
Amidine- and Amidoxime-Substituted Heterocycles: Synthesis, Antiproliferative Evaluations and DNA Binding

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
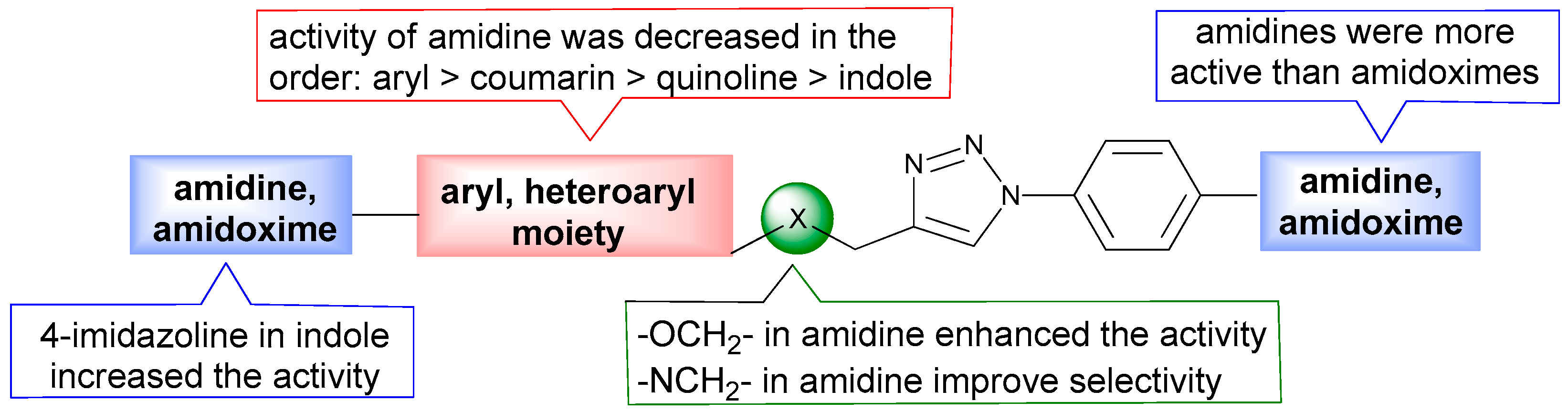
2.2. Biological Evaluation of Aromatic Amidines and Amidoximes
2.2.1. Evaluation of Antiproliferative Activity
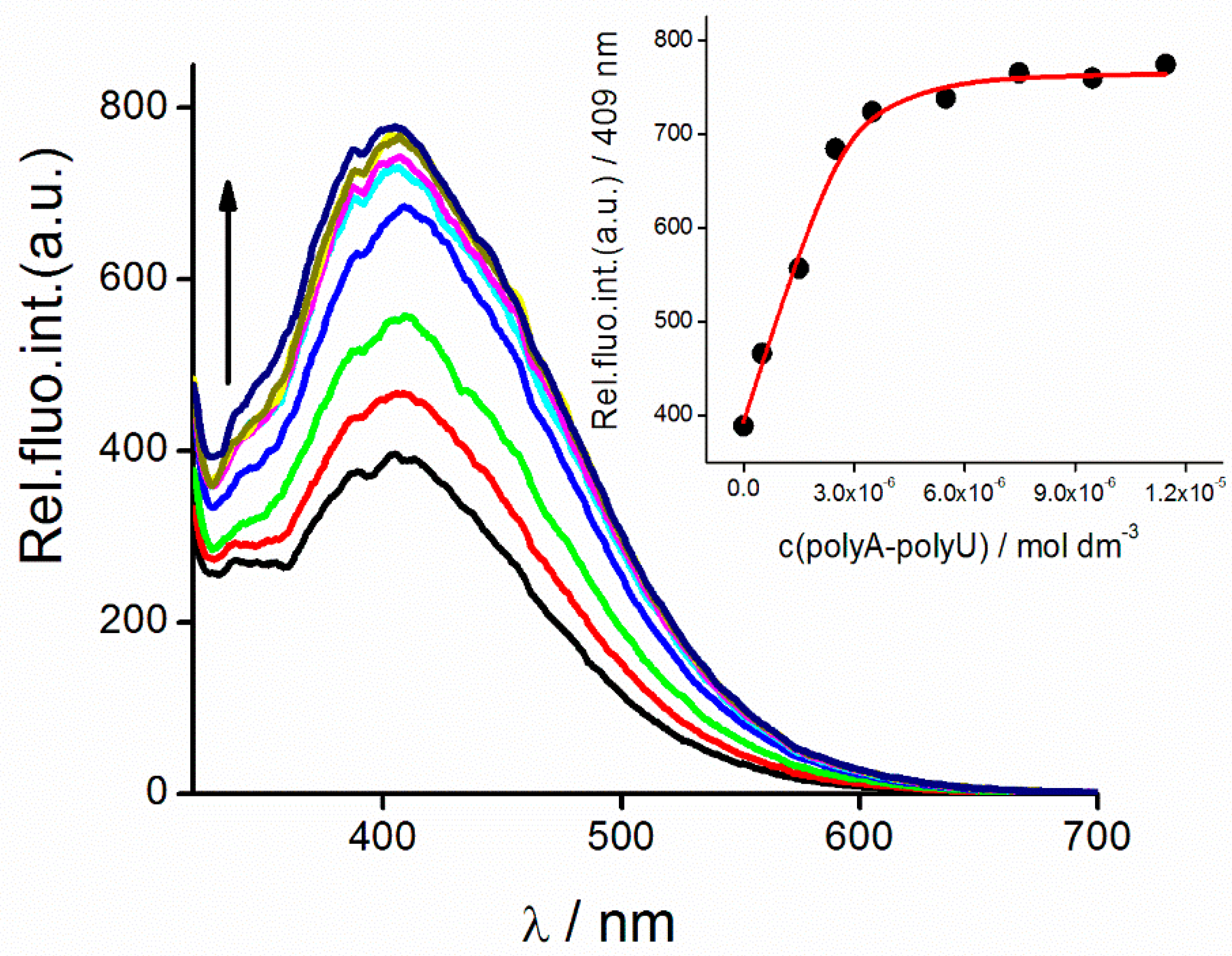
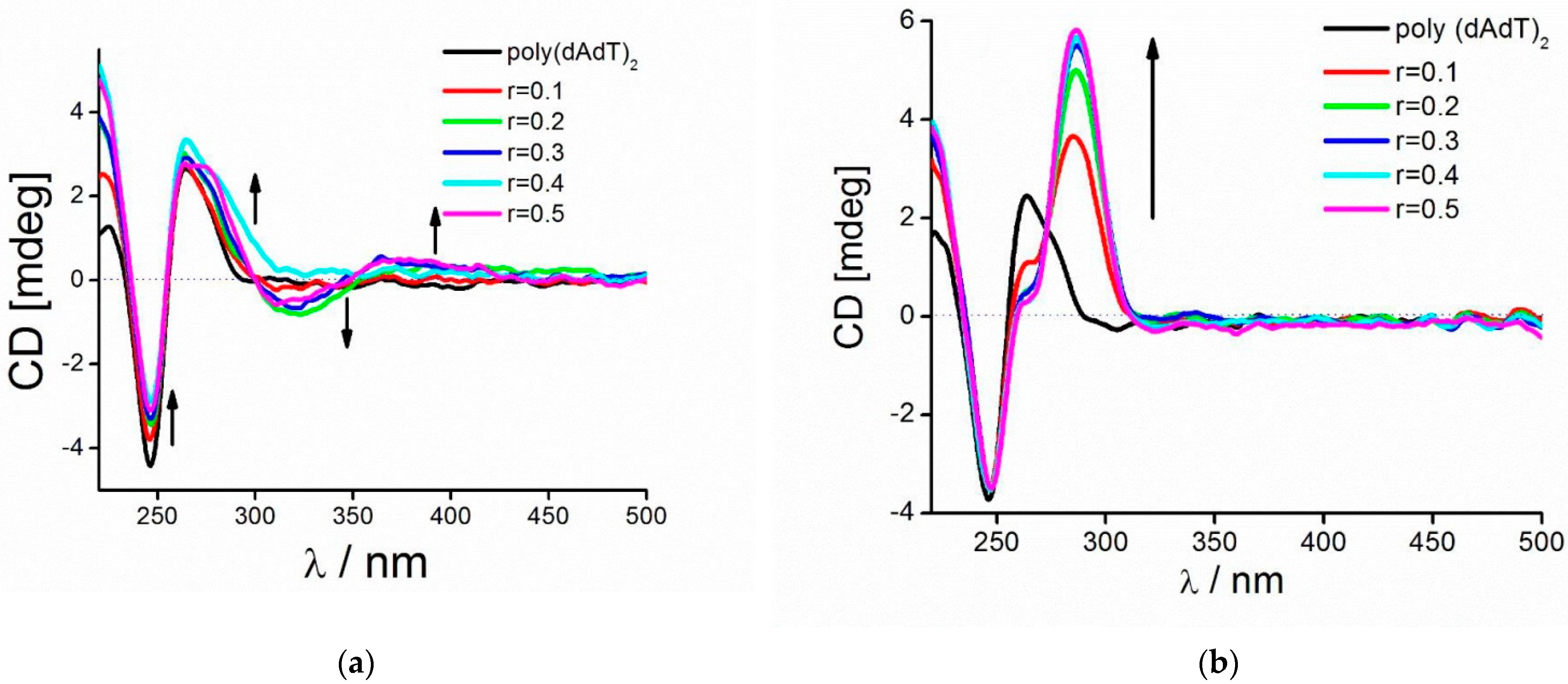
2.2.2. DNA Binding Study
3. Experimental Part
3.1. Materials and Methods
3.2. Synthetic Procedures
3.3. Preparation of Alkynyl Derivatives 1c, 1d and 1g
3.3.1. 4-Cyano-1-((prop-2-ynyl)amino)-1H-indole (1c)
3.3.2. 5-Cyano-1-((prop-2-ynyl)amino)-1H-indole (1d)
3.3.3. 2-Cyano-8-(prop-2-ynyloxy)-quinoline (1g)
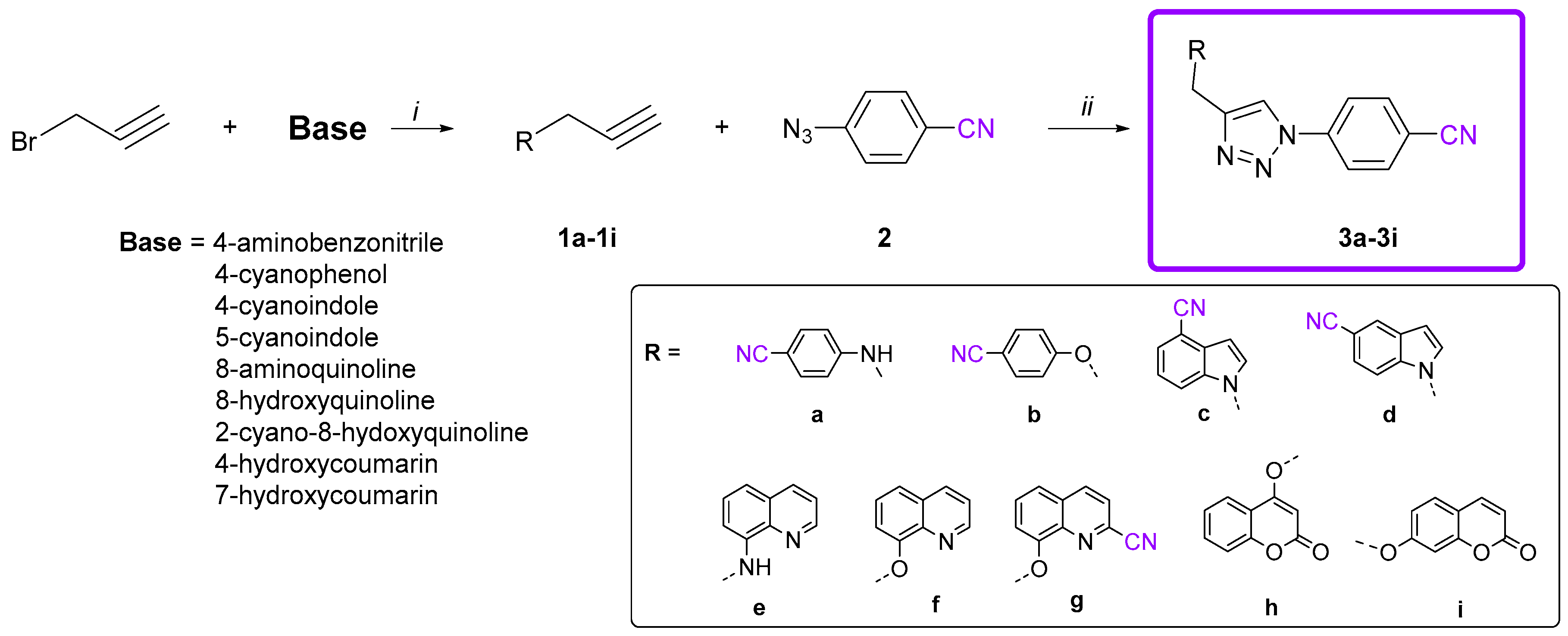
3.4. General Procedure for Synthesis of Cyano-Substituted 1,2,3-Triazole Derivatives 3a–3i
3.4.1. 4-((1-(4-Cyanophenyl)-1H-1,2,3-triazole-4-yl)methylamino)benzonitrile (3a)
3.4.2. 4-((1-(4-Cyanophenyl)-1H-1,2,3-triazol-4-yl)methyloxy)benzonitrile (3b)
3.4.3. 4-Cyano-1-((1-(4-cyanophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1H-indole (3c)
3.4.4. 5-Cyano-1-((1-(4-cyanophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1H-indole (3d)
3.4.5. 8-((1-(4-Cyanophenyl)-1H-1,2,3-triazol-4-yl)methylamino)quinoline (3e)
3.4.6. 8-((1-(4-Cyanophenyl)-1H-1,2,3-triazol-4-yl)methyloxy)quinoline (3f)
3.4.7. 2-Cyano-8-((1-(4-cyanophenyl)-1H-1,2,3-triazol-4-yl)methyloxy)quinoline (3g)
3.4.8. 4-((1-(4-Cyanophenyl)-1H-1,2,3-triazol-4-yl)methyloxy)coumarin (3h)
3.4.9. 7-((1-(4-Cyanophenyl)-1H-1,2,3-triazol-4-yl)methyloxy)coumarin (3i)
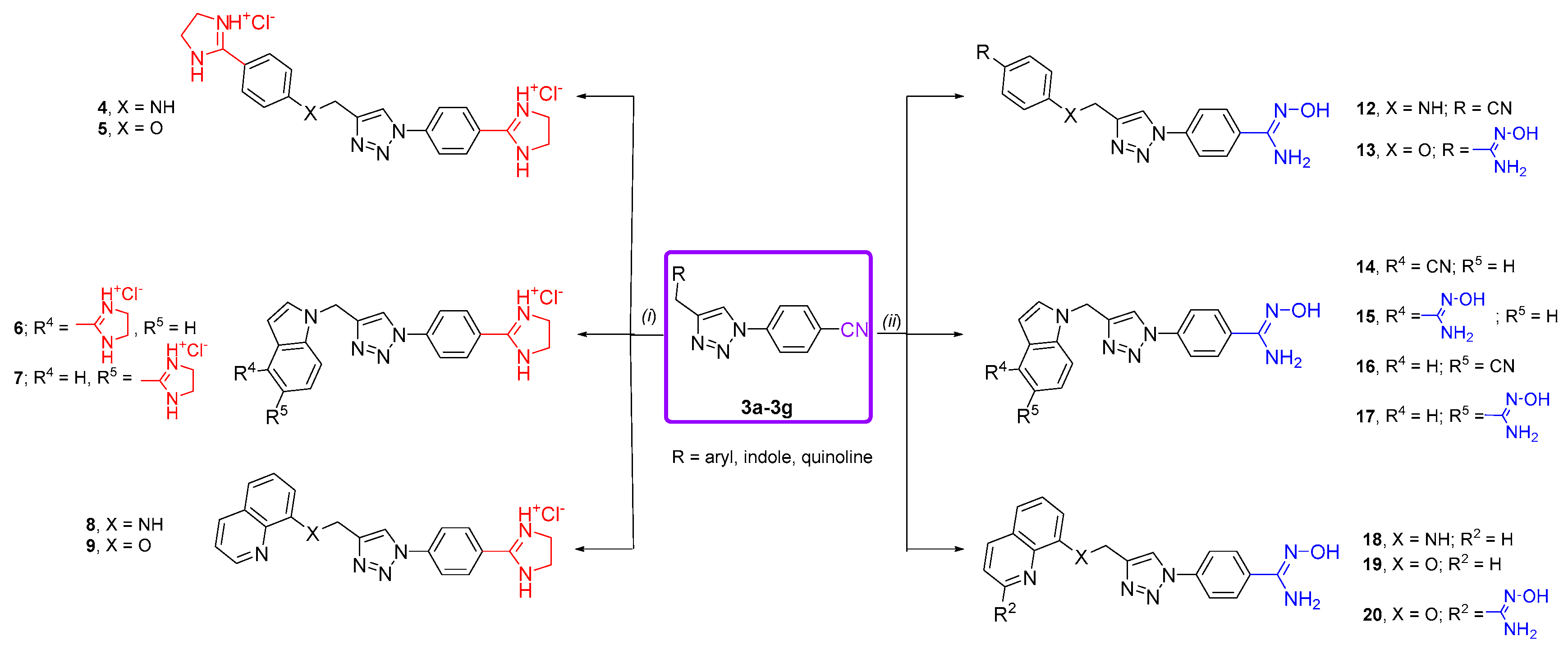
3.5. General Procedure for Synthesis of Imidazolinyl-Substituted 1,2,3-Triazole Derivatives 4–11
3.5.1. 1-(4-(4,5-Dihydro-1H-imidazol-2-yl)-phenyl)-4-((4-(4,5-dihydro-1H-imidazol-2-yl)phenyl)aminomethyl)-1H-1,2,3-triazole dihydrochloride (4)
3.5.2. 1-(4-(4,5-Dihydro-1H-imidazol-2-yl)-phenyl]-4-((4-(4,5-dihydro-1H-imidazol-2-yl) phenyloxy)methyl]-1H-1,2,3-triazole dihydrochloride (5)
3.5.3. 1-((1-(4-(Dihydro-1H-imidazol-2-yl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-4-(dihydro-1H-imidazol-2-yl)-1H-indole dihydrochloride (6)
3.5.4. 1-((1-(4-(Dihydro-1H-imidazol-2-yl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-5-(dihydro-1H-imidazol-2-yl)-1H-indole dihydrochloride (7)
3.5.5. 8-(((1-(4-(4,5-Dihydro-1H-imidazol-2-yl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-quinoline hydrochloride (8)
