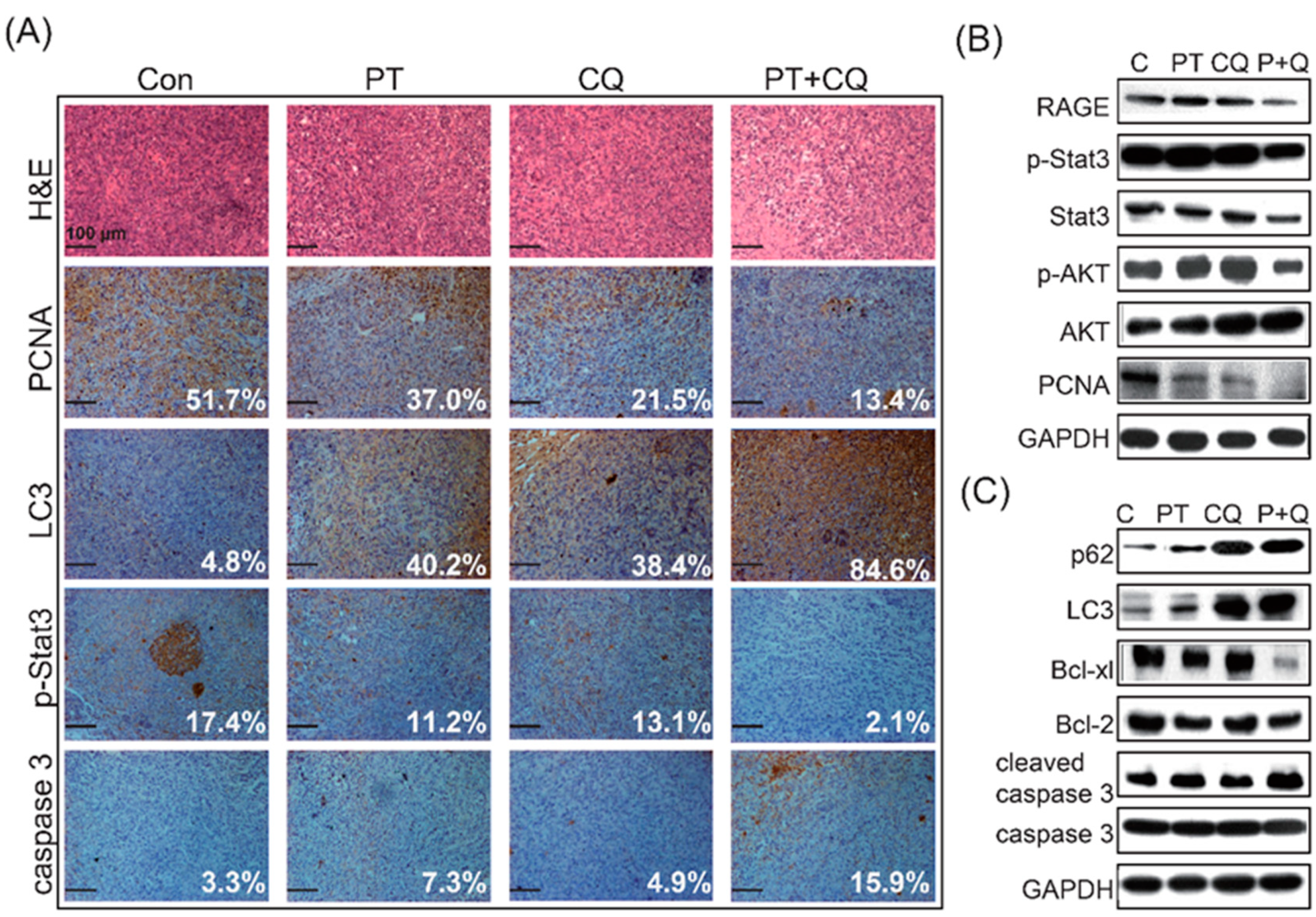
2.3. Pterostilbene Combined with Chloroquine Downregulates the RAGE/STAT3 Signaling Pathways and Increases Apoptosis
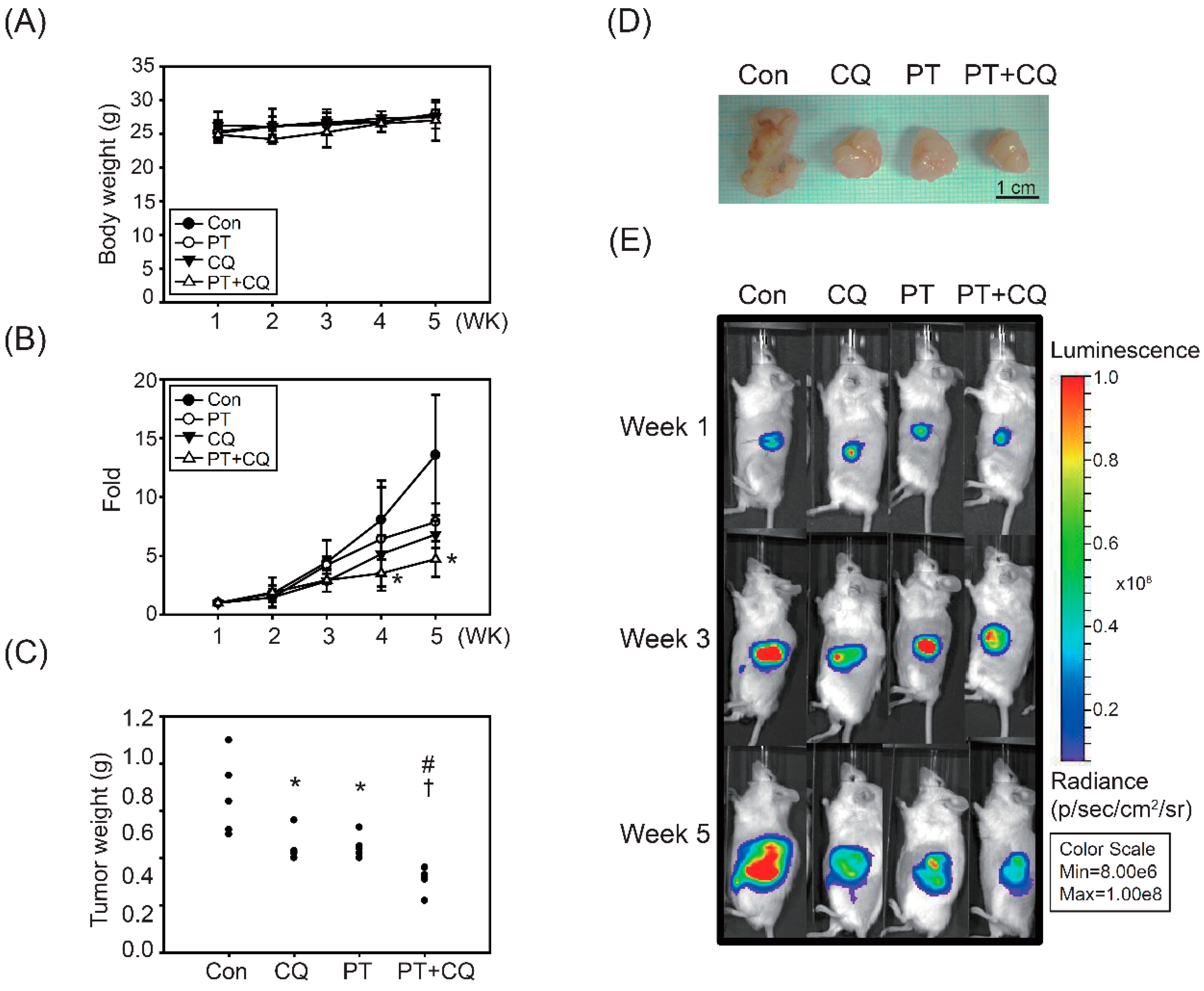
2.4. The Combination of PT and CQ Treatment Enhances Anticancer Effects in an Orthotopic Pancreatic Cancer Model
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Reagents
4.3. Cell Culture
4.4. Cell Viability Assay
4.5. Detection of SubG0/G1 and Apoptosis by Flow Cytometry
4.6. Immunofluorescence Staining and Detection of Autophagy
4.7. Western Blot Analysis
4.8. Transmission Electron Microscopy
4.9. Orthotopic Pancreatic Cancer Model
4.10. H&E Staining and Immunohistochemistry Staining
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Drubay, V.; Skrypek, N.; Cordiez, L.; Vasseur, R.; Schulz, C.; Boukrout, N.; Duchene, B.; Coppin, L.; Van Seuningen, I.; Jonckheere, N. TGF-betaRII knock-down in pancreatic cancer cells promotes tumor growth and gemcitabine resistance. Importance of STAT3 phosphorylation on S727. Cancers 2018, 10, 254. [Google Scholar] [CrossRef] [Green Version]
- Pusceddu, S.; Ghidini, M.; Torchio, M.; Corti, F.; Tomasello, G.; Niger, M.; Prinzi, N.; Nichetti, F.; Coinu, A.; Di Bartolomeo, M.; et al. Comparative effectiveness of gemcitabine plus Nab-Paclitaxel and FOLFIRINOX in the first-line setting of metastatic pancreatic cancer: A systematic review and meta-analysis. Cancers 2019, 11, 484. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, A.; Pappalardo, A.; Pompella, L.; Tirino, G.; Calabrese, F.; Laterza, M.M.; Caterino, M.; Ventriglia, A.; Orditura, M.; Conzo, G.; et al. Nab-paclitaxel plus gemcitabine as first line therapy in metastatic pancreatic cancer patients relapsed after gemcitabine adjuvant treatment. Med. Oncol. 2019, 36, 1–8. [Google Scholar] [CrossRef]
- Caparello, C.; Meijer, L.L.; Garajova, I.; Falcone, A.; Le Large, T.Y.; Funel, N.; Kazemier, G.; Peters, G.J.; Vasile, E.; Giovannetti, E. FOLFIRINOX and translational studies: Towards personalized therapy in pancreatic cancer. World J. Gastroenterol. 2016, 22, 6987–7005. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Castellanos, J.A.; Lamichhane, P.; Messaggio, F.; Shi, C.; Dai, X.; Rai, P.; Chen, X.; VanSaun, M.N.; Merchant, N.B. Inverse correlation of STAT3 and MEK signaling mediates resistance to RAS pathway inhibition in pancreatic cancer. Cancer Res. 2018, 78, 6235–6246. [Google Scholar] [CrossRef] [Green Version]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS mutant pancreatic cancer: No lone path to an effective treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; Devasthali, V.; Cheng, J.H.; Castillo, J.; Metcalfe, C.; Clermont, A.C.; Otter, D.D.; Chan, E.; Bou-Reslan, H.; Cao, T.; et al. Modeling targeted inhibition of MEK and PI3 kinase in human pancreatic cancer. Mol. Cancer Ther. 2015, 14, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.; Iyer, R.; Fountzilas, C. Poly(ADP-Ribose) Polymerase inhibitors in pancreatic cancer: A new treatment paradigms and future implications. Cancers 2019, 11, 1980. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol. Cancer 2020, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Murthy, P.; Miller-Ocuin, J.; Doerfler, W.R.; Ellis, J.T.; Liang, X.; Ross, M.A.; Wallace, C.T.; Sperry, J.L.; Lotze, M.T.; et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer 2018, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.J.; Lee, Y.H.; Yeh, Y.L.; Wang, Y.J.; Wang, B.J. The Roles of Autophagy and the inflammasome during environmental stress-triggered skin inflammation. Int. J. Mol. Sci. 2016, 17, 2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J., 3rd. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biancur, D.E.; Kimmelman, A.C. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.J.; Lee, Y.H.; Yeh, Y.L.; Wu, W.S.; Ho, C.T.; Li, C.Y.; Wang, B.J.; Wang, Y.J. Autophagy-inducing effect of pterostilbene: A prospective therapeutic/preventive option for skin diseases. J. Food Drug Anal. 2017, 25, 125–133. [Google Scholar] [CrossRef]
- Chen, R.J.; Wu, P.H.; Ho, C.T.; Way, T.D.; Pan, M.H.; Chen, H.M.; Ho, Y.S.; Wang, Y.J. P53-dependent downregulation of hTERT protein expression and telomerase activity induces senescence in lung cancer cells as a result of pterostilbene treatment. Cell Death Dis. 2017, 8, e2985. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.P.; Lu, C.C.; Chiang, J.H.; Tsai, F.J.; Juan, Y.N.; Tsao, J.W.; Chiu, H.Y.; Yang, J.S. Pterostilbene modulates the suppression of multidrug resistance protein 1 and triggers autophagic and apoptotic mechanisms in cisplatin-resistant human oral cancer CAR cells via AKT signaling. Int. J. Oncol. 2018, 52, 1504–1514. [Google Scholar]
