3.2. Synthesis
(−
)-Methyl (S)-2-((S)-3,3-diethyl-9,9-dimethyl-8,8-diphenyl-4,7-dioxa-3,8-disiladecan-5-yl)-2-methylpent-4-enoate (
17). To a solution of secondary alcohol
5 [
14] (3.90 g, 9.14 mmol, 1.00 equiv.) in anhydrous DCM (45 mL, 0.20 M), imidazole (1.56 g, 22.9 mmol, 2.50 equiv.) was added, immediately followed by triethylchlorosilane (2.30 mL, 13.7 mmol, 1.50 equiv.). The resulting mixture was stirred at room temperature for 16 h. The mixture was diluted with DCM (25 mL) and saturated NH
4Cl solution (25 mL). The aqueous phase was extracted with DCM (3 × 25 mL) and the combined organic phases were washed with brine, dried over MgSO
4, filtered and concentrated under reduced pressure. Purification by flash chromatography on silica gel (Hexanes/EtOAc, 97:3) provided the TES-protected methyl ester
17 (4.3 g, 87%) as a colorless oil. R
f = 0.43 (Hexanes/EtOAc, 95:5); [α]
25D −27 (
c 0.7, CH
2Cl
2); Formula: C
31H
48O
4Si
2; MW: 540.82 g/mol; IR (neat) ν
max 3073, 3051, 2954, 2879, 1738 cm
−1;
1H NMR (500 MHz, CDCl
3) δ 7.66–7.63 (m, 4H), 7.44–7.36 (m, 6H), 5.71–5.63 (m, 1H), 5.02 (s, 1H), 5.01–4.98 (m, 1H), 4.15 (t,
J = 5.9 Hz, 1H), 3.52 (d,
J = 5.9 Hz, 2H), 3.46 (s, 3H), 2.39 (dd,
J = 13.6, 7.3 Hz, 1H), 2.25 (dd,
J = 13.3, 7.7 Hz, 1H), 1.06 (s, 3H), 1.04 (s, 9H), 0.89 (t,
J = 7.9 Hz, 9H), 0.56 (q,
J = 8.0 Hz, 6H) ppm;
13C NMR (126 MHz, CDCl
3) δ 175.5, 135.83 (2C), 135.78 (2C), 134.3, 133.5, 133.3, 129.80 (2C), 129.76 (2C), 127.80, 127.75, 118.0, 77.3, 66.4, 51.5, 50.6, 42.1, 27.0 (3C), 19.3, 14.6, 7.0 (3C), 5.3 (3C) ppm; HRMS (ESI)
m/z [M + Na]
+ calcd for C
31H
48O
4Si
2Na: 563.2983; found 563.2985 (+0.4 ppm).
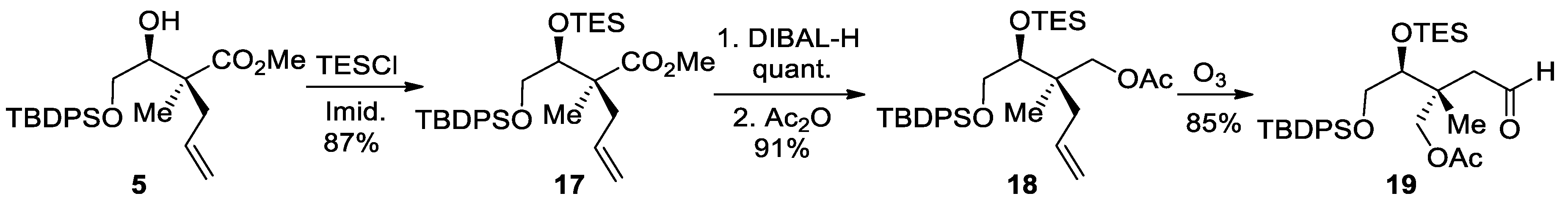
(−)-(R)-2-((S)-3,3-Diethyl-9,9-dimethyl-8,8-diphenyl-4,7-dioxa-3,8-disiladecan-5-yl)-2-methylpent-4-en-1-ol (S1). To a solution of methyl ester 17 (4.30 g, 7.95 mmol, 1.00 equiv.) in anhydrous DCM (40 mL, 0.20 M) at −40 °C, DIBAL-H (20 mL, 20 mmol, 2.5 equiv., 1.0 M in hexanes) was added dropwise. The resulting mixture was stirred at −40 °C for 2 h. Methanol (1.3 mL, 32 mmol, 4.0 equiv.) was added at −40 °C and the reaction was stirred for 10 min followed by addition of Et2O (50 mL) and saturated Rochelle salt solution (50 mL). The solution was stirred vigorously until separation of both layers. The aqueous phase was extracted with Et2O (2 × 50 mL), and the combined organic layers were washed with water, dried over MgSO4, filtered and concentrated under reduced pressure. The 1H NMR analysis indicated that alcohol S1 (4.08 g, quant., colorless oil) was clean enough to be directly used in the next step without further purification. Rf = 0.45 (Hexanes/EtOAc, 90:10); [α]25D −7.4 (c 1.1, CH2Cl2); Formula: C30H48O3Si2; MW: 512.88 g/mol; IR (neat) νmax 3461 (br), 3072, 3051, 2955, 2934, 2877 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.68–7.66 (m, 4H), 7.46–7.39 (m, 6H), 5.83–5.74 (m, 1H), 5.03–4.97 (m, 2H), 3.78 (dd, J = 10.9, 5.8 Hz, 1H), 3.70 (dd, J = 5.5, 4.4 Hz, 1H), 3.56 (dd, J = 10.9, 4.3 Hz, 1H), 3.52 (dd, J = 11.5, 6.3 Hz, 1H), 3.44 (dd, J = 11.4, 6.2 Hz, 1H), 3.18 (t, J = 6.3 Hz, 1H), 2.14 (dd, J = 13.6, 7.4 Hz, 1H), 1.97 (dd, J = 13.6, 7.5 Hz, 1H), 1.07 (s, 9H), 0.87 (t, J = 8.0 Hz, 9H), 0.85 (s, 3H), 0.52 (q, J = 7.9 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 135.90 (2C), 135.85 (2C), 134.7, 132.94, 132.87, 130.04, 130.02, 127.92 (2C), 127.91 (2C), 117.7, 79.6, 68.1, 66.0, 42.1, 39.1, 27.0 (3C), 19.2, 18.4, 7.0 (3C), 5.1 (3C) ppm; HRMS (ESI) m/z [M + Na]+ calcd for C30H48O3Si2Na: 535.3034; found 535.3042 (+1.4 ppm).
(+)-(R)-2-((S)-3,3-Diethyl-9,9-dimethyl-8,8-diphenyl-4,7-dioxa-3,8-disiladecan-5-yl)-2-methylpent-4-en-1-yl acetate (18). To a solution of primary alcohol S1 (2.25 g, 4.39 mmol, 1.00 equiv.), a 1:2 mixture of pyridine and Ac2O (18 mL, 0.25 M) was added at 0 °C and the resulting mixture was stirred at room temperature for 1 h. The solution was concentrated under reduced pressure and the crude product was purified by silica gel flash chromatography (Hexanes/EtOAc, 95:5), providing the acetate product 18 as a colorless oil (2.21 g, 91% over two steps). Rf = 0.65 (Hexanes/EtOAc, 90:10); [α]25D +0.4 (c 0.6, CH2Cl2); Formula: C32H50O4Si2; MW: 554.92 g/mol; IR (neat) νmax 3073, 3051, 2956, 2934, 2877, 2859, 1744 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.68–7.66 (m, 4H), 7.45–7.37 (m, 6H), 5.76–5.69 (m, 1H), 5.00–4.92 (m, 2H), 3.95–3.90 (m, 2H), 3.79–3.74 (m, 2H), 3.54 (dd, J = 10.0, 4.7 Hz, 1H), 2.15 (dd, J = 13.9, 7.4 Hz, 1H), 2.01 (dd, J = 13.8, 7.6 Hz, 1H), 1.97 (s, 3H), 1.07 (s, 9H), 0.91 (t, J = 7.9 Hz, 9H), 0.83 (s, 3H), 0.62–0.54 (m, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 171.0, 135.9 (2C), 135.8 (2C), 134.5, 133.5, 133.4, 129.9, 129.8, 127.8 (4C), 117.7, 77.4, 68.3, 66.5, 41.1, 38.5, 27.0 (3C), 21.0, 19.3, 18.9, 7.1 (3C), 5.3 (3C) ppm; HRMS (ESI) m/z [M + H]+ calcd for C32H51O4Si2: 555.3320; found 555.3328 (+1.4 ppm).
(−)-(2R,3S)-4-((tert-Butyldiphenylsilyl)oxy)-2-methyl-2-(2-oxoethyl)-3-((triethylsilyl)oxy) butyl acetate (19). To a solution of alkene 18 (2.21 g, 3.98 mmol, 1.00 equiv.) in anhydrous DCM (60 mL, 0.070 M) at −78 °C, ozone was bubbled under vacuum until the solution turned pale blue (about 25 min). The reaction was then purged with nitrogen to remove excess ozone. After addition of Et3N (1.67 mL, 11.9 mmol, 3.00 equiv.), the solution was kept at −78 °C for 30 min and then warmed to room temperature for 1 h. MgSO4 was added, and the resulting mixture was filtered and concentrated in vacuo. The crude product was purified by silica gel flash chromatography (Hexanes/EtOAc, 90:10) to provide aldehyde 19 as a colorless oil (1.89 g, 85%). Rf = 0.42 (Hexanes/EtOAc, 90:10); [α]25D −5.5 (c 0.7, CH2Cl2); Formula: C31H48O5Si2; MW: 556.89 g/mol; IR (neat) νmax 3072, 3050, 2955, 2935, 2877, 2859, 1745, 1720 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.78 (t, J = 2.9 Hz, 1H), 7.66–7.65 (m, 4H), 7.44–7.38 (m, 6H), 4.15 (d, J = 11.0 Hz, 1H), 4.10 (d, J = 11.0 Hz, 1H), 3.79–3.74 (m, 2H), 3.56 (dd, J = 10.1, 3.7 Hz, 1H), 2.45 (dd, J = 15.3, 3.2 Hz, 1H), 2.32 (dd, J = 15.3, 2.6 Hz, 1H), 2.03 (s, 3H), 1.07 (s, 9H), 1.03 (s, 3H), 0.86 (t, J = 8.0 Hz, 9H), 0.51 (q, J = 7.9 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 201.6, 170.8, 135.9 (2C), 135.8 (2C), 133.12, 133.09, 130.01, 129.99, 127.9 (4C), 76.6, 68.9, 66.0, 48.5, 42.4, 27.0 (3C), 20.9, 19.6, 19.2, 7.0 (3C), 5.1 (3C) ppm; HRMS (ESI) m/z [M − H]− calcd for C31H47O5Si2: 555.2968; found 555.2953 (−2.7 ppm).