3.5.6. 8-((1-(4-(4,5-Dihydro-1H-imidazol-2-yl)phenyl)-1H-1,2,3-triazol-4-yl)methyloxy)-quinoline hydrochloride (9)
3.5.7. 4-((1-(4-(4,5-Dihydro-1H-imidazol-2-yl)phenyl)-1H-1,2,3-triazol-4-yl)methyloxy)-coumarin hydrochloride (10)
3.5.8. 7-((1-(4-(4,5-Dihydro-1H-imidazol-2-yl)phenyl)-1H-1,2,3-triazol-4-yl)methyloxy)-coumarin hydrochloride (11)
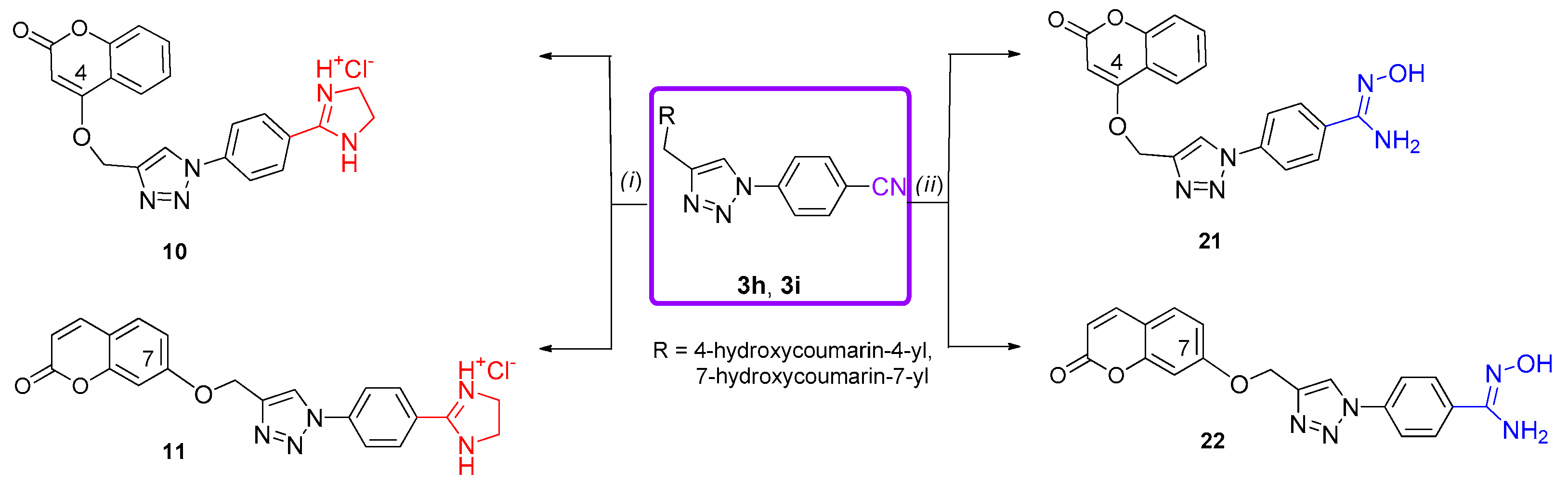
3.6. General Procedure for Synthesis of Amidoxime-Substituted 1,2,3-Triazole Derivatives 12–22
3.6.1. 4-(((1-(4-((Z)-N′-Hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)amino)benzonitrile (12)
3.6.2. (Z)-N′-Hydroxy-4-(4-((4-((Z)-N′-hydroxycarbamimidoyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)benzimidamide (13)
3.6.3. 1-((1-(4-((Z)-N′-Hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-4-cyanoindole (14) and (Z)-N′-hydroxy-1-((1-(4-((Z)-N′-hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-4-carboximidamide (15)
3.6.4. 1-((1-(4-((Z)-N′-Hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-5-cyanoindole (16) and (Z)-N′-hydroxy-1-((1-(4-((Z)-N′-hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-indole-5-carboximidamide (17)
3.6.5. 8-(((1-(4-((Z)-N′-Hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)amino)quinoline (18)
3.6.6. 8-((1-(4-((Z)-N′-Hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)quinoline (19)
3.6.7. (Z)-N′-Hydroxy-8-((1-(4-((Z)-N′-hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)quinoline-2-carboximidamide (20)
3.6.8. 4-((1-(4-((Z)-N′-Hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)coumarin (21)
3.6.9. 7-((1-(4-((Z)-N′-Hydroxycarbamimidoyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)coumarin (22)
3.7. Cell Culturing
3.8. Proliferation Assays
3.9. DNA Binding Study
3.9.1. UV–Vis Measurements
3.9.2. Fluorometric Measurements
3.9.3. CD Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Shahar Yar, M. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Silakari, O. The Current Status of O-Heterocycles: A Synthetic and Medicinal Overview. ChemMedChem 2018, 13, 1071–1087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, Z. Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem. 2019, 181, 111587. [Google Scholar] [CrossRef] [PubMed]
- Singla, P.; Luxami, V.; Paul, K. Benzimidazole-biologically attractive scaffold for protein kinase inhibitors. RSC Adv. 2014, 4, 12422–12440. [Google Scholar] [CrossRef]