- Lin, W.S.; Leland, J.V.; Ho, C.T.; Pan, M.H. Occurrence, bioavailability, anti-inflammatory, and anticancer effects of pterostilbene. J. Agric. Food Chem. 2020, 68, 12788–12799. [Google Scholar] [CrossRef]
- Chen, R.J.; Kuo, H.C.; Cheng, L.H.; Lee, Y.H.; Chang, W.T.; Wang, B.J.; Wang, Y.J.; Cheng, H.C. Apoptotic and nonapoptotic activities of pterostilbene against cancer. Int. J. Mol. Sci. 2018, 19, 287. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Chen, Y.-Y.; Hsiao, C.-M.; Pan, M.-H.; Wang, B., Jr.; Chen, Y.-C.; Ho, C.-T.; Huang, K.-C.; Chen, R.-J. Induction of autophagy by pterostilbene contributes to the prevention of renal fibrosis via attenuating NLRP3 inflammasome activation and epithelial-mesenchymal transition. Front. Cell Dev. Biol. 2020, 8, 436. [Google Scholar] [CrossRef]
- Sireesh, D.; Ganesh, M.R.; Dhamodharan, U.; SAKThivadivel, M.; Sivasubramanian, S.; Gunasekaran, P.; Ramkumar, K.M. Role of pterostilbene in attenuating immune mediated devastation of pancreatic beta cells via Nrf2 signaling cascade. J. Nutr. Biochem. 2017, 44, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Chiou, Y.S.; Ho, C.T.; Pan, M.H. Chemoprevention by resveratrol and pterostilbene: Targeting on epigenetic regulation. Biofactors 2018, 44, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.Y.; Liu, Z.S.; Yan, H.J.; Yuan, Y.F.; Levenson, A.S.; Li, K. Pterostilbene inhibits MTA1/HDAC1 complex leading to PTEN acetylation in hepatocellular carcinoma. Biomed. Pharm. 2018, 101, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Guo, H.; Yang, H.; Wang, D.; Gao, P.; Wei, W. Pterostilbene, An active constituent of blueberries, suppresses proliferation potential of human cholangiocarcinoma via enhancing the autophagic flux. Front. Pharmacol. 2019, 10, 1238. [Google Scholar] [CrossRef] [PubMed]
- Riche, D.M.; McEwen, C.L.; Riche, K.D.; Sherman, J.J.; Wofford, M.R.; Deschamp, D.; Griswold, M. Analysis of safety from a human clinical trial with pterostilbene. J. Toxicol. 2013, 2013, 463595. [Google Scholar] [CrossRef]