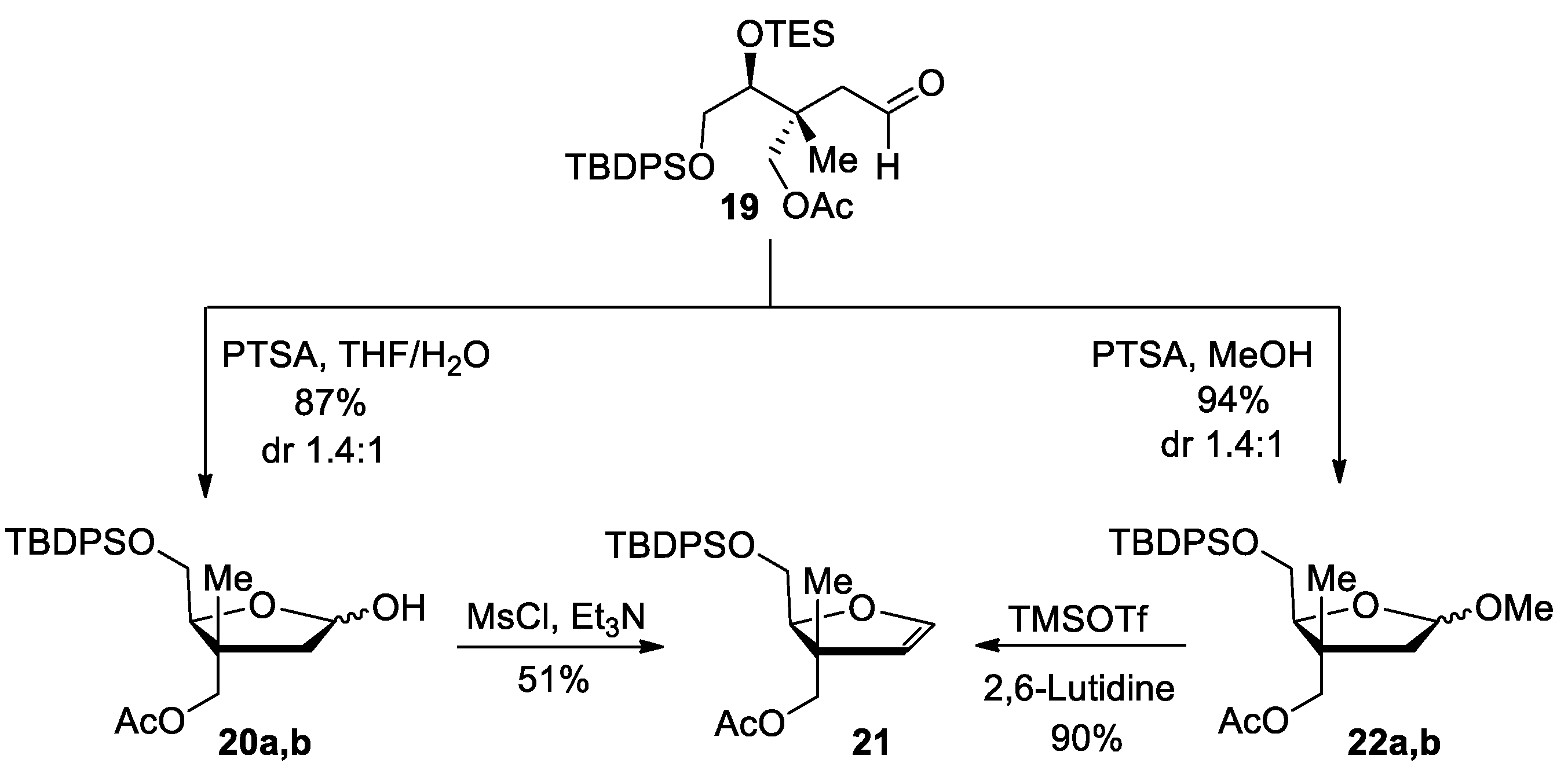
((2S,3R)-2-(((tert-Butyldiphenylsilyl)oxy)methyl)-5-hydroxy-3-methyltetrahydrofuran-3-yl)methyl acetate (20a,b). Aldehyde 19 (1.88 g, 3.38 mmol, 1.00 equiv.) was dissolved in a THF and H2O mixture (4:1) (34 mL, 0.1 M) followed by the addition of PTSA (963 mg, 5.06 mmol, 1.50 equiv.). The resulting solution was stirred at 50 °C for 1.5 h. The solution was diluted with DCM (30 mL) and a saturated solution of NaHCO3 (20 mL) was added. The aqueous layer was extracted with DCM (3 × 25 mL) and the combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography (Hexanes/EtOAc, 80:20) to provide lactols 20a,b (1.3 g, 87%, dr 1.4:1) as a colorless oil. Rf = 0.38 (Hexanes/EtOAc, 70:30); Formula: C25H34O5Si; MW: 442.63 g/mol; IR (neat) νmax 3428 (br), 3071, 3050, 2955, 2932, 2887, 2858, 1742 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.71–7.67 (m, 8H, major and minor), 7.46–7.37 (m, 12H, major and minor), 5.58–5.55 (m, 1H, major), 5.46–5.43 (m, 1H, minor), 4.11–4.09 (m, 3H, major), 3.92–3.85 (m, 3H, minor), 3.73–3.66 (m, 4H, major and minor), 3.23 (d, J = 7.5 Hz, 1H, minor), 2.74 (d, J = 4.3 Hz, 1H, major), 2.21 (dd, J = 13.7, 6.1 Hz, 1H, minor), 2.06 (dd, J = 13.0, 5.8 Hz, 1H, major), 2.033 (s, 3H, minor), 2.027 (s, 3H, major), 1.90 (dd, J = 13.6, 2.7 Hz, 1H, major), 1.81 (dd, J = 14.0, 3.1 Hz, 1H, minor), 1.18 (s, 3H, minor), 1.09 (s, 9H, minor), 1.06 (s, 3H, major), 1.05 (s, 9H, major) ppm; 13C NMR (126 MHz, CDCl3) δ 171.10 (minor), 171.07 (major), 135.9 (minor, 2C), 135.8 (major, 2C), 135.7 (major and minor, 4C), 133.4 (major), 133.3 (major), 132.81 (minor), 132.77 (minor), 130.1 (minor), 130.0 (minor), 129.9 (major, 2C), 127.98 (minor, 2C), 127.95 (minor, 2C), 127.86 (major, 2C), 127.84 (major, 2C), 98.7 (minor), 97.8 (major), 83.5 (minor), 82.4 (major), 70.8 (minor), 70.4 (major), 64.8 (minor), 63.9 (major), 45.3 (major), 45.2 (minor), 44.4 (minor), 44.2 (major), 27.1 (minor, 3C), 26.9 (major, 3C), 21.0 (major and minor), 19.3 (minor), 19.2 (major), 18.6 (major), 18.4 (minor) ppm; HRMS (ESI) m/z [M + Na]+ calcd for C25H34O5SiNa+: 465.2068; found 465.2070 (+0.4 ppm).
(+)-((2S,3R)-2-(((tert-Butyldiphenylsilyl)oxy)methyl)-3-methyl-2,3-dihydrofuran-3-yl)methyl acetate (21). To a solution of lactols 20a,b (1.27 g, 2.87 mmol, 1.00 equiv.) in anhydrous DCE (72 mL, 0.04 M), MsCl (0.78 mL, 10 mmol, 3.5 equiv.) was added. The resulting solution was stirred 3 min at room temperature, followed by 3 min at 75 °C. Triethylamine (3.0 mL, 22 mmol, 7.5 equiv.) was then added and the resulting solution was stirred 3 min at 75 °C. After cooling to room temperature, a saturated solution of NaHCO3 (20 mL) was added and the aqueous layer was extracted with Et2O (3 × 25 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. Purification by silica gel flash chromatography (Hexanes/EtOAc, 90:10) provided glycal 21 (620 mg, 51%) as a colorless oil. Rf = 0.65 (Hexanes/EtOAc, 80:20); [α]25D +59 (c 0.8, CH2Cl2); Formula: C25H32O4Si; MW: 424.61 g/mol; IR (neat) νmax 3071, 3050, 2958, 2932, 2887, 2858, 1743 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.70–7.68 (m, 4H), 7.45–7.37 (m, 6H), 6.28 (d, J = 2.7 Hz, 1H), 4.74 (d, J = 2.7 Hz, 1H), 4.33 (t, J = 6.1 Hz, 1H), 4.06 (d, J = 10.8 Hz, 1H), 3.89 (d, J = 10.8 Hz, 1H), 3.87–3.80 (m, 2H), 2.02 (s, 3H), 1.08 (s, 3H), 1.07 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 171.1, 145.6, 135.76 (2C), 135.75 (2C), 133.5, 133.4, 129.89, 129.87, 127.9 (4C), 107.4, 85.7, 70.9, 63.0, 48.1, 27.0 (3C), 21.0, 19.3, 18.0 ppm; HRMS (ESI) m/z [M + H]+ calcd for C25H33O4Si+: 425.2143; found 425.2148 (+1.2 ppm).
((2S,3R)-2-(((tert-Butyldiphenylsilyl)oxy)methyl)-5-methoxy-3-methyltetrahydrofuran-3-yl)methyl acetate (22a,b). To a solution of aldehyde 19 (1.12 g, 2.00 mmol, 1.00 equiv.) in anhydrous MeOH (10 mL, 0.20 M), PTSA (0.19 g, 1.0 mmol, 0.50 equiv.) was added at room temperature. The resulting mixture was stirred for 20 min until completion as indicated by TLC. The reaction was neutralized by addition of anhydrous Et3N (0.56 mL, 4.0 mmol, 2.0 equiv.) and the resulting mixture was concentrated under reduced pressure. Purification by silica gel flash chromatography (Hexanes/EtOAc, 90:10) provided methoxy lactols 22a,b (0.86 g, 94%, dr 1.4:1) as a colorless oil. Rf = 0.48 (Hexanes/EtOAc, 80:20); Formula: C26H36O5Si; MW: 456.65 g/mol; IR (neat) νmax 3071, 3049, 2954, 2931, 2890, 2858, 1743 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.69–7.68 (m, 8H, major and minor), 7.45–7.36 (m, 12H, major and minor), 5.03 (dd, J = 5.7, 2.9 Hz, 1H, major), 4.94 (dd, J = 6.0, 2.3 Hz, 1H, minor), 4.10 (d, J = 10.8 Hz, 1H, major), 4.00–3.93 (m, 5H, major and minor), 3.80–3.72 (m, 4H, major and minor), 3.36 (s, 3H, major), 3.30 (s, 3H, minor), 2.12 (dd, J = 13.5, 5.9 Hz, 1H, minor), 2.05 (s, 3H, minor), 1.99 (s, 3H, major), 1.97 (dd, J = 13.6, 5.8 Hz, 1H, major), 1.88 (dd, J = 13.6, 2.9 Hz, 1H, major), 1.74 (dd, J = 13.5, 2.2 Hz, 1H, minor), 1.18 (s, 3H, minor), 1.06 (s, 18H, major and minor), 1.01 (s, 3H, major) ppm; 13C NMR (126 MHz, CDCl3) δ 171.12 (minor), 171.05 (major), 135.77 (major and minor, 4C), 135.75 (minor, 2C), 135.73 (major, 2C), 133.65 (minor), 133.56 (major), 133.52 (minor), 133.47 (major), 129.9 (major), 129.84 (minor), 129.82 (major and minor), 127.8 (major and minor, 8C), 105.0 (minor), 104.2 (major), 83.6 (minor), 82.0 (major), 70.8 (minor), 70.3 (major), 64.4 (minor), 63.8 (major), 55.4 (minor), 55.2 (major), 44.5 (major), 43.9 (minor), 43.7 (minor), 43.5 (major), 26.9 (major and minor, 6C), 20.99 (minor), 20.96 (major), 19.33 (minor), 19.30 (major), 18.6 (major), 18.4 (minor) ppm; HRMS (ESI) m/z [M + Na]+ calcd for C26H36O5SiNa: 479.2224; found 479.2220 (−0.9 ppm).
(+)-((2S,3R)-2-(((tert-Butyldiphenylsilyl)oxy)methyl)-3-methyl-2,3-dihydrofuran-3-yl)methyl acetate (
21). To a solution of methoxy lactols
22a,
b (0.12 g, 0.25 mmol, 1.0 equiv.) in anhydrous DCM (1.3 mL, 0.2 M), 2,6-lutidine (0.12 mL, 1.0 mmol, 4.0 equiv.) and TMSOTf (0.09 mL, 0.5 mmol, 2 equiv.) were added at 0 °C [
32].The resulting solution was stirred for 30 min at room temperature. The mixture was then diluted in DCM (5 mL) followed by washing with water (5 mL), and the aqueous layer was extracted with DCM (3 × 5 mL). The combined organic layers were dried over MgSO
4, filtered and concentrated under reduced pressure. Purification by silica gel flash chromatography (Hexanes/EtOAc, 90:10) provided glycal
21 (96 mg, 90%) as a colorless oil, which was confirmed to be identical to the compound formed from
20a,
b (see above).