- Carneiro, A.; Matos, M.J.; Uriarte, E.; Santana, L. Trending topics on coumarin and its derivatives in 2020. Molecules 2021, 26, 501. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Skouta, R. Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef] [Green Version]
- Meščić Macan, A.; Perin, N.; Jakopec, S.; Mioč, M.; Stojković, M.R.; Kralj, M.; Hranjec, M.; Raić-Malić, S. Synthesis, antiproliferative activity and DNA/RNA-binding properties of mono- and bis-(1,2,3-triazolyl)-appended benzimidazo[1,2-a]quinoline derivatives. Eur. J. Med. Chem. 2020, 185, 111845. [Google Scholar] [CrossRef] [PubMed]
- Bistrović, A.; Krstulović, L.; Harej, A.; Grbčić, P.; Sedić, M.; Koštrun, S.; Pavelić, S.K.; Bajić, M.; Raić-Malić, S. Design, synthesis and biological evaluation of novel benzimidazole amidines as potent multi-target inhibitors for the treatment of non-small cell lung cancer. Eur. J. Med. Chem. 2018, 143, 1616–1634. [Google Scholar] [CrossRef] [PubMed]
- Bistrović, A.; Harej, A.; Grbčić, P.; Sedić, M.; Pavelić, S.K.; Cetina, M.; Raić-Malić, S. Synthesis and anti-proliferative effects of mono-and bis-purinomimetics targeting Kinases. Int. J. Mol. Sci. 2017, 18, 2292. [Google Scholar] [CrossRef] [Green Version]
- Martorana, A.; la Monica, G.; Lauria, A. Quinoline-based molecules targeting c-Met, EGF, and VEGF receptors and the proteins involved in related carcinogenic pathways. Molecules 2020, 25, 4279. [Google Scholar] [CrossRef]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem. 2019, 183, 111691. [Google Scholar] [CrossRef] [PubMed]
- Dadashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Al-Warhi, T.; Sabt, A.; Elkaeed, E.B.; Eldehna, W.M. Recent advancements of coumarin-based anticancer agents: An up-to-date review. Bioorg. Chem. 2020, 103, 104163. [Google Scholar] [CrossRef] [PubMed]
- Fotopoulos, I.; Hadjipavlou-Litina, D. Hybrids of Coumarin Derivatives as Potent and Multifunctional Bioactive Agents: A Review. Med. Chem. 2019, 16, 272–306. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, S.J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Hu, J.; Tian, J.L.; Yan, H.; Zheng, C.G.; Hu, W. Le Novel hybrids from N-hydroxyarylamide and indole ring through click chemistry as histone deacetylase inhibitors with potent antitumor activities. Chin. Chem. Lett. 2015, 26, 675–680. [Google Scholar] [CrossRef]