- Mannal, P.W.; Alosi, J.A.; Schneider, J.G.; McDonald, D.E.; McFadden, D.W. Pterostilbene inhibits pancreatic cancer in vitro. J Gastrointest. Surg. 2010, 14, 873–879. [Google Scholar] [CrossRef]
- Kostin, S.F.; McDonald, D.E.; McFadden, D.W. Inhibitory effects of (-)-epigallocatechin-3-gallate and pterostilbene on pancreatic cancer growth in vitro. J. Surg. Res. 2012, 177, 255–262. [Google Scholar] [CrossRef]
- McCormack, D.; McDonald, D.; McFadden, D. Pterostilbene ameliorates tumor necrosis factor alpha-induced pancreatitis in vitro. J. Surg. Res. 2012, 178, 28–32. [Google Scholar] [CrossRef]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology 2017, 152, 1492–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Benlloch, M.; Obrador, E.; Valles, S.L.; Rodriguez, M.L.; Sirerol, J.A.; Alcacer, J.; Pellicer, J.A.; Salvador, R.; Cerda, C.; Saez, G.T.; et al. Pterostilbene Decreases the Antioxidant Defenses of Aggressive Cancer Cells In Vivo: A Physiological Glucocorticoids- and Nrf2-Dependent Mechanism. Antioxid. Redox Signal. 2016, 24, 974–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battista, R.A.; Resnati, M.; Facchi, C.; Ruggieri, E.; Cremasco, F.; Paradiso, F.; Orfanelli, U.; Giordano, L.; Bussi, M.; Cenci, S.; et al. Autophagy mediates epithelial cancer chemoresistance by reducing p62/SQSTM1 accumulation. PLoS ONE 2018, 13, e0201621. [Google Scholar] [CrossRef]
- Hu, F.; Zhao, Y.; Yu, Y.; Fang, J.M.; Cui, R.; Liu, Z.Q.; Guo, X.L.; Xu, Q. Docetaxel-mediated autophagy promotes chemoresistance in castration-resistant prostate cancer cells by inhibiting STAT3. Cancer Lett. 2018, 416, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharm. 2019, 119, 109415. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Zhao, Y.; Zhang, M.; He, R.Z.; Shi, X.H.; Guo, X.J.; Shi, C.J.; Peng, F.; Wang, M.; Shen, M.; et al. Inhibition of autophagy by deguelin sensitizes pancreatic cancer cells to doxorubicin. Int. J. Mol. Sci. 2017, 18, 370. [Google Scholar] [CrossRef] [Green Version]
- Bigelsen, S. Evidence-based complementary treatment of pancreatic cancer: A review of adjunct therapies including paricalcitol, hydroxychloroquine, intravenous vitamin C, statins, metformin, curcumin, and aspirin. Cancer Manag. Res. 2018, 10, 2003–2018. [Google Scholar] [CrossRef] [Green Version]