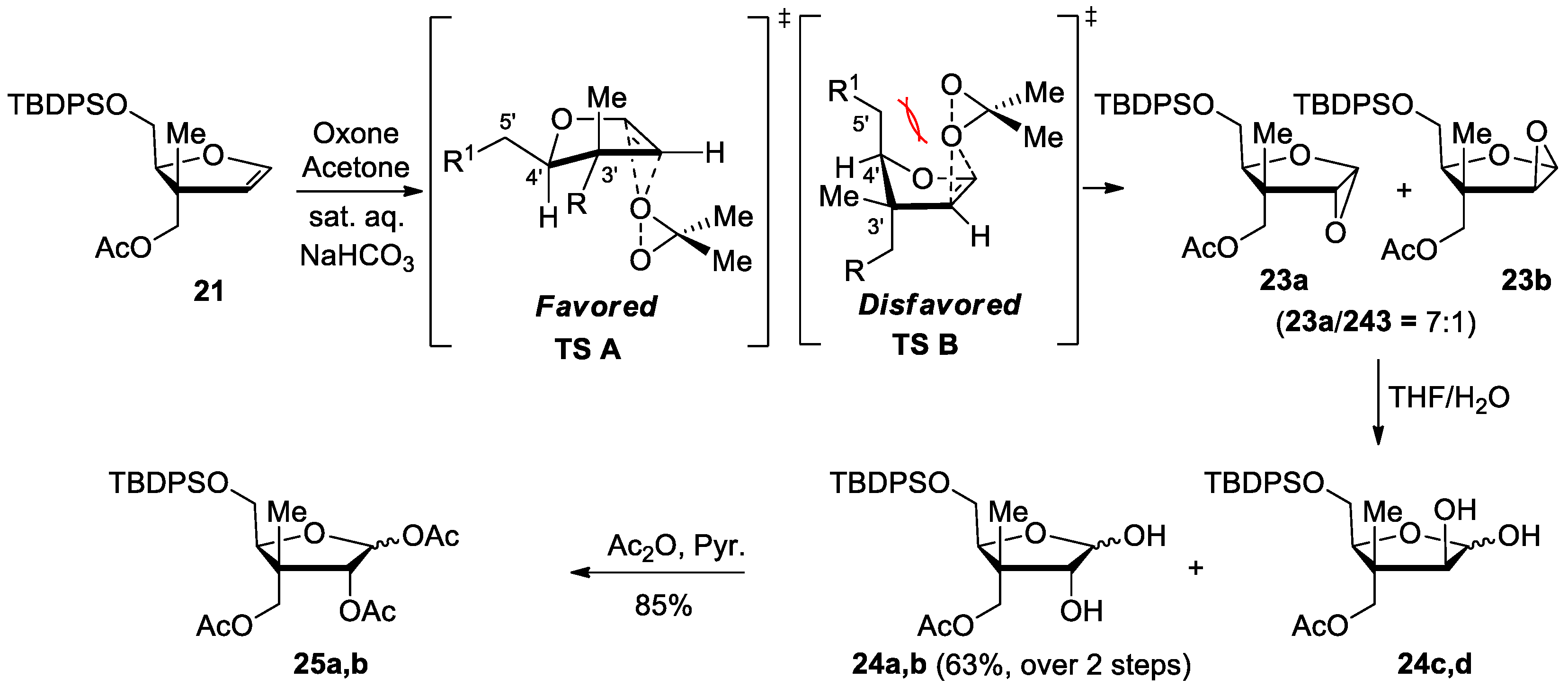
((2S,3S,4R)-2-(((tert-Butyldiphenylsilyl)oxy)methyl)-4,5-dihydroxy-3-methyltetra hydrofuran-3-yl)methyl acetate (24a,b). To a solution of glycal 21 (118 mg, 0.278 mmol, 1.00 equiv.) in anhydrous DCM (1.3 mL, 0.22 M) at 0 °C, acetone (0.13 mL, 1.6 mmol, 6.0 equiv.) and a saturated NaHCO3 solution (2.5 mL) were added. To the resulting biphasic mixture, a 0.37 mM solution of oxone in water (1.5 mL) was added. After sealing the flask, the resulting mixture was stirred for 30 min at 0 °C and 3 h at room temperature. After degassing the flask, the aqueous phase was extracted with DCM (3 × 5 mL). The organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The resulting crude epoxide 23a and 23b (dr 7:1, determined by 1H NMR of crude epoxide) was stirred in a THF and H2O (1:1) mixture (5.5 mL, 0.05 M) for 1 h. The aqueous phase was extracted with EtOAc (3 × 5 mL), and the resulting organic phase was dried over MgSO4, filtered and concentrated under reduced pressure. Purification by flash chromatography on silica gel (DCM/EtOAc, 80:20) provided lactols 24a,b (80 mg, 63%, 4:1 mixture) as a white foam. Rf = 0.33 (DCM/EtOAc, 85:15); Formula: C25H34O6Si; MW: 458.63 g/mol; IR (Neat) νmax 3421 (br), 2931, 2857, 1741, 1720 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.68–7.63 (m, 8H, major and minor), 7.45–7.37 (m, 12H, major and minor), 5.42 (dd, J = 9.6, 3.9 Hz, 1H, major), 5.23 (dd, J = 5.4, 1.1 Hz, 1H, minor), 4.53 (d, J = 11.4 Hz, 1H, major), 4.39 (d, J = 11.4 Hz, 1H, minor), 4.18 (d, J = 11.4 Hz, 1H, major), 4.06 (d, J = 11.4 Hz, 1H, minor), 4.11 (dd, J = 7.5, 4.9 Hz, 1H, minor), 4.02 (dd, J = 6.0, 4.9 Hz, 1H, minor), 3.89 (d, J = 4.2 Hz, 1H, major), 3.87 (d, J = 2.9 Hz, 1H, minor), 3.80–3.73 (m, 3H, major and minor), 3.64 (dd, J = 10.6, 7.5 Hz, 1H, major), 2.10 (s, 3H, major), 2.09 (s, 3H, minor), 1.18 (s, 3H, minor), 1.08 (s, 9H, minor), 1.05 (s, 9H, major), 1.03 (s, 3H, major) ppm; OH signals are missing possibly due to exchange in CDCl3 ppm; 13C NMR (126 MHz, CDCl3) δ 172.3 (major), 172.0 (minor), 135.74 (minor, 2C), 135.69 (minor, 2C), 135.67 (major, 2C), 135.65 (major, 2C), 133.1 (major), 133.0 (major), 132.9 (minor), 132.8 (minor), 130.04 (minor), 130.02 (minor), 129.95 (major, 2C), 128.0 (minor, 2C), 127.94 (minor, 2C), 127.90 (major, 2C), 127.89 (major, 2C), 103.9 (minor), 96.8 (major), 83.6 (minor) 83.0 (minor), 80.1 (major), 77.3 (major), 66.8 (minor), 66.6 (major), 63.9 (minor), 63.0 (major), 48.4 (major), 47.4 (minor), 27.0 (minor, 3C), 26.9 (major, 3C), 21.0 (major and minor), 19.3 (minor), 19.2 (major), 15.7 (minor), 15.4 (major) ppm; HRMS (ESI) m/z [M + NH4]+ calcd for C25H38O6NSi: 476.2463; found 476.2462 (−0.2 ppm).
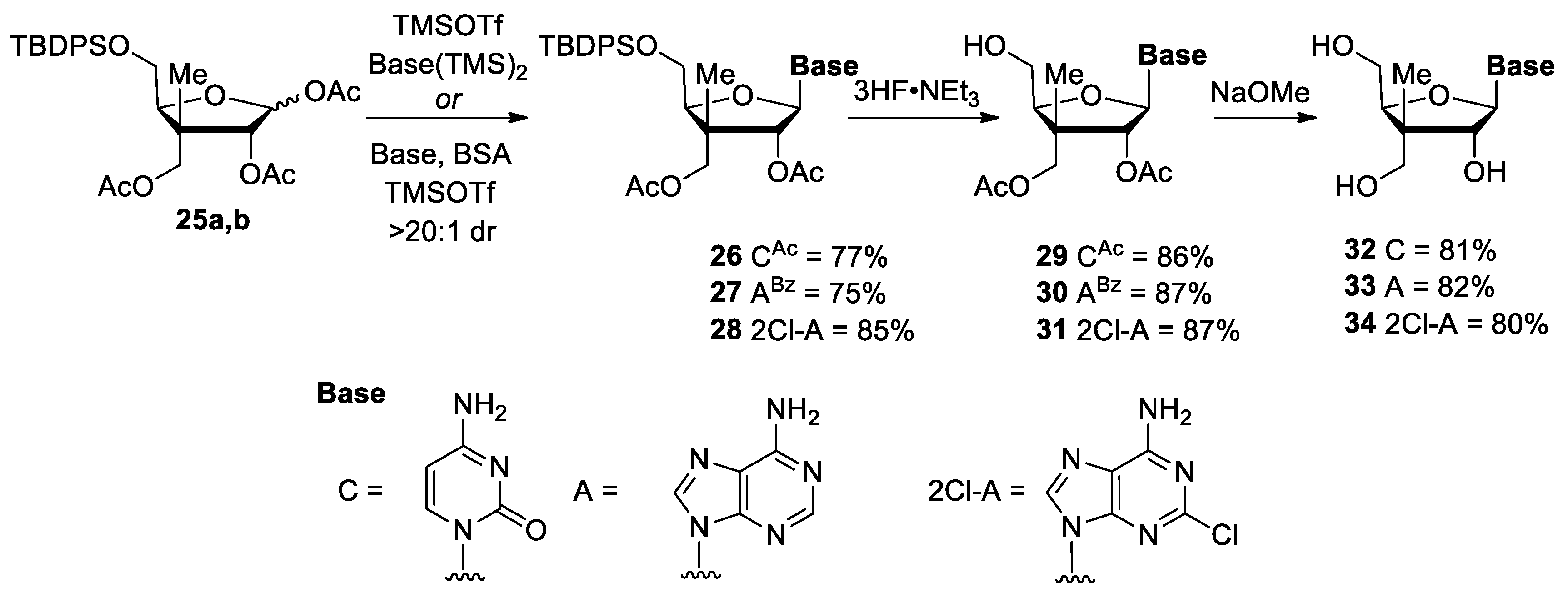
(3R,4R,5S)-4-(Acetoxymethyl)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-4-methyltetra hydrofuran-2,3-diyl diacetate (25a,b). Lactols 24a,b (65 mg, 0.14 mmol) were stirred in a solution of Ac2O:Pyr (2:1, 1.0 mL, 0.14 M) at room temperature for 18 h and then concentrated. Purification by flash chromatography on silica gel (Hexanes/EtOAc, 60:40), provided acetates 25a,b (65 mg, 85%, 4:1 mixture) as a colorless oil. Rf = 0.53 (Hexanes/EtOAc, 70:30); Formula: C29H38O8Si; MW: 542.70 g/mol; IR (Neat) νmax 2933, 2858, 1746 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.70–7.65 (m, 8H, major and minor), 7.46–7.37 (m, 12H, major and minor), 6.41 (d, J = 4.8 Hz, 1H, major), 6.00 (d, J = 1.6 Hz, 1H, minor), 5.35 (d, J = 4.8 Hz, 1H, major), 5.31 (d, J = 1.6 Hz, 1H, minor), 4.29 (d, J = 11.2 Hz, 1H, major), 4.25–4.23 (m, 2H, major and minor), 4.24 (d, J = 11.2 Hz, 1H, major), 4.16–4.10 (m, 2H, minor), 3.77–3.74 (m, 3H, major and minor), 3.68 (dd, J = 11.3, 3.5 Hz, 1H, major), 2.11 (s, 3H, minor), 2.10 (s, 3H, major), 2.07 (s, 3H, major), 2.03 (s, 3H, minor), 2.03 (s, 3H, major), 1.98 (s, 3H, minor), 1.28 (s, 3H, minor), 1.25 (s, 3H, major), 1.08 (s, 9H, minor), 1.08 (s, 9H, major) ppm; 13C NMR (126 MHz, CDCl3) δ 170.9 (major), 170.7 (minor), 169.8 (major), 169.7 (minor), 169.64 (minor), 169.61 (major), 135.8 (major, 2C), 135.72 (major, 2C), 135.71 (minor, 2C), 135.69 (minor, 2C), 133.03 (minor), 132.97 (major), 132.8 (minor), 132.7 (major), 130.01 (minor), 129.98 (major, 2C), 129.95 (minor), 127.94 (major, 2C), 127.92 (major and minor, 4C), 127.89 (minor, 2C), 100.0 (minor), 94.2 (major), 84.9 (minor), 82.9 (major), 82.0 (minor), 77.9 (major), 66.8 (major), 66.4 (minor), 63.4 (major), 63.2 (minor), 46.1 (minor), 45.2 (major), 26.90 (minor, 3C), 26.88 (major, 3C), 21.21 (minor), 21.19 (major), 20.93 (major), 20.89 (minor), 20.85 (minor), 20.6 (major), 19.3 (minor), 19.2 (major), 16.8 (major), 15.8 (minor) ppm; HRMS (ESI) m/z [M + NH4]+ calcd for C29H42O8NSi: 560.2674; found 560.2673 (−0.2 ppm).
(+)-(2R,3R,4R,5S)-2-(4-Acetamido-2-oxopyrimidin-1(2H)-yl)-4-(acetoxymethyl)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-4-methyltetrahydrofuran-3-yl acetate (26). To a solution of acetates 25a,b (70 mg, 0.13 mmol, 1.0 equiv.) in anhydrous MeCN (0.6 mL, 0.2 M), silylated N4-acetylcytosine (0.52 mL, 0.21 mmol, 1.6 equiv. 0.40 M in DCE) was added at room temperature. The resulting mixture was stirred for 10 min and TMSOTf (0.10 mL, 0.52 mmol, 4.0 equiv.) was added. The reaction was stirred at 60 °C for 3.5 h and then cooled to room temperature and quenched with saturated aqueous NaHCO3 (1 mL). The aqueous layer was extracted with DCM (3 × 5 mL) and the combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. Purification by flash chromatography on silica gel (DCM/MeOH, 95:5) provided the 1′,2′-trans ribo-like nucleoside analogue 26 (63 mg, 77%, dr 20:1) as a white foam. Rf = 0.34 (DCM/MeOH, 95:5); [α]25D +35 (c 0.2, MeOH); Formula: C33H41N3O8Si; MW: 635.79 g/mol; IR (neat) νmax 2957, 2932, 2893, 2858, 1746, 1671 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.17 (d, J = 7.6 Hz, 1H), 7.72–7.68 (m, 4H), 7.50–7.41 (m, 6H), 7.11 (d, J = 7.5 Hz, 1H), 6.06 (d, J = 5.3 Hz, 1H), 5.37 (d, J = 5.3 Hz, 1H), 4.27 (t, J = 3.9 Hz, 1H), 4.18 (d, J = 11.2 Hz, 1H), 4.13 (d, J = 11.2 Hz, 1H), 4.09 (dd, J = 11.9, 4.0 Hz, 1H), 3.94 (dd, J = 11.8, 3.9 Hz, 1H), 2.16 (s, 3H), 2.09 (s, 3H), 2.04 (s, 3H), 1.18 (s, 3H), 1.11 (s, 9H) ppm; NH signals are missing possibly due to exchange in CD3OD; 13C NMR (126 MHz, CD3OD) δ 173.0, 172.2, 171.3, 164.3, 158.1, 145.5, 136.9 (2C), 136.7 (2C), 134.1, 133.5, 131.35, 131.32, 129.12 (2C), 129.09 (2C), 98.2, 90.1, 85.4, 82.5, 67.5, 64.9, 47.2, 27.6 (3C), 24.5, 20.7, 20.6, 20.1, 16.8 ppm; HRMS (ESI) m/z [M + H]+ calcd for C33H42N3O8Si: 636.2736; found 636.2726 (−1.5 ppm).