- Narsimha, S.; Satheesh Kumar, N.; Kumara Swamy, B.; Vasudeva Reddy, N.; Althaf Hussain, S.K.; Srinivasa Rao, M. Indole-2-carboxylic acid derived mono and bis 1,4-disubstituted 1,2,3-triazoles: Synthesis, characterization and evaluation of anticancer, antibacterial, and DNA-cleavage activities. Bioorg. Med. Chem. Lett. 2016, 26, 1639–1644. [Google Scholar] [CrossRef]
- Aneja, B.; Arif, R.; Perwez, A.; Napoleon, J.V.; Hasan, P.; Rizvi, M.M.A.; Azam, A.; Rahisuddin; Abid, M. N-Substituted 1,2,3-Triazolyl-Appended Indole-Chalcone Hybrids as Potential DNA Intercalators Endowed with Antioxidant and Anticancer Properties. ChemistrySelect 2018, 3, 2638–2645. [Google Scholar] [CrossRef]
- Praveena, K.S.S.; Shivaji Ramarao, E.V.V.; Murthy, N.Y.S.; Akkenapally, S.; Ganesh Kumar, C.; Kapavarapu, R.; Pal, S. Design of new hybrid template by linking quinoline, triazole and dihydroquinoline pharmacophoric groups: A greener approach to novel polyazaheterocycles as cytotoxic agents. Bioorg. Med. Chem. Lett. 2015, 25, 1057–1063. [Google Scholar] [CrossRef]
- De O Freitas, L.B.; Borgati, T.F.; De Freitas, R.P.; Ruiz, A.L.T.G.; Marchetti, G.M.; De Carvalho, J.E.; Da Cunha, E.F.F.; Ramalho, T.C.; Alves, R.B. Synthesis and antiproliferative activity of 8-hydroxyquinoline derivatives containing a 1,2,3-triazole moiety. Eur. J. Med. Chem. 2014, 84, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hou, Y.; Yin, W.; Zhou, S.; Qian, P.; Guo, Z.; Xu, L.; Zhao, Y. Discovery of a novel 6,7-disubstituted-4-(2-fluorophenoxy)quinolines bearing 1,2,3-triazole-4-carboxamide moiety as potent c-Met kinase inhibitors. Eur. J. Med. Chem. 2016, 119, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Akkol, E.K.; Genç, Y.; Karpuz, B.; Sobarzo-Sánchez, E.; Capasso, R. Coumarins and coumarin-related compounds in pharmacotherapy of cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef]
- Peterson, L.B.; Blagg, B.S.J. Click chemistry to probe Hsp90: Synthesis and evaluation of a series of triazole-containing novobiocin analogues. Bioorg. Med. Chem. Lett. 2010, 20, 3957–3960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Hou, Z.; Tian, Y.; Mou, Y.; Guo, C. Design, synthesis, cytotoxicity and mechanism of novel dihydroartemisinin-coumarin hybrids as potential anti-cancer agents. Eur. J. Med. Chem. 2018, 151, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Pingaew, R.; Saekee, A.; Mandi, P.