- Balic, A.; Sorensen, M.D.; Trabulo, S.M.; Sainz, B., Jr.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [Green Version]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Samaras, P.; Tusup, M.; Nguyen-Kim, T.D.L.; Seifert, B.; Bachmann, H.; von Moos, R.; Knuth, A.; Pascolo, S. Phase I study of a chloroquine-gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother. Pharmacol. 2017, 80, 1005–1012. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, L.; Hui, K.; Hu, C.; Wen, Y.; Yang, S.; Zhu, P.; Wang, L.; Xia, Y.; Qiao, Y.; Sun, W.; et al. Autophagy inhibition potentiates the anti-angiogenic property of multikinase inhibitor anlotinib through JAK2/STAT3/VEGFA signaling in non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef]
- Liu, X.; Sun, K.; Wang, H.; Dai, Y. Inhibition of autophagy by chloroquine enhances the antitumor efficacy of sorafenib in glioblastoma. Cell. Mol. Neurobiol. 2016, 36, 1197–1208. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Cao, L.; Zhang, Q.; Song, Q.; Meng, Z.; Wu, X.; Xu, K. Inhibition of autophagy potentiates the anti-metastasis effect of phenethyl isothiocyanate through JAK2/STAT3 pathway in lung cancer cells. Mol. Carcinog. 2018, 57, 522–535. [Google Scholar] [CrossRef]
- Zai, W.; Chen, W.; Luan, J.; Fan, J.; Zhang, X.; Wu, Z.; Ding, T.; Ju, D.; Liu, H. Dihydroquercetin ameliorated acetaminophen-induced hepatic cytotoxicity via activating JAK2/STAT3 pathway and autophagy. Appl. Microbiol. Biotechnol. 2018, 102, 1443–1453. [Google Scholar] [CrossRef]
- Fu, C.; Liu, P.; Li, P.; Liu, W.; Huang, X.; Liang, Y. FSP1 promotes the biofunctions of adventitial fibroblast through the crosstalk among RAGE, JAK2/STAT3 and Wnt3a/β-catenin signalling pathways. J. Cell Mol. Med. 2019, 23, 7246–7260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Huo, Y.; Zheng, H.; Zhao, J.; Jia, L.; Wang, P. Ethyl pyruvate suppresses the growth, invasion and migration and induces the apoptosis of non-small cell lung cancer cells via the HMGB1/RAGE axis and the NF-κB/STAT3 pathway. Oncol. Rep. 2019, 42, 817–825. [Google Scholar] [CrossRef]