(−)-((2S,3R,4R,5R)-4-Acetoxy-5-(6-benzamido-9H-purin-9-yl)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-methyltetrahydrofuran-3-yl)methyl acetate (
27). To a suspension of
N6-benzoyladenine (44 mg, 0.18 mmol, 2.5 equiv.) in anhydrous DCE (0.9 mL, 0.08 M), bis(trimethylsilyl)acetamide (0.10 mL, 0.40 mmol, 5.5 equiv.) was added at room temperature [
33]. The resulting mixture was vigorously stirred for 2 h until a clear solution was obtained. The solution was concentrated under high vacuum and treated with a solution of
25a,b (40 mg, 74 μmol, 1.0 equiv.) in anhydrous DCE (0.9 mL, 0.08 M). After adding TMSOTf (27 μL, 0.15 mmol, 2.0 equiv.), the resulting solution was heated at reflux for 4 h. The solution was diluted in DCM (5 mL) and washed with a saturated NaHCO
3 solution (2 mL). The aqueous phase was extracted with DCM (3 × 5 mL) and the combined organic layers were washed with brine, dried over MgSO
4, filtered and concentrated under reduced pressure. The
1H NMR spectrum of the crude reaction mixture indicated a >20:1 dr and only the N
9-regioisomer. Purification by flash chromatography on silica gel (EtOAc/MeOH, 97:3) provided
N6-benzoyladenine nucleoside analogue
27 (40 mg, 75%) as a white foam. R
f = 0.37 (DCM/MeOH, 95:5); [α]
25D −15 (
c 0.8, MeOH); Formula: C
39H
43N
5O
7Si; MW: 721.89 g/mol; IR (neat) ν
max 3268 (br), 3070, 3053, 2956, 2931, 2892, 2857, 1745 cm
−1;
1H NMR (500 MHz, CD
3OD) δ 8.66 (s, 1H), 8.44 (s, 1H), 8.08 (d,
J = 7.9 Hz, 2H), 7.69–7.63 (m, 5H), 7.56 (t,
J = 7.7 Hz, 2H), 7.47–7.38 (m, 4H), 7.31 (t,
J = 7.5 Hz, 2H), 6.28 (d,
J = 6.2 Hz, 1H), 6.02 (d,
J = 6.2 Hz, 1H), 4.34 (t,
J = 4.4 Hz, 1H), 4.30 (d,
J = 11.3 Hz, 1H), 4.22 (d,
J = 11.3 Hz, 1H), 4.08 (dd,
J = 11.6, 4.3 Hz, 1H), 3.99 (dd,
J = 11.6, 4.7 Hz, 1H), 2.11 (s, 3H), 2.07 (s, 3H), 1.34 (s, 3H), 1.07 (s, 9H) ppm; NH signals are missing possibly due to exchange in CD
3OD;
13C NMR (126 MHz, CD
3OD) δ 172.3, 171.5, 168.0, 153.4, 153.3, 151.1, 143.9, 136.73 (2C), 136.65 (2C), 135.0, 134.3, 133.9, 133.8, 131.2, 131.1, 129.8 (2C), 129.4 (2C), 129.0 (2C), 128.9 (2C), 125.0, 88.5, 85.4, 81.4, 67.7, 65.2, 47.4, 27.5 (3C), 20.8, 20.4, 20.1, 17.1 ppm; HRMS (ESI)
m/z [M + H]
+ calcd for C
39H
44N
5O
7Si: 722.3005; found 722.3011 (+0.9 ppm).
(+)-((2S,3R,4R,5R)-4-Acetoxy-5-(6-amino-2-chloro-9H-purin-9-yl)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-3-methyltetrahydrofuran-3-yl)methyl acetate (28). To a suspension of 2-chloroadenine (47 mg, 0.28 mmol, 2.5 equiv.) in anhydrous DCE (1.4 mL, 0.08 M), bis(trimethylsilyl)acetamide (0.15 mL, 0.61 mmol, 5.5 equiv.) was added at room temperature. The resulting mixture was vigorously stirred for 2 h until a clear solution was obtained. The solution was concentrated under high vacuum and treated with a solution of 25a,b (60 mg, 0.11 mmol, 1.0 equiv.) in anhydrous DCE (1.4 mL, 0.08 M). After adding TMSOTf (40 μL, 0.22 mmol, 2.0 equiv.), the resulting solution was heated at reflux for 4 h. The solution was diluted in DCM (5 mL) and washed with a saturated NaHCO3 solution (2 mL). The aqueous phase was extracted with DCM (3 × 5 mL) and the combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The 1H NMR spectrum of the crude reaction mixture indicated a >20:1 dr and only the N9-regioisomer. Purification by flash chromatography on silica gel (DCM/MeOH, 95:5) provided 2-chloroadenine nucleoside analogue 28 (61 mg, 85%) as a white foam. Rf = 0.32 (DCM/MeOH, 95:5); [α]25D +6.7 (c 1.0, MeOH); Formula: C32H38ClN5O6Si; MW: 652.22 g/mol; IR (neat) νmax 3319 (br), 3170 (br), 2957, 2932, 2893, 2859, 1744, 1646 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.10 (s, 1H), 7.68–7.63 (m, 4H), 7.46–7.31 (m, 6H), 6.08 (d, J = 6.3 Hz, 1H), 5.89 (d, J = 6.4 Hz, 1H), 4.30–4.27 (m, 2H), 4.19 (d, J = 11.3 Hz, 1H), 4.06 (dd, J = 11.6, 4.1 Hz, 1H), 3.94 (dd, J = 11.7, 4.3 Hz, 1H), 2.10 (s, 3H), 2.08 (s, 3H), 1.31 (s, 3H), 1.06 (s, 9H) ppm; NH signals are missing possibly due to exchange in CD3OD; 13C NMR (126 MHz, CD3OD) δ 172.4, 171.6, 158.0, 155.4, 151.8, 140.8, 136.8 (2C), 136.7 (2C), 134.2, 133.7, 131.12, 131.11, 129.0 (2C), 128.9 (2C), 119.3, 88.2, 85.4, 81.5, 67.7, 65.2, 47.4, 27.6 (3C), 20.8, 20.5, 20.1, 17.0 ppm; HRMS (ESI) m/z [M + H]+ calcd for C32H39ClN5O6Si+: 652.2353; found 652.2344 (–1.3 ppm).
(+)-(2R,3R,4R,5S)-2-(4-Acetamido-2-oxopyrimidin-1(2H)-yl)-4-(acetoxymethyl)-5-(hydroxymethyl)-4-methyltetrahydrofuran-3-yl acetate (29). Representative Procedure A: To a solution of 1′,2′-trans ribo-like nucleoside 26 (50 mg, 77 μmol, 1.0 equiv.) in anhydrous THF (0.8 mL, 0.1 M), 3HF‧NEt3 (0.13 mL, 0.79 mmol, 10 equiv.) was added at room temperature and the resulting mixture was stirred for 16 h. Triethylamine (1.10 mL, 7.90 mmol, 100 equiv.) was then added and the mixture was concentrated under reduced pressure. Purification by flash chromatography on silica gel (DCM/MeOH, 95:5) provided the 1′,2′-trans ribo-like nucleoside 29 (27 mg, 86%) as a white foam. Rf = 0.30 (DCM/MeOH, 95:5); [α]25D +26 (c 0.2, MeOH); Formula: C17H23N3O8; MW: 397.38 g/mol; IR (neat) νmax 3307 (br), 2927, 1742, 1650 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.61 (d, J = 7.6 Hz, 1H), 7.45 (d, J = 7.5 Hz, 1H), 6.13 (d, J = 5.7 Hz, 1H), 5.35 (d, J = 5.7 Hz, 1H), 4.22 (t, J = 3.6 Hz, 1H), 4.18 (s, 2H), 3.89 (dd, J = 12.1, 3.4 Hz, 1H), 3.79 (dd, J = 12.0, 3.8 Hz, 1H), 2.18 (s, 3H), 2.09 (s, 3H), 2.07 (s, 3H), 1.19 (s, 3H) ppm; OH and NH signals are missing possibly due to exchange in CD3OD; 13C NMR (126 MHz, CD3OD) δ 173.0, 172.4, 171.5, 164.4, 158.3, 146.3, 98.2, 90.0, 85.9, 82.6, 67.7, 62.5, 47.3, 24.5, 20.8, 20.6, 16.4 ppm; HRMS (ESI) m/z [M + H]+ calcd for C17H24N3O8: 398.1558; found 398.1552 (–1.6 ppm).
(−)-((2S,3R,4R,5R)-4-Acetoxy-5-(6-benzamido-9H-purin-9-yl)-2-(hydroxymethyl)-3-methyltetrahydrofuran-3-yl)methyl (30). Following the Representative Procedure A, N6-benzoyladenine nucleoside analogue 27 (40 mg, 55 μmol, 1.0 equiv.) in anhydrous THF (0.55 mL, 0.10 M), 3HF·NEt3 (0.14 mL, 0.83 mmol, 15 equiv.) was added and the mixture was stirred for 16 h. Triethylamine (0.77 mL, 5.5 mmol, 100 equiv.) was then added. Purification by flash chromatography on silica gel (DCM/MeOH, 95:5), provided the 1′,2′-trans ribo-like N6-benzoyladenine nucleoside analogue 30 (23 mg, 86%) as a white foam. Rf = 0.54 (DCM/MeOH, 90:10); [α]25D −51 (c 0.7, MeOH); Formula: C23H25N5O7; MW: 483.48 g/mol; IR (neat) νmax 3300 (br), 3069, 2927, 2855, 1742 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.82 (s, 1H), 8.70 (s, 1H), 8.09–8.08 (m, 2H), 7.67–7.64 (m, 1H), 7.58–7.55 (m, 2H), 6.35 (d, J = 6.7 Hz, 1H), 5.92 (d, J = 6.7 Hz, 1H), 4.31–4.25 (m, 3H), 3.93 (dd, J = 12.2, 3.1 Hz, 1H), 3.84 (dd, J = 12.2, 3.3 Hz, 1H), 2.16 (s, 3H), 2.03 (s, 3H), 1.34 (s, 3H) ppm; OH and NH signals are missing possibly due to exchange in CD3OD; 13C NMR (126 MHz, CD3OD) δ 172.4, 171.5, 168.1, 153.4, 153.1, 151.2, 144.5, 135.0, 133.9, 129.8 (2C), 129.4 (2C), 125.2, 88.4, 86.2, 81.6, 67.9, 63.0, 47.3, 20.8, 20.4, 16.6 ppm; HRMS (ESI) m/z [M + H]+ calcd for C23H26N5O7: 484.1827; found 484.1831 (+0.8 ppm).
(−)-((2S,3R,4R,5R)-4-Acetoxy-5-(6-amino-2-chloro-9H-purin-9-yl)-2-(hydroxymethyl)-3-methyltetrahydrofuran-3-yl)methyl acetate (31). Following the Representative Procedure A, Nucleoside analogue 28 (60 mg, 92 μmol, 1.0 equiv.) in anhydrous THF (0.9 mL, 0.1 M), 3HF·NEt3 (0.23 mL, 1.4 mmol, 15 equiv.) was added and the mixture was stirred for 16 h. Triethylamine (1.3 mL, 9.2 mmol, 100 equiv.) was then added. Purification by flash chromatography on silica gel (DCM/MeOH, 95:5), provided the 1′,2′-trans ribo-like 2-chloroadenine nucleoside analogue 31 (33 mg, 87%) as a white foam. Rf = 0.50 (DCM/MeOH, 90:10); [α]25D −75 (c 0.2, MeOH); Formula: C16H20ClN5O6; MW: 413.82 g/mol; IR (neat) νmax 3326 (br), 2942, 1743, 1618 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.41 (s, 1H), 6.12 (d, J = 6.8 Hz, 1H), 5.80 (d, J = 6.8 Hz, 1H), 4.27 (d, J = 11.2 Hz, 1H), 4.24–4.21 (m, 2H), 3.91 (dd, J = 12.4, 3.0 Hz, 1H), 3.80 (dd, J = 12.4, 3.2 Hz, 1H), 2.15 (s, 3H), 2.05 (s, 3H), 1.32 (s, 3H) ppm; OH and NH signals are missing possibly due to exchange in CD3OD; 13C NMR (126 MHz, CD3OD) δ 172.5, 171.5, 158.1, 155.2, 151.7, 141.6, 119.4, 88.4, 86.0, 81.4, 67.9, 63.0, 47.2, 20.8, 20.4, 16.6 ppm; HRMS (ESI) m/z [M + H]+ calcd for C16H21ClN5O6+: 414.1175; found 414.1169 (−1.5 ppm).