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis, biological evaluation and molecular docking of novel chalcone-coumarin hybrids as anticancer and antimalarial agents. Eur. J. Med. Chem. 2014, 85, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kraljević, T.G.; Harej, A.; Sedić, M.; Pavelić, S.K.; Stepanić, V.; Drenjančević, D.; Talapko, J.; Raić-Malić, S. Synthesis, in vitro anticancer and antibacterial activities and in silico studies of new 4-substituted 1,2,3-triazole–coumarin hybrids. Eur. J. Med. Chem. 2016, 124, 794–808. [Google Scholar] [CrossRef] [PubMed]
- An, R.; Hou, Z.; Li, J.T.; Yu, H.N.; Mou, Y.H.; Guo, C. Design, synthesis and biological evaluation of novel 4-substituted coumarin derivatives as antitumor agents. Molecules 2018, 23, 2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakeel-u-Rehman; Masood-ur-Rahman; Tripathi, V.K.; Singh, J.; Ara, T.; Koul, S.; Farooq, S.; Kaul, A. Synthesis and biological evaluation of novel isoxazoles and triazoles linked 6-hydroxycoumarin as potent cytotoxic agents. Bioorg. Med. Chem. Lett. 2014, 24, 4243–4246. [Google Scholar] [CrossRef] [PubMed]
- Demeunynck, M.; Bailly, C.; Wilson, W.D. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2002. [Google Scholar]
- Munde, M.; Kumar, A.; Peixoto, P.; Depauw, S.; Ismail, M.A.; Farahat, A.A.; Paul, A.; Say, M.V.; David-Cordonnier, M.H.; Boykin, D.W.; et al. The unusual monomer recognition of guanine-containing mixed sequence DNA by a dithiophene heterocyclic diamidine. Biochemistry 2014, 53, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Reeh, C.; Wundt, J.; Clement, B. N,N′-dihydroxyamidines: A new prodrug principle to improve the oral bioavailability of amidines. J. Med. Chem. 2007, 50, 6730–6734. [Google Scholar] [CrossRef] [PubMed]
- Berger, O.; Ortial, S.; Wein, S.; Denoyelle, S.; Bressolle, F.; Durand, T.; Escale, R.; Vial, H.J.; Vo-Hoang, Y. Evaluation of amidoxime derivatives as prodrug candidates of potent bis-cationic antimalarials. Bioorg. Med. Chem. Lett. 2019, 29, 2203–2207. [Google Scholar] [CrossRef] [PubMed]
- Maračić, S.; Lapić, J.; Djaković, S.; Opačak-Bernardi, T.; Glavaš-Obrovac, L.; Vrček, V.; Raić-Malić, S. Quinoline and ferrocene conjugates: Synthesis, computational study and biological evaluations. Appl. Organomet. Chem. 2019, 33, e4628. [Google Scholar] [CrossRef] [Green Version]