- Liu, Y.; Hou, B.; Zhang, Y.; Fan, Y.; Peng, B.; Liu, W.; Han, S.; Yin, J.; He, X. Anticonvulsant agent DPP4 inhibitor sitagliptin downregulates CXCR3/RAGE pathway on seizure models. Exp. Neurol. 2018, 307, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Rabie, M.A.; Abd El Fattah, M.A.; Nassar, N.N.; Abdallah, D.M.; El-Abhar, H.S. Correlation between angiotensin 1-7-mediated Mas receptor expression with motor improvement, activated STAT3/SOCS3 cascade, and suppressed HMGB-1/RAGE/NF-κB signaling in 6-hydroxydopamine hemiparkinsonian rats. Biochem. Pharmacol. 2020, 171, 113681. [Google Scholar] [CrossRef] [PubMed]
- Wazea, S.A.; Wadie, W.; Bahgat, A.K.; El-Abhar, H.S. Galantamine anti-colitic effect: Role of alpha-7 nicotinic acetylcholine receptor in modulating Jak/STAT3, NF-κB/HMGB1/RAGE and p-AKT/Bcl-2 pathways. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Loze, M.T.; Zeh, H.J.; Kang, R. The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy 2010, 6, 1181–1183. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D.; Lotze, M.T.; Zeh, H.J., 3rd. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy 2012, 8, 989–991. [Google Scholar] [CrossRef] [Green Version]
- Boone, B.A.; Zeh, H.J., 3rd; Bahary, N. Autophagy inhibition in pancreatic adenocarcinoma. Clin. Colorectal. Cancer 2018, 17, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotto-Filho, A.; Braganhol, E.; Klafke, K.; Figueiro, F.; Terra, S.R.; Paludo, F.J.; Morrone, M.; Bristot, I.J.; Battastini, A.M.; Forcelini, C.M.; et al. Autophagy inhibition improves the efficacy of curcumin/temozolomide combination therapy in glioblastomas. Cancer Lett. 2015, 358, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Chen, S.Y.; Wang, S.Y.; Lin, J.A.; Yen, G.C. Pterostilbene enhances cytotoxicity and chemosensitivity in human pancreatic cancer cells. Biomolecules 2020, 10, 709. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Lowe, G.; Roberts, C.M.; Finlay, J.; Han, E.S.; Glackin, C.A.; Dellinger, T.H. Pterostilbene suppresses ovarian cancer growth via induction of apoptosis and blockade of cell cycle progression involving inhibition of the STAT3 pathway. Int. J. Mol. Sci. 2018, 19, 1983. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.F.; Bu, L.L.; Wang, W.M.; Ma, S.R.; Liu, J.F.; Deng, W.W.; Mao, L.; Yu, G.T.; Huang, C.F.; Liu, B.; et al. Tumor growth suppression by inhibiting both autophagy and STAT3 signaling in HNSCC. Oncotarget 2015, 6, 43581–43593. [Google Scholar] [CrossRef]