(+)-4-Amino-1-((2R,3R,4S,5S)-3-hydroxy-4,5-bis(hydroxymethyl)-4-methyltetrahydro furan-2-yl)pyrimidin-2(1H)-one (
32). Representative procedure B: To a solution of nucleoside
29 (13 mg, 32 μmol, 1.0 equiv.) in anhydrous MeOH (0.3 mL, 0.1 M), NaOMe (32 μL, 32 μmol, 1.0 equiv., 1.0 M in MeOH) was added at room temperature. The reaction was stirred for 3 h, quenched with amberlite acidic resin (~50 mg) and stirred for 10 min. The mixtrue was filtered with MeOH (~5 mL) and concentrated under reduced pressure. Purification by C18 reverse phase flash chromatography (MeOH/H
2O) provided 1′,2′-
trans ribo-like cytosine nucleoside analogue
32 [
14] (7 mg, 81%) as a white foam. [α]
25D +28 (
c 0.4, CH
3OH); Formula: C
11H
17N
3O
5; MW: 271.27 g/mol; IR (neat) ν
max 3345, 3217, 2967, 2949, 1649, 1607 cm
−1;
1H NMR (500 MHz, CD
3OD) δ 8.03 (d,
J = 7.5 Hz, 1H), 5.91 (d,
J = 7.5 Hz, 1H), 5.83 (d,
J = 5.5 Hz, 1H), 4.18 (dd,
J = 4.5, 3.5 Hz, 1H), 4.11 (d,
J = 5.5 Hz, 1H), 3.78 (dd,
J = 11.5, 3.5 Hz, 1H), 3.70 (d,
J = 11.1 Hz, 1H), 3.67 (dd,
J = 11.8, 4.7 Hz, 1H), 3.63 (d,
J = 11.1 Hz, 1H), 1.09 (s, 3H) ppm;
OH and
NH signals are missing possibly due to exchange in CD
3OD;
13C NMR (126 MHz, CD
3OD) δ 167.7, 159.1, 143.1, 95.9, 92.8, 85.5, 83.1, 66.3, 63.1, 48.4, 16.6 ppm; HRMS (ESI)
m/z [M + Na]
+ calcd for C
11H
17O
5N
3Na
+: 294.1060; found 294.1063 (+0.8 ppm).
(−)-((2S,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-4-hydroxy-3-methyltetrahydrofuran-2,3-diyl)dimethanol (33). Following Representative Procedure B, NaOMe (25 μL, 25 μmol, 1.0 equiv., 1.0 M in MeOH) was added to a solution of nucleoside 30 (12 mg, 25 μmol, 1.0 equiv.) in MeOH (0.25 mL, 0.10 M). Purification by C18 reverse phase flash chromatography (MeOH/H2O) provided 1′,2′-trans ribo-like adenine nucleoside analogue 33 (6 mg, 82%) as a white foam. Rf = 0.30 (DCM/MeOH, 80:20); [α]25D −42 (c 0.2, MeOH); Formula: C12H17N5O4; MW: 295.30 g/mol; IR (neat) νmax 3332 (br), 3193 (br), 2928, 2882, 1649 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.30 (s, 1H), 8.18 (s, 1H), 6.03 (d, J = 7.4 Hz, 1H), 4.66 (d, J = 7.5 Hz, 1H), 4.23 (t, J = 2.6 Hz, 1H), 3.92 (dd, J = 12.6, 2.7 Hz, 1H), 3.80 (d, J = 11.0 Hz, 1H), 3.73 (dd, J = 12.6, 2.6 Hz, 1H), 3.61 (d, J = 11.0 Hz, 1H), 1.26 (s, 3H) ppm; OH and NH signals are missing possibly due to exchange in CD3OD; 13C NMR (126 MHz, CD3OD) δ 157.6, 153.3, 149.9, 142.4, 121.1, 91.4, 85.9, 80.9, 66.5, 63.9, 48.1, 16.4 ppm; HRMS (ESI) m/z [M + H]+ calcd for C12H18N5O4: 296.1353; found 296.1356 (+1.0 ppm).
(−)-((2S,3S,4R,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-hydroxy-3-methyltetrahydro furan-2,3-diyl)dimethanol (34). Following Representative Procedure B, NaOMe (24 μL, 24 μmol, 1.0 equiv., 1.0 M in MeOH) was added to a solution of nucleoside 31 (10 mg, 24 μmol, 1.0 equiv.) in MeOH (0.24 mL, 0.10 M). Purification by C18 reverse phase flash chromatography (MeOH/H2O) provided 1′,2′-trans ribo-like 2-chloroadenine nucleoside analogue 34 (6.4 mg, 80%) as a white foam. Rf = 0.50 (DCM/MeOH, 80:20); [α]25D −25 (c 0.2, MeOH); Formula: C12H16ClN5O4; MW: 329.74 g/mol; IR (neat) νmax 3322 (br), 3186 (br), 2940, 2884, 1653 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.30 (s, 1H), 5.98 (d, J = 7.3 Hz, 1H), 4.61 (d, J = 7.3 Hz, 1H), 4.22 (t, J = 2.9 Hz, 1H), 3.90 (dd, J = 12.5, 2.9 Hz, 1H), 3.79 (d, J = 11.0 Hz, 1H), 3.74 (dd, J = 12.5, 3.0 Hz, 1H), 3.60 (d, J = 11.0 Hz, 1H), 1.24 (s, 3H) ppm; OH and NH signals are missing possibly due to exchange in CD3OD; 13C NMR (126 MHz, CD3OD) δ 158.2, 155.0, 151.4, 142.4, 119.9, 91.1, 85.8, 81.1, 66.4, 63.8, 48.1, 16.4 ppm; HRMS (ESI) m/z [M + H]+ calcd for C12H17ClN5O4: 330.0964; found 330.0958 (−1.7 ppm).
(−)-(3R,4S,5R)-4-((tert-Butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3-methyl-3-vinyldihydrofuran-2(3H)-one (S2). To a solution of secondary alcohol 40 (4.00 g, 14.0 mmol, 1.00 equiv.) in anhydrous DCM (42 mL, 0.33 M), 2,6 Lutidine (4.04 mL, 34.9 mmol, 2.50 equiv.) and TBSOTf (4.81 mL, 2.09 mmol, 1.50 equiv.) were added at 0 °C. The resulting mixture was gradually warmed to room temperature and stirring was continued for overnight. A saturated aqueous solution of NaHCO3 (~20 mL) was added and the mixture was extracted with CH2Cl2 (3 × 40 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered and condensed under reduced pressure. The residue was purified by flash chromatography on silica gel (Hexanes/EtOAc, 90:10) to provide bis-silylated ethers S2 (4.2 g, 75%) as a colorless oil. Rf= 0.28 (Hexanes/EtOAc, 9:1); [α]25D −60 (c 1.2, CH2Cl2); IR (neat) νmax 2954, 2930, 2858, 1786 cm−1; Formula: C20H40O4Si2; MW: 400.70 g/mol; 1H NMR (500 MHz, CDCl3): δ 5.94 (dd, J = 17.7, 10.7 Hz, 1H), 5.24 (dd, J = 10.7, 0.9 Hz, 1H), 5.18 (d, J = 17.6 Hz, 1H), 4.27 (d, J = 8.1 Hz, 1H), 4.28–3.99 (m, 1H), 3.98 (dd, J = 12.3, 1.8 Hz, 1H), 3.74 (dd, J = 12.3, 2.5 Hz, 1H), 1.33 (s, 3H), 0.90 (s, 9H), 0.88 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H), 0.07 (s, 3H), 0.06 (s, 3H) ppm. 13C NMR (126 MHz, CDCl3) δ 176.6, 134.1, 116.8, 82.1, 75.0, 59.6, 51.6, 25.9 (3C), 25.8 (3C), 21.3, 18.4, 18.1, −4.2, −4.7, −5.2, −5.4 ppm; HRMS (ESI) m/z [M + H]+ calcd for C20H41O4Si2: 401.2543; found 401.2539 (+0.2 ppm) and [M+NH4]+ calcd for C20H44NO4Si2: 418.2809; found 418.2800 (−0.7 ppm).
(+)-(3S,4S,5R)-4-((tert-Butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3-methyl-2-oxotetrahydrofuran-3-carbaldehyde (41). The alkene S2 (761 mg, 1.90 mmol, 1.00 equiv.) was dissolved in CH2Cl2 (114 mL, 0.02 M) and the mixture was cooled to −78 °C. Ozone was bubbled through the reaction mixture until a blue color appeared at which point the ozone inlet was changed for a N2 inlet and bubbling was continued for 15 min. The solution was then purged with nitrogen to remove excess of ozone. Triethylamine (0.265 mL, 1.90 mmol, 1.00 equiv.) was added and the mixture was stirred for 30 min at −78 °C. Then, it was gradually warmed to room temperature and stirring was continued for 1 h. The reaction mixture was filtered over MgSO4 and condensed under reduced pressure. The crude was purified by flash chromatography on silica gel (Hexanes/EtOAc, 100:0 to 70:30) to provide the aldehyde 41 (663 mg, 87%) as colourless oil. Rf = 0.67 (EtOAc/Hexanes, 1:4); [α]25D: +37 (c 5.4, in CH2Cl2); IR: (neat) νmax 2954, 2930, 2886, 2858, 1789, 1729, 1472, 1463, 1254 cm−1. Formula: C19H38O5Si2; MW: 402.67 g.mol−1; NMR 1H: (500 MHz, CDCl3) δ 9.57 (s, 1H), 4.54 (d, J = 6.8 Hz, 1H), 4.35 (appdt, J = 6.8, 2.2 Hz, 1H), 4.00 (dd, J = 12.3, 2.1 Hz, 1H), 3.77 (dd, J = 12.3, 2.3 Hz, 1H), 1.49 (s, 3H), 0.88 (s, 9H), 0.86 (s, 9H), 0.11 (s, 3H), 0.09 (s, 3H), 0.08 (s, 3H), 0.06 (s, 3H) ppm; NMR 13C: (126 MHz, CDCl3) δ 195.9, 172.7, 83.7, 76.8, 60.5, 59.9, 25.9 (3C), 25.6 (3C), 18.4, 17.9, 15.4, −4.5, −4.8, −5.2, −5.4 ppm; HRMS (ESI): m/z [M + H]+ calcd for C19H39O5Si2: 403.2331, found: 403.2329 (−0.50 ppm).
(3R,4S,5R)-4-((tert-Butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3-(hydroxymethyl)-3-methyltetrahydrofuran-2-ol (42a,b). To a solution of aldehyde 41 (2.30 g, 5.71 mmol, 1.00 equiv.) in anhydrous THF (57 mL, 0.10 M), Red-Al (3.57 mL, 11.4 mmol, 2.00 equiv.) was added at −40 °C and stirring was continued for 40 min. at −40 °C. The reaction was then cooled to −78 °C and was quenched by the addition of few drops (~0.5 mL) of saturated Rochelle salt solution. The stirring was continued for 10 min at −78 °C and it was then gradually warmed to room temperature. THF (~20 mL) and saturated Rochelle salt solution (~15 mL) were added. The resulting biphasic mixture was stirred vigorously for an hour. The layers were separated, the aqueous layer was extracted with Et2O (3 × 40 mL), and the combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (Hexanes/EtOAc, 4:1) to give the lactol 42a,b (1.6 g, 69%, dr 1:2) as a colorless oil. Rf = 0.24 (Hexanes/EtOAc, 4:1); IR (neat) νmax 3329, 3200, 2930, 1645, 1600 cm−1; Formula: C19H42O5Si2; MW: 406.70 g/mol; 1H NMR (500 MHz, CDCl3): δ 5.36 (dd, J = 8.9, 2.3 Hz, 1H, minor (OH, D2O exchange), 5.04 (d, J = 8.6 Hz, 1H, minor), 4.92 (d, J = 8.8 Hz, 1H, major), 4.25 (d, J = 5.5 Hz, 1H, major), 4.08 (d, J = 6.3 Hz, 1H, minor), 4.02–3.95 (m, 2H, major and minor), 3.89 (d, J = 11.8 Hz, 1H, minor), 3.83 (dd, J = 11.6, 2.8 Hz, 1H, minor), 3.80–3.74 (m, 2H, major and minor), 3.74–3.67 (m, 2H, minor and major), 3.65 (dd, J = 11.0, 2.3 Hz, 1H, major), 3.59–3.52 (m, 2H, major and OH (D2O exchange)), 1.11 (s, 3H, major), 1.01 (s, 3H, minor), 0.93 (s, 9H, major), 0.90 (s, 9H, minor), 0.89 (s, 18H, major and minor), 0.12 (s, 6H, major), 0.11 (s, 12H, minor), 0.07 (s, 3H, major), 0.06 (s, 3H, major) ppm; OH signals are missing possibly due to exchange in CDCl3 ppm. 13C NMR (126 MHz, CDCl3) δ 105.7 (minor), 102.2 (major), 85.4 (major), 84.6 (minor), 78.9 (minor), 78.7 (major), 66.7 (major), 65.8 (minor), 62.6 (minor), 61.6 (major), 51.4 (major), 49.0 (minor), 26.00 (3C, minor), 25.96 (3C, major), 25.84 (3C, minor), 25.80 (3C, major), 21.2 (minor), 18.43 (minor), 18.40 (major), 18.0 (minor), 17.9 (major), 16.0 (major), −4.3 (major), −4.39 (minor), −4.43 (major), −4.6 (major), −5.1 (minor), −5.39 (major), −5.40 (2C, minor) ppm; HRMS (ESI) m/z [M+NH4]+ calcd for C19H46NO5Si2: 424.2915; found 424.2904 (−1.6 ppm).