- Bistrović, A.; Stipaničev, N.; Opačak-Bernardi, T.; Jukić, M.; Martinez, S.; Glavaš-Obrovac, L.; Raić-Malić, S. Synthesis of 4-aryl-1,2,3-triazolyl appended natural coumarin-related compounds with antiproliferative and radical scavenging activities and intracellular ROS production modification. New J. Chem. 2017, 41, 7531–7543. [Google Scholar] [CrossRef]
- Maračić, S.; Kraljević, T.G.; Paljetak, H.Č.; Perić, M.; Matijašić, M.; Verbanac, D.; Cetina, M.; Raić-Malić, S. 1,2,3-Triazole pharmacophore-based benzofused nitrogen/sulfur heterocycles with potential anti-Moraxella catarrhalis activity. Bioorg. Med. Chem. 2015, 23, 7448–7463. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.B.; Krstulović, L.; Koštrun, S.; Jelić, D.; Bokulić, A.; Stojković, M.R.; Zonjić, I.; Taylor, M.C.; Kelly, J.M.; Bajić, M.; et al. Design, synthesis, antitrypanosomal activity, DNA/RNA binding and in vitro ADME profiling of novel imidazoline-substituted 2-arylbenzimidazoles. Eur. J. Med. Chem. 2020, 207, 112802. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.B.; Stolić, I.; Krstulović, L.; Taylor, M.C.; Kelly, J.M.; Tomić, S.; Tumir, L.; Bajić, M.; Raić-Malić, S. Novel symmetric bis-benzimidazoles: Synthesis, DNA/RNA binding and antitrypanosomal activity. Eur. J. Med. Chem. 2019, 173, 63–75. [Google Scholar] [CrossRef]
- Barral, K.; Moorhouse, A.D.; Moses, J.E. Efficient Conversion of Aromatic Amines into Azides: A One-Pot Synthesis of Triazole Linkages. Org. Lett. 2007, 9, 1809–1811. [Google Scholar] [CrossRef]
- Hranjec, M.; Starčević, K.; Zamola, B.; Mutak, S.; Derek, M.; Karminski-Zamola, G. New amidino-benzimidazolyl derivatives of tylosin and desmycosin. J. Antibiot. 2002, 55, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Scatchard, G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- Eriksson, M.; Nordén, B. Linear and circular dichroism of drug-nucleic acid complexes. Methods Enzymol. 2001, 340, 68–98. [Google Scholar]
- Šmidlehner, T.; Piantanida, I.; Pescitelli, G. Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids—Tutorial. Beilstein J. Org. Chem. 2018, 14, 84–105. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Hori, H.; Ogiwara, Y. Copper(II)-Catalyzed [4+1] Annulation of Propargylamines with N,O-Acetals: Entry to the Synthesis of Polysubstituted Pyrrole Derivatives. Eur. J. Org. Chem. 2015, 2015, 1905–1909. [Google Scholar] [CrossRef]