- Zhang, H.; McCarty, N. Tampering with cancer chemoresistance by targeting the TGM2-IL6-autophagy regulatory network. Autophagy 2017, 13, 627–628. [Google Scholar] [CrossRef]
- Saleem, M.Z.; Alshwmi, M.; Zhang, H.; Din, S.R.U.; Nisar, M.A.; Khan, M.; Alam, S.; Alam, G.; Jin, L.; Ma, T. Inhibition of JNK-mediated autophagy promotes proscillaridin A- induced apoptosis via ROS generation, intracellular Ca(+2) oscillation and inhibiting STAT3 signaling in breast cancer cells. Front. Pharmacol. 2020, 11, 1055. [Google Scholar] [CrossRef]
- Kong, C.; Li, Y.; Liu, Z.; Ye, J.; Wang, Z.; Zhang, L.; Kong, W.; Liu, H.; Liu, C.; Pang, H.; et al. Targeting the Oncogene KRAS Mutant Pancreatic Cancer by Synergistic Blocking of Lysosomal Acidification and Rapid Drug Release. ACS Nano. 2019, 13, 4049–4063. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Number of Mice | Inhibition (%) | TVQTs | TGDTs | p-Value (t-Test) | ||
|---|---|---|---|---|---|---|---|
| Control | CQ | PT | |||||
| Control | 5 | -- | 17.6 ± 2.3 | -- | |||
| CQ | 5 | 29.9 ± 12.8 | 24.1 ± 4.2 | 6.5 ± 4.2 | * <0.05 | ||
| PT | 5 | 19.7 ± 13.4 | 21.3 ± 3.4 | 3.6 ± 3.4 | 0.08 | 0.27 | |
| CQ + PT | 5 | 39.4 ± 17.2 | 33.5 ± 12.0 | 15.9 ± 12.0 | * <0.05 | 0.14 | # <0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.-J.; Lyu, Y.-J.; Chen, Y.-Y.; Lee, Y.-C.; Pan, M.-H.; Ho, Y.-S.; Wang, Y.-J. Chloroquine Potentiates the Anticancer Effect of Pterostilbene on Pancreatic Cancer by Inhibiting Autophagy and Downregulating the RAGE/STAT3 Pathway. Molecules 2021, 26, 6741. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216741
Chen R-J, Lyu Y-J, Chen Y-Y, Lee Y-C, Pan M-H, Ho Y-S, Wang Y-J. Chloroquine Potentiates the Anticancer Effect of Pterostilbene on Pancreatic Cancer by Inhibiting Autophagy and Downregulating the RAGE/STAT3 Pathway. Molecules. 2021; 26(21):6741. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216741
Chicago/Turabian StyleChen, Rong-Jane, Yi-Jhen Lyu, Yu-Ying Chen, Yen-Chien Lee, Min-Hsiung Pan, Yuan-Soon Ho, and Ying-Jan Wang. 2021. "Chloroquine Potentiates the Anticancer Effect of Pterostilbene on Pancreatic Cancer by Inhibiting Autophagy and Downregulating the RAGE/STAT3 Pathway" Molecules 26, no. 21: 6741. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26216741