((3R,4S,5R)-2-(Benzoyloxy)-4-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3-methyltetrahydrofuran-3-yl)methyl benzoate (43a,b). To a solution of lactol 42a,b (3.30 g, 8.11 mmol, 1.00 equiv.) in CH2Cl2 (45 mL, 0.18 M), pyridine (3.94 mL, 48.7 mmol, 6.00 equiv.) and DMAP (99 mg, 0.81 mmol, 0.10 equiv.) were added. After cooling the resulting mixture to 0 °C, BzCl (4.71 mL, 40.6 mmol, 5.00 equiv.) was added dropwise. The reaction mixture was gradually warmed to room temperature and stirred for 16 h. The mixture was cooled to 0 °C and ethylenediamine (1.36 mL, 20.3 mmol, 2.50 equiv.) was added and stirring was continued for 1 h at 0 °C. The reaction mixture was then diluted with hexanes (40 mL) and passed through a celite using Et2O. The filtrates were concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (Hexanes/EtOAc, 4:1) to afford the 43a,b (3.8 g, 76%, dr 1.2:1) as a mixture of anomers as a white foam. Rf= 0.34 (Hexanes/EtOAc, 4:1); IR (neat) νmax 2954, 2929, 2885, 2857, 1722, 1261 cm−1; Formula: C33H50O7Si2; MW: 614.92 g/mol; 1H NMR (500 MHz, CDCl3): δ 8.11–8.03 (m, 4H, major), 8.00–7.96 (m, 2H, minor), 7.62–7.50 (m, 5H, minor), 7.49–7.36 (m, 9H, major and minor), 6.54 (s, 1H, major), 6.37 (s, 1H, minor), 4.61 (d, J = 11.1 Hz, 1H, minor), 4.56 (d, J = 11.1 Hz, 1H, minor), 4.58–4.54 (m, 2H, major and minor), 4.40 (d, J = 7.4 Hz, 1H, major), 4.27–4.23 (m, 1H, minor), 4.19 (d, J = 3.1 Hz, 1H, minor), 4.09–4.03 (m, 1H, major), 3.91 (dd, J = 11.6, 2.4 Hz, 1H, major), 3.88–3.84 (m, 2H, major and minor), 3.72 (dd, J = 11.6, 4.4 Hz, 1H, major), 1.32 (s, 6H, major and minor), 0.97 (s, 9H, minor), 0.94 (s, 9H, major), 0.93 (s, 9H, minor), 0.79 (s, 9H, major), 0.19 (s, 3H, major), 0.14 (s, 6H, major and minor), 0.13 (s, 3H, minor), 0.11 (s, 6H, major and minor), −0.01 (s, 3H, major), −0.08 (s, 3H, minor) ppm. 13C NMR (126 MHz, CDCl3) δ 166.6 (minor), 166.5 (major), 165.6 (major), 165.5 (minor), 133.34 (major), 133.31 (minor), 133.2 (major), 133.0 (minor), 130.3 (minor), 130.13 (major), 130.06 (minor), 130.0 (3C, 2minor and 1major), 129.9 (2C, major), 129.8 (2C, major), 129.61 (2C, minor), 128.6 (2C, major), 128.5 (4C, major and minor), 128.4 (2C, minor), 102.4 (minor), 100.5 (major), 89.9 (minor), 85.5 (major), 77.8 (minor), 76.7 (major), 66.3 (major), 65.4 (minor), 63.1 (major), 62.6 (minor), 50.1 (minor), 49.8 (major), 26.1 (3C, minor), 26.0 (3C, major), 25.9 (3C, major), 25.8 (3C, minor), 20.8 (minor), 18.6 (minor), 18.5 (major), 18.08 (major), 18.07 (minor), 16.9 (major), −4.1 (major), −4.2 (minor), −4.3 (major), −4.8 (major), −5.2 (minor), −5.28 (minor), −5.34 (2C major and minor) ppm HRMS (ESI) m/z [M-OBz]+ calcd for C26H45O5Si2: 493.2806; found 493.2805; and [M+NH4]+ calcd for C33H54NO7Si2: 632.3439; found 632.3439 (−0.2 ppm).
(−)((2R,3R,4S,5R)-4-((tert-Butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-2-(2,6-dichloro-9H-purin-9-yl)-3-methyltetrahydrofuran-3-yl)methyl benzoate (44). To a solution of the benzoylated furanosides 43a,b (434 mg, 0.706 mmol, 1.00 equiv.) in anhydrous MeCN (2.8 mL, 0.25 M), 2,6-dichloropurine (147 mg, 0.776 mmol, 1.10 equiv.) was added at room temperature. The resulting mixture was cooled to −10 °C and DBU (0.316 mL, 2.12 mmol, 3.00 equiv.) was added followed by dropwise addition of TMSOTf (0.520 mL, 2.82 mmol, 4.00 equiv.). The stirring was continued at −10 °C for 3 h. The mixture was warmed to room temperature, and a saturated solution of NaHCO3 (~8 mL) was added. The aqueous layer was extracted with CH2Cl2 (3 × 20 mL) and the combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (CH2Cl2/MeOH, 100:0 to 70:30) to provide the pure product 44 (375 mg, 78%, β:α = 10:1) as brown gum. Rf = 0.87 (MeOH/CH2Cl2, 1:9); [α]25D: −10 (c 4.9, MeOH); IR: (neat) νmax 2955, 2930, 2898, 2858, 1724, 1590, 1553, 1470, 1464, 1452, 1356, 1255, 1214 cm−1; Formula: C31H46Cl2N4O5Si2; MW: 680.80 g.mol−1; NMR 1H: (500 MHz, CDCl3) δ 8.90 (s, 1H), 8.20 (s, 2H), 7.61 (t, J = 7.4 Hz, 1H), 7.52 (t, J = 7.7 Hz, 2H), 6.61 (s, 1H), 4.60 (d, J = 11.7 Hz, 1H), 4.50 (d, J = 7.2 Hz, 1H), 4.48 (d, J = 11.6 Hz, 1H), 4.15 (d, J = 12.1 Hz, 1H), 4.07 (appd, J = 8.4 Hz, 1H), 3.90 (d, J = 12.1 Hz, 1H), 1.01 (s, 9H), 0.93 (s, 9H), 0.87 (s, 3H), 0.21 (s, 3H), 0.21 (s, 3H), 0.14 (s, 3H), 0.12 (s, 3H) ppm; NMR 13C: (126 MHz, CDCl3) δ 166.7, 153.1, 152.8, 151.9, 145.0, 133.5, 131.0, 129.9 (2C), 129.7, 128.8 (2C), 88.2, 84.3, 74.6, 66.5, 61.0, 50.1, 26.4 (3C), 25.9 (3C), 18.8, 18.1, 17.5, −4.1, −4.3, −5.0, −5.1 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C31H47Cl2N4O5Si2: 681.2457, found: 681.2450 (−1.0 ppm).
(+)((2R,3R,4S,5R)-2-(6-Amino-2-chloro-9H-purin-9-yl)-4-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3-methyltetrahydrofuran-3-yl)methanol (S3). To a stirred solution of the nucleoside 44 (199 mg, 0.292 mmol, 1.00 equiv.) in anhydrous MeOH (1.2 mL, 0.20 M) in a high-pressure flask, NH3(g) was bubbled until saturation at room temperature. The reaction mixture was then stirred at 80 °C for 24 h. The mixture was then diluted with MeOH and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (CH2Cl2/MeOH, 100/0 to 94/6) to provide the nucleoside analogue S3 (141 mg, 87%) as white foam. Rf = 0.66 (EtOAc/Hexanes, 1:4); [α]25D: + 12 (c 0.8, in MeOH); IR: (neat) νmax 3324, 3189, 2955, 2930, 2896, 2858, 1642, 1594, 1463, 1346, 1316, 1253, 1215 cm−1; Formula: C24H44ClN5O4Si2; MW: 558.26 g.mol−1; NMR 1H: (500 MHz, CDCl3) δ 8.40 (s, 1H), 6.30 (s, 1H), 4.37 (d, J = 7.6 Hz, 1H), 4.06 (dd, J = 11.8, 2.0 Hz, 1H), 4.01 (dt, J = 7.6, 2.3 Hz, 1H), 3.89 (s, 2H), 3.86 (dd, J = 11.8, 2.5 Hz, 1H), 0.97 (s, 9H), 0.93 (s, 9H), 0.71 (s, 3H), 0.17 (s, 3H), 0.16 (s, 3H), 0.14 (s, 3H), 0.13 (s, 3H) ppm; OH and NH signals are missing possibly due to exchange in CDCl3; NMR 13C: (126 MHz, CDCl3) δ 155.6, 150.3, 145.8, 139.3, 114.2, 89.3, 85.0, 77.5, 66.2, 61.1, 50.6, 26.3 (3C), 25.9 (3C), 18.7, 18.0, 17.3, −4.1, −4.4, −5.1, −5.1 ppm; HRMS (ESI): m/z [M + H]+ calcd for C24H45ClN5O4Si2: 558.2693, found: 558.2686 (−1.25 ppm).
(−)-((2R,3S,4R,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-3-hydroxy-4-methyltetra hydrofuran-2,4-diyl)dimethanol (45). To a solution of nucleoside S3 (68 mg, 0.12 mmol, 1.0 equiv) in THF (1.2 mL, 0.1 M) was added HF⸱Et3N (0.30 mL, 1.8 mmol, 15 equiv.) at room temperature. The reaction mixture was stirred for 18 h. It was then cooled to 0 °C and triethylamine (1.70 mL, 12.2 mmol, 100 equiv.) was added. The mixture was concentrated under reduced pressure. The crude was purified by reverse phase C18 flash chromatography (MeOH/H2O, 0:100 to 4:6) to provide nucleoside analogue 45 (30 mg, 75%) as white foam. Rf = 0.17 (CH2Cl2/CH3OH, 90:10); [α]25D –10 (c 1.0, CH3OH); Formula: C12H16ClN5O; MW: 329.74 g/mol; IR (neat) νmax 3339, 3199, 2936, 1653, 1594 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.48 (s, 1H), 6.27 (s, 1H), 4.36 (d, J = 8.8 Hz, 1H), 4.07 (ddd, J = 8.8, 3.6, 2.2 Hz, 1H), 3.97 (dd, J = 12.4, 2.2 Hz, 1H), 3.89–3.84 (m, 2H), 3.77 (d, J = 11.3 Hz, 1H), 0.70 (s, 3H) ppm; OH and NH signals are missing possibly due to exchange in CD3OD; 13C NMR (125 MHz, CD3OD) δ 158.1, 155.3, 151.5, 140.9, 119.0, 90.6, 85.6, 76.3, 65.4, 61.7, 51.7, 17.3 ppm; HRMS (ESI): m/z [M + Na]+ calcd for: C12H16ClN5NaO4: 352.0783; found 352.0783 (−0.096 ppm).