- Ferroni, C.; Pepe, A.; Kim, Y.S.; Lee, S.; Guerrini, A.; Parenti, M.D.; Tesei, A.; Zamagni, A.; Cortesi, M.; Zaffaroni, N.; et al. 1,4-Substituted Triazoles as Nonsteroidal Anti-Androgens for Prostate Cancer Treatment. J. Med. Chem. 2017, 60, 3082–3093. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jin, J.; Zhao, L.; Shen, H.; Shen, C.; Zhang, P. Synthesis of C-glycosyl triazolyl quinoline-based fluorescent sensors for the detection of mercury ions. Carbohydr. Res. 2016, 433, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Maity, D.; Govindaraju, T. A differentially selective sensor with fluorescence turn-on response to Zn2+ and dual-mode ratiometric response to Al3+ in aqueous media. Chem. Commun. (Camb.) 2012, 48, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Qi, P.-P.; Shi, X.-J.; Huang, R.; Guo, H.; Zheng, Y.-C.; Yu, D.-Q.; Liu, H.-M. Efficient synthesis of new antiproliferative steroidal hybrids using the molecular hybridization approach. Eur. J. Med. Chem. 2016, 117, 241–255. [Google Scholar] [CrossRef]
- Gazivoda, T.; Raić-Malić, S.; Krištafor, V.; Makuc, D.; Plavec, J.; Bratulić, S.; Kraljević-Pavelić, S.; Pavelić, K.; Naesens, L.; Andrei, G.; et al. Synthesis, cytostatic and anti-HIV evaluations of the new unsaturated acyclic C-5 pyrimidine nucleoside analogues. Bioorg. Med. Chem. 2008, 16, 5624–5634. [Google Scholar] [CrossRef]
- Chaires, J.B.; Dattagupta, N.; Crothers, D.M. Studies on Interaction of Anthracycline Antibiotics and Deoxyribonucleic Acid: Equilibrium Binding Studies on Interaction of Daunomycin with Deoxyribonucleic Acid. Biochemistry 1982, 21, 3933–3940. [Google Scholar] [CrossRef] [PubMed]
- Bresloff, J.L.; Crothers, D.M. Equilibrium Studies of Ethidium-Polynucleotide Interactions. Biochemistry 1981, 20, 3547–3553. [Google Scholar] [CrossRef]
- Chalikian, T.V.; Völker, J.; Plum, G.E.; Breslauer, K.J. A more unified picture for the thermodynamics of nucleic acid duplex melting: A characterization by calorimetric and volumetric techniques. Proc. Natl. Acad. Sci. USA 1999, 96, 7853–7858. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 4–22 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | R | R′ | IC50 a (µM) | ||||
| A549 | HeLa | HepG2 | SW620 | HFF | |||
| 4 |  |  | 46.68 | 41.23 | 48.87 | 61.87 | NA |
| 5 |  |  | 15.67 | 0.80 | 0.64 | 0.22 | <0.01 |
| 6 |  |  | 49.87 | 13.54 | 2.37 | 18.02 | <0.01 |
| 7 |  |  | >100 | >100 | 69.07 | 49.77 | NA |