(−)-((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3-hydroxy-4-methyltetrahydrofuran-2,4-diyl)dimethanol (46). To a solution of nucleoside 45 (70 mg, 0.21 mmol, 1.0 equiv) in methanol (10 mL, 0.021 M), palladium (10 wt.%) on activated carbon (90 mg, 85 μmol, 0.40 equiv.) was added. The reaction mixture was degassed and flushed using a hydrogen-filled balloon. The resulting reaction was stirred for overnight at 40 °C. The reaction mixture was filtered through Celite®, washed with methanol, and filtrates were concentrated under reduced pressure to provide 46 (49 mg, 78%). The crude was pure by NMR and used for the next step without purification. Rf = 0.27 (CH2Cl2/CH3OH, 4:1); [α]25D −60 (c 0.4, CH3OH); IR (neat) νmax 3427, 2954, 2929, 2886, 2857, 1472, 1252 cm−1; Formula: C12H17N5O4; MW: 295.29 g/mol; 1H NMR (500 MHz, CD3OD): δ 8.51 (s, 1H), 8.18 (s, 1H), 6.35 (s, 1H), 4.37 (d, J = 8.7 Hz, 1H), 4.08 (ddd, J = 8.7, 3.1, 2.3 Hz, 1H), 3.99 (dd, J = 12.4, 2.3 Hz, 1H), 3.89–3.82 (m, 2H), 3.77 (d, J = 11.2 Hz, 1H), 0.67 (s, 3H) OH and NH signals are missing possibly due to exchange in CD3OD ppm; 13C NMR (126 MHz, CD3OD) δ 157.4, 153.7, 150.3, 141.6, 120.1, 90.7, 85.6, 76.1, 65.5, 61.5, 51.7, 17.3 ppm.; HRMS (ESI) m/z [M + H]+ calcd for C12H18N5O4: 296.1359; found 296.1348 (−1.8 ppm).
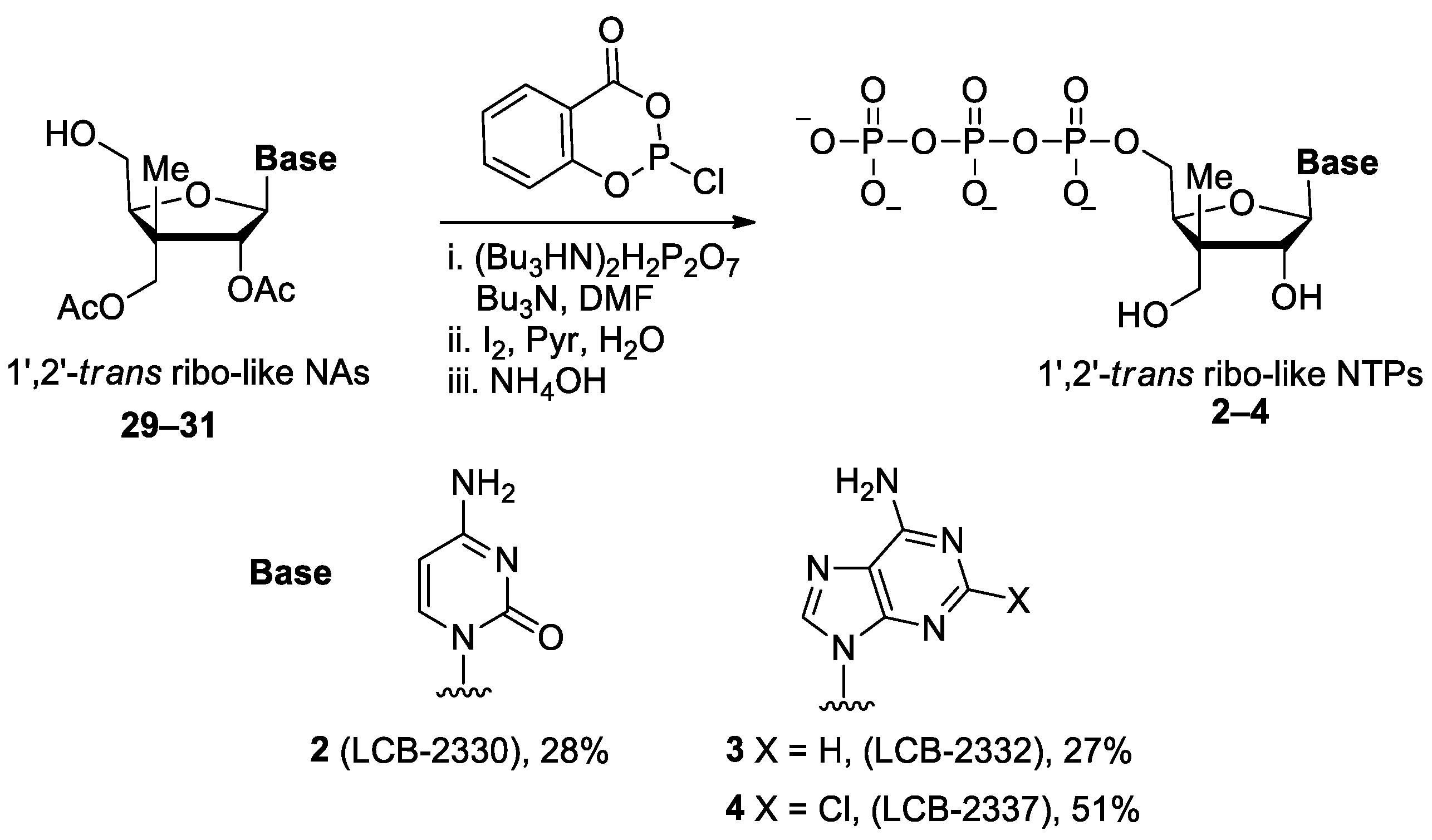
General Procedure C: Prior to the reaction, nucleoside analogue (29–31 and 45–47), salicyl phosphorochloridite (SalPCl) and tributylammonium pyrophosphate [(Bu3HN)2H2P2O7] were respectively dried under reduced pressure in 10 mL, 5 mL, 5 mL flasks for 1 h. To a solution of (Bu3HN)2H2P2O7 (1.2–2.5 equiv.) in anhydrous DMF (0.1 M), NBu3 (0.25 M) was added under nitrogen atmosphere and the mixture was stirred until (5 min) it became homogenious. The reaction mixture then was injected into a 5 mL flask containing SalPCl (1.2–2.5 equiv.) and the resulting mixture was stirred at room temperature for 30 min. The mixture was then transferred to a flask containing nucleoside 29–31 and 45–47 (1.0 equiv.) and the resulting mixture was stirred for 1.5 h. A solution of iodine (3% in Pyr: H2O 9:1 wV) was injected dropwise into the solution until a permanent brown color was persisted (~0.5 mL) and the resulting mixture was stirred for 15 min. Water (1.5 mL) was added and the solution was stirred for 1.5 h to provide the desired C5′-triphosphate which was detected by TLC (i-PrOH: NH4OH: H2O, 5:3:2). The reaction mixture was transferred into a centrifuge tube using 15 mL of EtOH. A solution of 3M NaCl was added dropwise until the reaction mixture became cloudy (~0.5 mL) and was cooled to –78 °C for 1 h. Centrifugation was conducted at 10 °C with 3200 rpm for 20 min and the resulting liquid phase was then transferred to a 50 mL Erlenmeyer flask. The resulting solid (residue) inside the centrifuge tube was air dried for 15 min. The residue was purified by reverse phase C18 flash chromatography (MeCN in 20 mM triethylammonium acetate (TEAAc) buffer, pH = 7) to provide corresponding nucleoside triphosphate triethylammonium salt, which was then lyophilized to provide pure solid nucleoside triphosphate as a white powder.
General Procedure D: If the nucleside analogues hydroxyl or amine functional groups are protected with acetyl (Ac) or benzoyl (Bz) follow the Gerenaral Procedure C until centrifiguation. Residue (solid) inside the centrifuge tube was dissolved in NH4OH (0.02 M) and was stirred for overnight. The mixture was concentrated under reduced pressure. The residue was purified by reverse phase C18 flash chromatography (MeCN in 20 mM triethylammonium acetate buffer, pH = 7) to provide corresponding nucleoside triphosphate triethylammonium salt, which was then lyophilized to provide pure solid nucleoside triphosphate as a white powder.
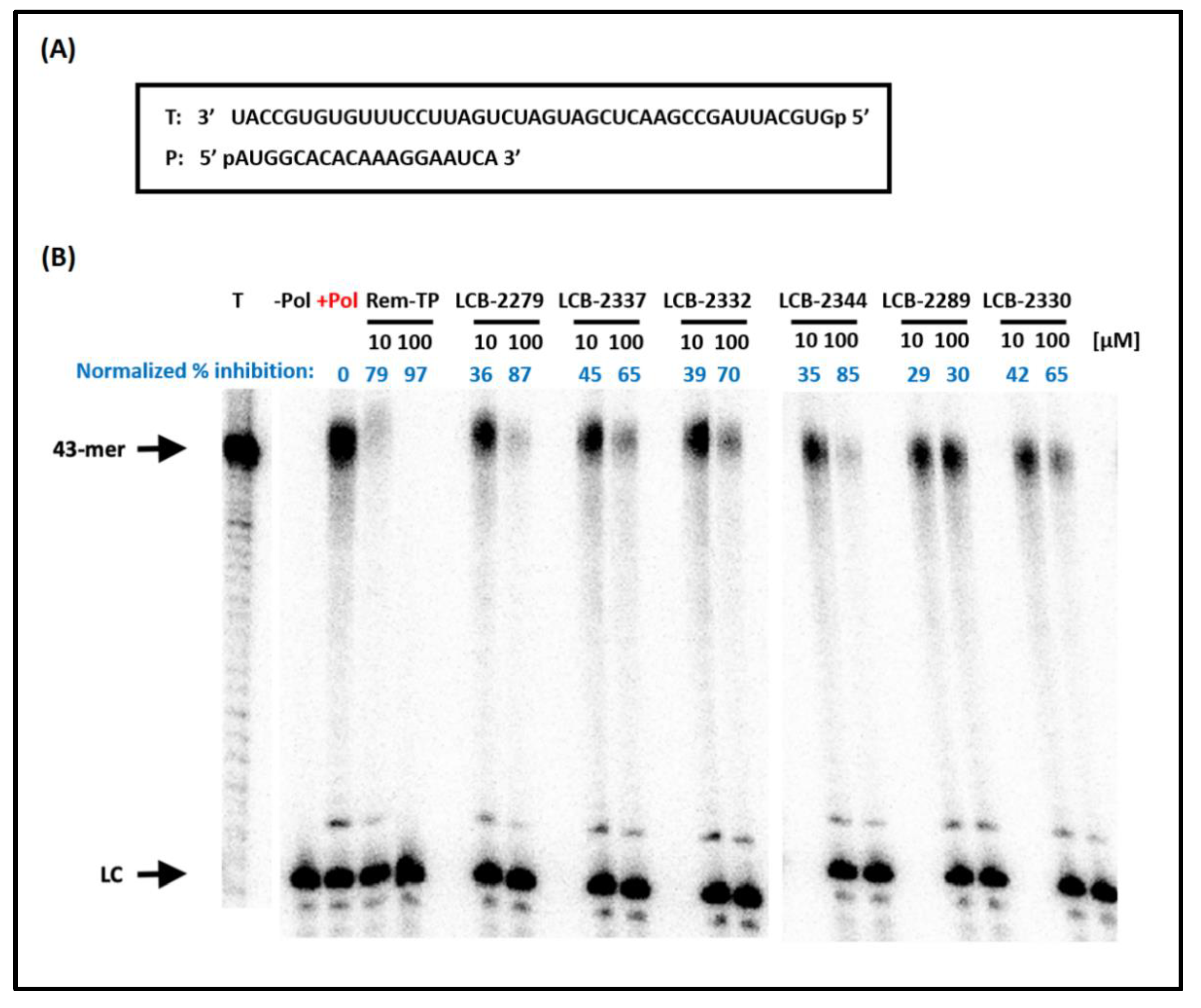
((2S,3S,4R,5R)-5-(4-Amino-2-oxopyrimidin-1(2H)-yl)-4-hydroxy-3-(hydroxymethyl)-3-methyltetrahydrofuran-2-yl)methyl tetrahydrogen triphosphate 2 (LCB-2330). Following general procedure D, (Bu3HN)2H2P2O7 (0.11 g, 0.20 mmol, 2.5 equiv.), NBu3 (0.28 mL, 0.25 M), SalPCl (40 mg, 0.20 mmol, 2.5 equiv.) and nucleoside analogue 29 (36.0 mg, 0.079 mmol, 1.00 equiv.) in anhydrous DMF (0.8 mL, 0.1 M) were employed in the phosphorylation. The final mixture was concentrated under reduced pressure. Purification by reverse phase C18 flash chromatography (flow rate of 10 mL/min., gradient run of acetonitrile from 0 to 10% in 20 mM TEAAc, pH = 7) provided nucleoside triphosphate triethylammonium salt 2 (LCB-2330) (20 mg, 28%) as a white powder. Formula: C11H20N3O14P3; MW: 511.21 g/mol; 1H NMR (500 MHz, D2O, signals for triethylammonium denoted by *) δ 8.38 (d, J = 8.0 Hz, 1H), 6.42 (d, J = 8.0 Hz, 1H), 6.11 (d, J = 6.5 Hz, 1H), 4.42–4.41 (m, 1H), 4.32 (d, J = 6.8 Hz, 1H), 4.30–4.27 (m, 1H), 4.16–4.13 (m, 1H), 3.78 (d, J = 11.6 Hz, 1H), 3.64 (d, J = 11.5 Hz, 1H), 3.22 (q, J = 7.3 Hz, 16H*), 1.29 (t, J = 7.3 Hz, 24H*), 1.17 (s, 3H) ppm; OH and NH2 signals are missing possibly due to exchange in D2O; 13C NMR (126 MHz, D2O, signals for triethylammonium denoted by *) δ 164.4, 155.9, 142.3, 88.9, 82.5 (d, J = 9.3 Hz), 80.4, 66.2, 64.5, 46.9, 46.6*, 42.2, 15.2, 8.2* ppm; 31P NMR (162 MHz, D2O) δ −10.69 (br s, 1P), −11.94 (s, 1P), −23.14 (br s, 1P) ppm; HRMS (ESI) m/z [M − H]− calcd for C11H19N3O14P3: 510.0085; found 510.0087 (+0.4 ppm).
((2S,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-4-hydroxy-3-(hydroxymethyl)-3-methyltetrahydrofuran-2-yl)methyl tetrahydrogen triphosphate 3 (LCB-2332). Following general procedure D, (Bu3HN)2H2P2O7 (0.10 g, 0.18 mmol, 2.5 equiv.), NBu3 (0.29 mL, 0.25 M), SalPCl (37 mg, 0.18 mmol, 2.5 equiv.) and nucleoside analogue 30 (35.0 mg, 0.072 mmol, 1.00 equiv.) in anhydrous DMF (0.6 mL, 0.1 M) were employed in the phosphorylation. The final mixture was concentrated under reduced pressure. Purification by reverse phase C18 flash chromatography (flow rate of 10 mL/min., gradient run of acetonitrile from 0 to 10% in 20 mM TEAAc, pH =7) provided nucleoside triphosphate 3 (LCB-2332) triethylammonium salt (18 mg, 27%) as a white powder. Formula: C12H20N5O13P3; MW: 535.24 g/mol; 1H NMR (500 MHz, D2O, signals for triethylammonium denoted by *) δ 8.66 (br s, 1H), 8.25 (s, 1H), 6.20 (d, J = 7.0 Hz, 1H), 4.76–4.73 (m, 1H), 4.46 (s, 1H), 4.25–4.14 (m, 1H), 3.85 (d, J = 11.5 Hz, 1H), 3.73 (d, J = 11.5 Hz, 1H), 3.19 (q, J = 7.3 Hz, 18H*), 1.27 (t, J = 7.3 Hz, 27H*), 1.24 (s, 3H) ppm; OH and NH2 signals are missing possibly due to exchange in D2O; 13C NMR (126 MHz, D2O, signals for triethylammonium denoted by *) δ 154.7, 151.6, 149.4, 140.5, 118.8, 86.9, 82.8 (d, J = 10.2 Hz), 80.1, 66.3 (d, J = 6.4 Hz), 64.5, 47.0, 46.5*, 15.1, 8.2* ppm; 31P NMR (162 MHz, D2O) δ –11.68 (br s, 2P), –23.60 (br s, 1P) ppm; HRMS (ESI) m/z [M − H]− calcd for C12H19N5O13P3: 534.0198; found 534.0203 (+1.0 ppm).
((2S,3S,4R,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-4-hydroxy-3-(hydroxymethyl)-3-methyltetrahydrofuran-2-yl)methyl tetrahydrogen triphosphate 4 (LCB-2337). Following general procedure D, (Bu3HN)2H2P2O7 (73.0 mg, 0.133 mmol, 2.20 equiv.), NBu3 (0.35 mL, 0.25 M), SalPCl (27.0 mg, 0.133 mmol, 2.20 equiv.) and nucleoside analogue 31 (25.0 mg, 0.06 mmol, 1.00 equiv.) in anhydrous DMF (0.5 mL, 0.1 M) were employed in the phosphorylation. The final mixture was concentrated under reduced pressure. Purification by reverse phase C18 flash chromatography (flow rate of 10 mL/min., gradient run of acetonitrile from 0 to 10% in 20 mM TEAAc, pH = 7) provided nucleoside triphosphate 4 (LCB-2337) triethylammonium salt (30 mg, 51%) as a white powder. Formula: C12H19ClN5O13P3; MW: 569.68 g/mol; 1H NMR (500 MHz, D2O, signals for triethylammonium denoted by *) δ 8.60 (s, 1H), 6.12 (d, J = 7.1 Hz, 1H), 4.75–4.70 (m, 1H), 4.46 (q, J = 3.3 Hz, 1H), 4.26–4.12 (m, 2H), 3.84 (d, J = 11.5 Hz, 1H), 3.72 (d, J = 11.5 Hz, 1H), 3.20 (q, J = 7.3 Hz, 22H*), 1.28 (t, J = 7.3 Hz, 31H*), 1.24 (s, 3H) ppm; OH and NH2 signals are missing possibly due to exchange in D2O; 13C NMR (126 MHz, D2O, signals for triethylammonium denoted by *) δ 156.3, 153.8, 150.7 (Brs), 140.2, 117.54 (Brs), 86.9, 82.9 (d, J = 10.1 Hz), 80.2, 66.23 (d, J = 6.2 Hz), 64.5, 47.1, 46.6*, 15.2, 8.2* ppm (J values result from 13C−31P coupling and were assigned when possible); 31P NMR (162 MHz, D2O) δ −6.34 (d, J = 18.7 Hz, 1P), −11.71 (d, J = 19.4 Hz, 1P), −22.58 (t, J = 19.0 Hz, 1P) ppm; HRMS (ESI) m/z [M − H]⁻ calcd for C12H18ClN5O13P3: 567.9808; found 567.9808 (−0.09 ppm).
((2R,3S,4R,5R)-5-(4-Amino-2-oxopyrimidin-1(2H)-yl)-3-hydroxy-4-(hydroxymethyl)-4-methyltetrahydrofuran-2-yl)methyl triphosphate: 5 (LCB-2289). Following general procedure C, (Bu
3HN)
2H
2P
2O
7 (162 mg, 295 μmol, 2.00 equiv.), NBu
3 (0.6 mL, 0.25 M), SalPCl (60.0 mg, 295 μmol, 2.00 equiv.) and nucleoside analogue
47 [
13] (40.0 mg, 147 μmol, 1.00 equiv.) in anhydrous DMF (1 mL, 0.1 M) were employed in the phosphorylation. The final mixture was concentrated under reduced pressure. Purification by reverse phase C18 flash chromatography (flow rate of 8 mL/min., gradient run of acetonitrile from 0 to 6% in 20 mM TEAAc, pH = 7) provided nucleoside triphosphate
5 (LCB-2289) triethylammonium salt (7 mg, 5%) as a white powder. Formula: C
11H
16N
3O
14P
3; MW: 511.21 g/mol;
1H NMR (700 MHz, D
2O, signals for triethylammonium denoted by *) δ 8.14 (d,
J = 7.8 Hz, 1H), 6.25 (d,
J = 9.5 Hz, 1H), 6.24 (brs, 1H), 4.37−4.33 (m, 1H), 4.29−4.20 (m, 3H), 3.84 (d,
J = 11.5 Hz, 1H), 3.72 (d,
J = 11.5 Hz, 1H), 3.20 (q,
J = 7.3 Hz, 24H*), 1.28 (t,
J = 7.3 Hz, 36H*), 0.88 (s, 3H) ppm, OH and NH
2 signals are missing possibly due to exchange in D
2O;
13C NMR (176 MHz, D
2O, signals for triethylammonium denoted by *) δ 163.1, 153.8, 143.4, 95.8, 89.3, 81.8 (d,
Jc-p = 8.9 Hz), 73.7, 64.0, 63.4 (d,
Jc-p = 5.0 Hz), 49.6, 46.6*, 16.0, 8.2* ppm;
31P NMR (162 MHz, D
2O) −10.88 (d,
J = 10.6 Hz, 1P), −11.34 (d,
J = 19.6 Hz, 1P), −23.24 (brs, 1P) ppm; HRMS (ESI)
m/z [M − H]⁻ calcd for C
11H
19N
3O
14P
3:510.0085; found 510.0081 (−0.87 ppm) and [M + Na − H]⁻ calcd for C
11H
18N
3NaO
14P
3: 531.9905; found 531.9900 (−0.88 ppm).
((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3-hydroxy-4-(hydroxymethyl)-4-methyltetrahydrofuran-2-yl)methyl tetrahydrogen triphosphate: 6 (LCB-2344). Following general procedure C, (Bu3HN)2H2P2O7 (51.0 mg, 0.092 mmol, 1.30 equiv.), NBu3 (0.28 mL, 0.25 M), SalPCl (17.0 mg, 0.085 mmol, 1.20 equiv.) and nucleoside analogue 46 (21.0 mg, 0.071 mmol, 1.00 equiv.) in anhydrous DMF (0.7 mL, 0.1 M) were employed in the phosphorylation. Purification by reverse phase C18 flash chromatography (flow rate of 8 mL/min., gradient run of acetonitrile from 0 to 8% in 20 mM TEAAc, pH =7) provided nucleoside triphosphate 6 (LCB-2344) triethylammonium salt (3.4 mg, 5%) as a white powder. Formula: C12H20N5O13P3; MW: 535.23 g/mol; 1H NMR (500 MHz, D2O, signals for triethylammonium denoted by *) δ 8.60 (Br s, 1H), 8.28 (s, 1H), 6.43 (s, 1H), 4.50 (Br d, J = 8.6 Hz, 1H), 4.43–4.30 (m, 3H), 3.93 (d, J = 11.9 Hz, 1H), 3.83 (d, J = 11.6 Hz, 1H), 3.21 (q, J = 7.3 Hz, 21H*), 1.29 (t, J = 7.3 Hz, 30H*), 0.72 (s, 3H) ppm; OH and NH2 signals are missing possibly due to exchange in D2O; 13C NMR (176 MHz, D2O, signals for triethylammonium denoted by *) δ 152.8, 148.7, 148.4, 118.2, 88.4, 82.11 (d, J = 7.5 Hz), 73.7, 63.9, 63.4, 50.0, 46.6*, 15.7, 8.2* ppm. One aromatic carbon signal could not be found, with the small amount of sample to isolate (J values result from 13C−31P coupling and were assigned when possible); 31P NMR (162 MHz, D2O) δ −6.43 (d, J = 21.2 Hz, 1P), −11.35 (d, J = 19.9 Hz, 1P), −22.54 (t, J = 20.6 Hz, 1P) ppm; HRMS (ESI) m/z [M − H]− calcd for C12H19N5O13P3: 534.0198; found 534.0197 (−0.08 ppm).
((2R,3S,4R,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-3-hydroxy-4-(hydroxymethyl)-4-methyltetrahydrofuran-2-yl)methyl triphosphate: 7 (LCB-2279). Following general procedure C, (Bu3HN)2H2P2O7 (100 mg, 0.182 mmol, 2.00 equiv.), NBu3 (0.36 mL, 0.25 M), SalPCl (37.0 mg, 0.182 mmol, 2.00 equiv.) and nucleoside analogue 45 (30.0 mg, 0.091 mmol, 1.00 equiv.) in anhydrous DMF (1 mL, 0.1 M) were employed in the phosphorylation. Purification by reverse phase C18 flash chromatography (flow rate of 12 mL/min., gradient run of acetonitrile from 0 to 20% in 20 mM TEAAc, pH = 7) provided nucleoside triphosphate 7 (LCB-2279) triethylammonium salt (10 mg, 11%) as a white powder. Formula: C12H19ClN5O13P3; MW: 569.68 g/mol; 1H NMR (500 MHz, D2O, signals for triethylammonium denoted by *) δ 8.52 (Br s, 1H), 6.29 (s, 1H), 4.48 (d, J = 8.4 Hz, 1H), 4.41–4.26 (m, 3H), 3.91 (d, J = 11.7 Hz, 1H), 3.82 (d, J = 11.6 Hz, 1H), 3.19 (q, J = 6.2 Hz, 20H*), 1.27 (t, J = 7.3 Hz, 30H*), 0.72 (s, 3H) ppm; OH and NH2 signals are missing possibly due to exchange in D2O; 13C NMR (176 MHz, D2O, signals for triethylammonium denoted by *) δ 156.3, 153.7, 149.7, 140.9, 117.3, 88.1, 82.1 (d, Jc-p = 8.8 Hz), 73.8, 63.9 (d, Jc-p = 5.3 Hz), 63.5, 49.9, 46.6*, 15.7, 8.2* ppm (J values result from 13C−31P coupling and were assigned when possible); 31P NMR (162 MHz, D2O) −6.42 (d, J = 21.5 Hz, 1P), −11.36 (d, J = 19.9 Hz, 1P), −22.50 (t, J = 20.6 Hz, 1P) ppm; HRMS (ESI) m/z [M − H]− calcd for C12H18ClN5O13P3: 567.9808; found 567.9819 (+1.86 ppm) and [M + Na − H] calcd for C12H17ClN5NaO13P3: 589.9627; found 589.9637 (+1.62 ppm). A minor and inseparable impurity was isolated with the compound. 31P NMR signals at −11.04 (d) and −6.49 (d) ppm suggest that this side product could be the corresponding diphosphate.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