| 8 |  |  | 29.13 | 23.75 | 4.84 | 35.55 | 11.66 |
| 9 |  |  | 25.22 | 10.27 | 9.07 | 2.69 | <0.01 |
| 10 |  |  | 7.86 | 1.90 | 3.55 | 1.75 | <0.01 |
| 11 |  |  | 4.49 | 2.13 | 0.28 | 4.77 | 3.14 |
| 12 |  |  | >100 | >100 | >100 | 65.14 | NA |
| 13 |  |  | >100 | >100 | >100 | >100 | NA |
| 14 |  |  | >100 | 65.75 | >100 | >100 | NA |
| 15 |  |  | >100 | >100 | >100 | >100 | NA |
| 16 |  |  | >100 | >100 | >100 | >100 | NA |
| 17 |  |  | >100 | 60.66 | >100 | >100 | NA |
| 18 |  |  | 6.52 | 25.55 | >100 | NA | 9.81 |
| 19 |  |  | 76.68 | 64.51 | >100 | 72.71 | NA |
| 20 |  |  | 31.35 | 7.15 | 51.31 | 7.24 | NA |
| 21 |  |  | >100 | 99.72 | >100 | >100 | NA |
| 22 |  |  | >100 | 87.38 | 91.40 | >100 | NA |
| 5-FU | 2.80 | 8.81 | 8.90 | 0.08 | / | ||
| ctDNA | p(dAdT)2 | poly A-poly U | |||||||
|---|---|---|---|---|---|---|---|---|---|
| logKs | n | I/I0 c | logKs | n | I/I0 c | logKs | n | I/I0 c | |
| 5 | 5.7 | 0.1 | 2.0 | 6.5 | <0.1 | 2.2 | 7.0 | 0.7 | 2.0 |
| 6 | 5.0 | 0.7 | 0.8 | 6.6 | 0.1 | 0.9 | 4.2 | 0.6 | 0.1 |
| 8 | - d | - d | - d | 5.5 | 0.5 | 2.3 | - d | - d | - d |
| 9 | 5.3 | 0.3 | 0.5 | 5.8 | 0.1 | 0.2 | 5.1 | <0.1 | <0.1 |
| 10 | - d | - d | - d | 6.7 | 0.8 | 1.3 | - d | - d | - d |
| 11 | 4.5 d | 0.2 | 0.5 | 4.9 | 0.2 | 0.6 | 4.9 | 0.2 | 0.7 |
| 20 | 4.3 d | 0.2 | - | 4.7 d | 0.1 | - | 4.6 d | 0.1 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maračić, S.; Grbčić, P.; Shammugam, S.; Radić Stojković, M.; Pavelić, K.; Sedić, M.; Kraljević Pavelić, S.; Raić-Malić, S. Amidine- and Amidoxime-Substituted Heterocycles: Synthesis, Antiproliferative Evaluations and DNA Binding. Molecules 2021, 26, 7060. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26227060
Maračić S, Grbčić P, Shammugam S, Radić Stojković M, Pavelić K, Sedić M, Kraljević Pavelić S, Raić-Malić S. Amidine- and Amidoxime-Substituted Heterocycles: Synthesis, Antiproliferative Evaluations and DNA Binding. Molecules. 2021; 26(22):7060. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26227060
Chicago/Turabian StyleMaračić, Silvija, Petra Grbčić, Suresh Shammugam, Marijana Radić Stojković, Krešimir Pavelić, Mirela Sedić, Sandra Kraljević Pavelić, and Silvana Raić-Malić. 2021. "Amidine- and Amidoxime-Substituted Heterocycles: Synthesis, Antiproliferative Evaluations and DNA Binding" Molecules 26, no. 22: 7060. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26227060