
A Review on Mechanistic Insight of Plant Derived Anticancer Bioactive Phytocompounds and Their Structure Activity Relationship

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Method
2.1. Search Scheme
2.2. Inclusion Criteria and Data Extraction
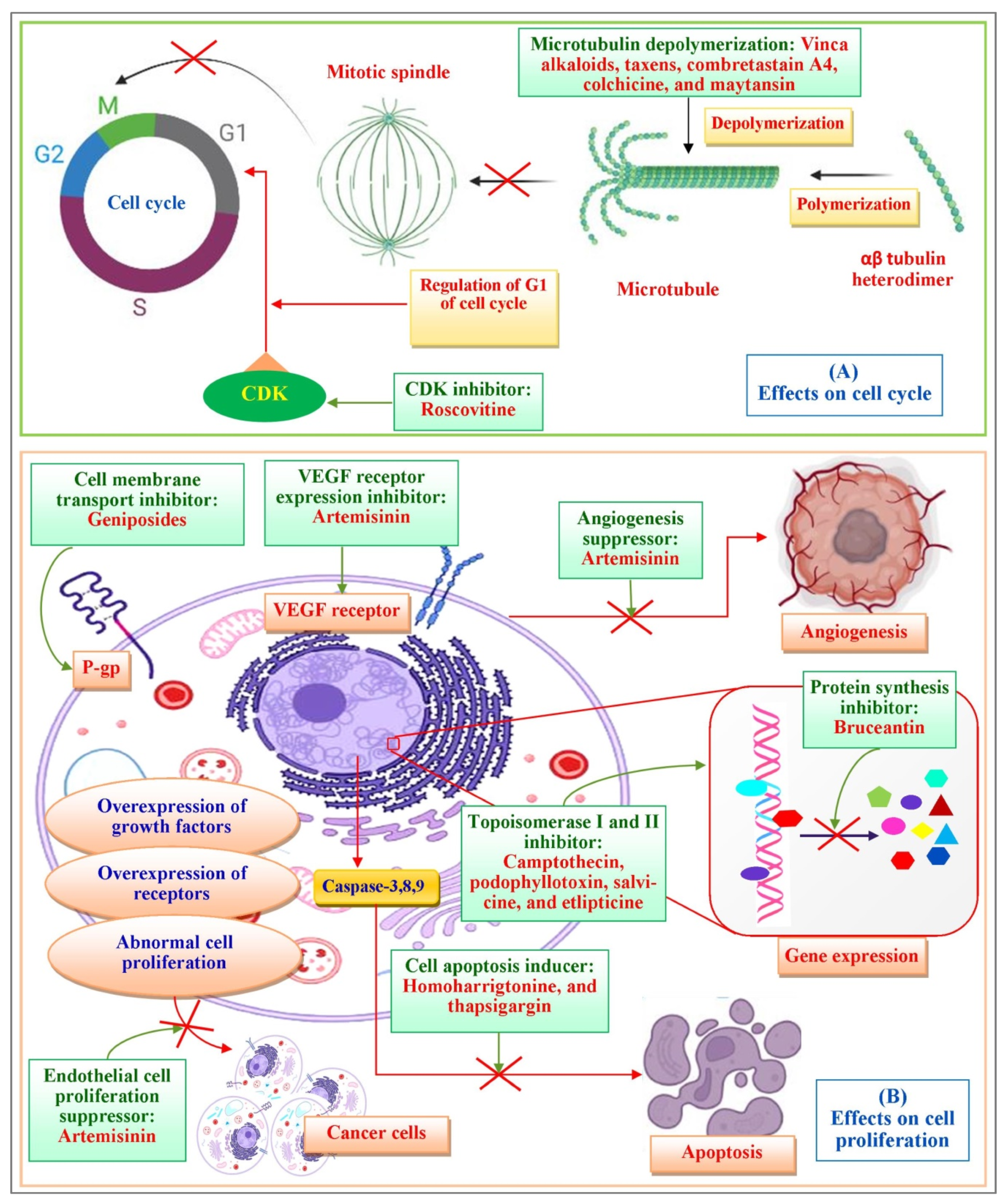
3. Plant Derivatives Compounds According to Their Chemical Structure
3.1. The Catharanthus Alkaloids
3.1.1. Relevance to Medicine
3.1.2. Viscum Album Extract
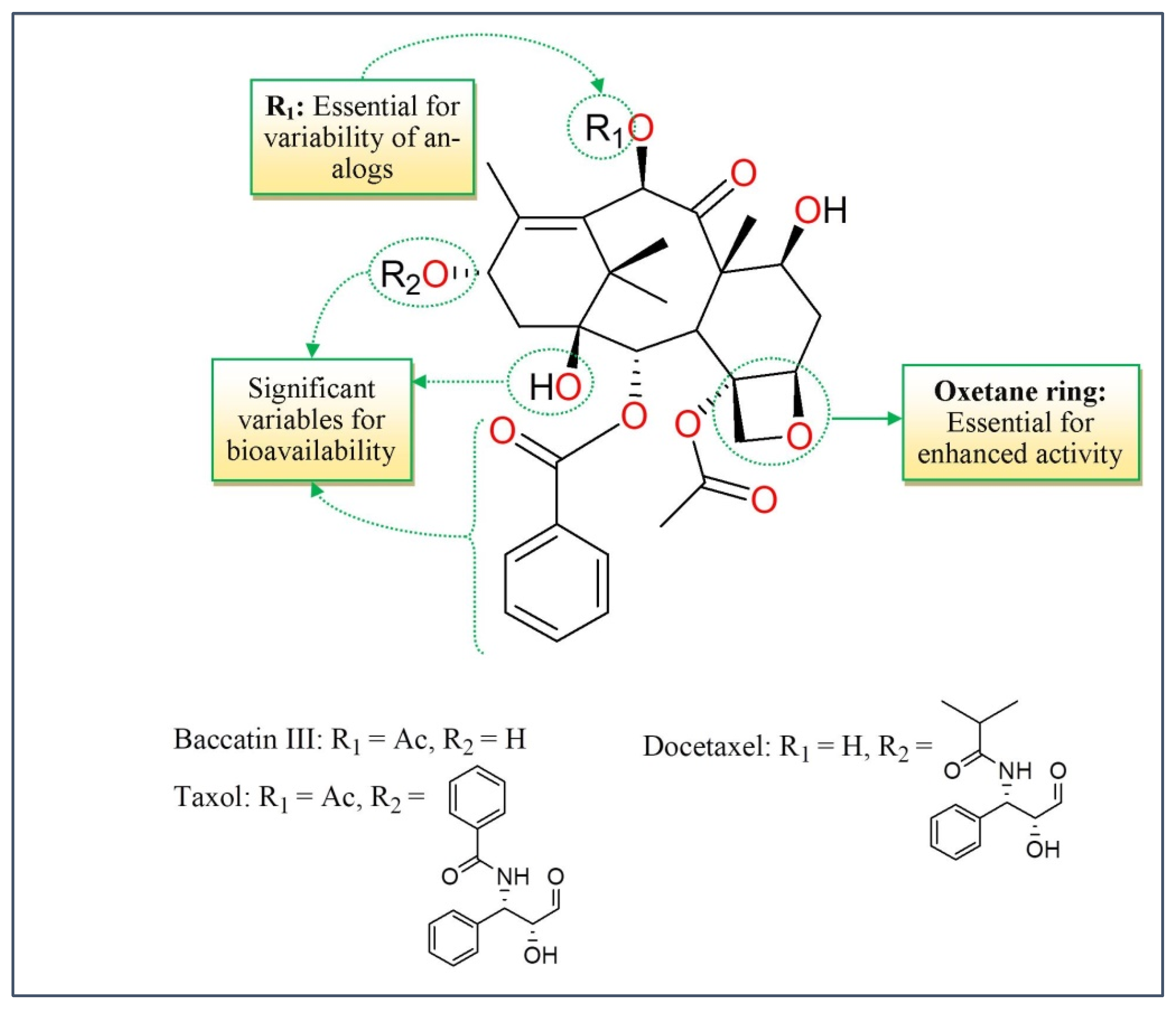
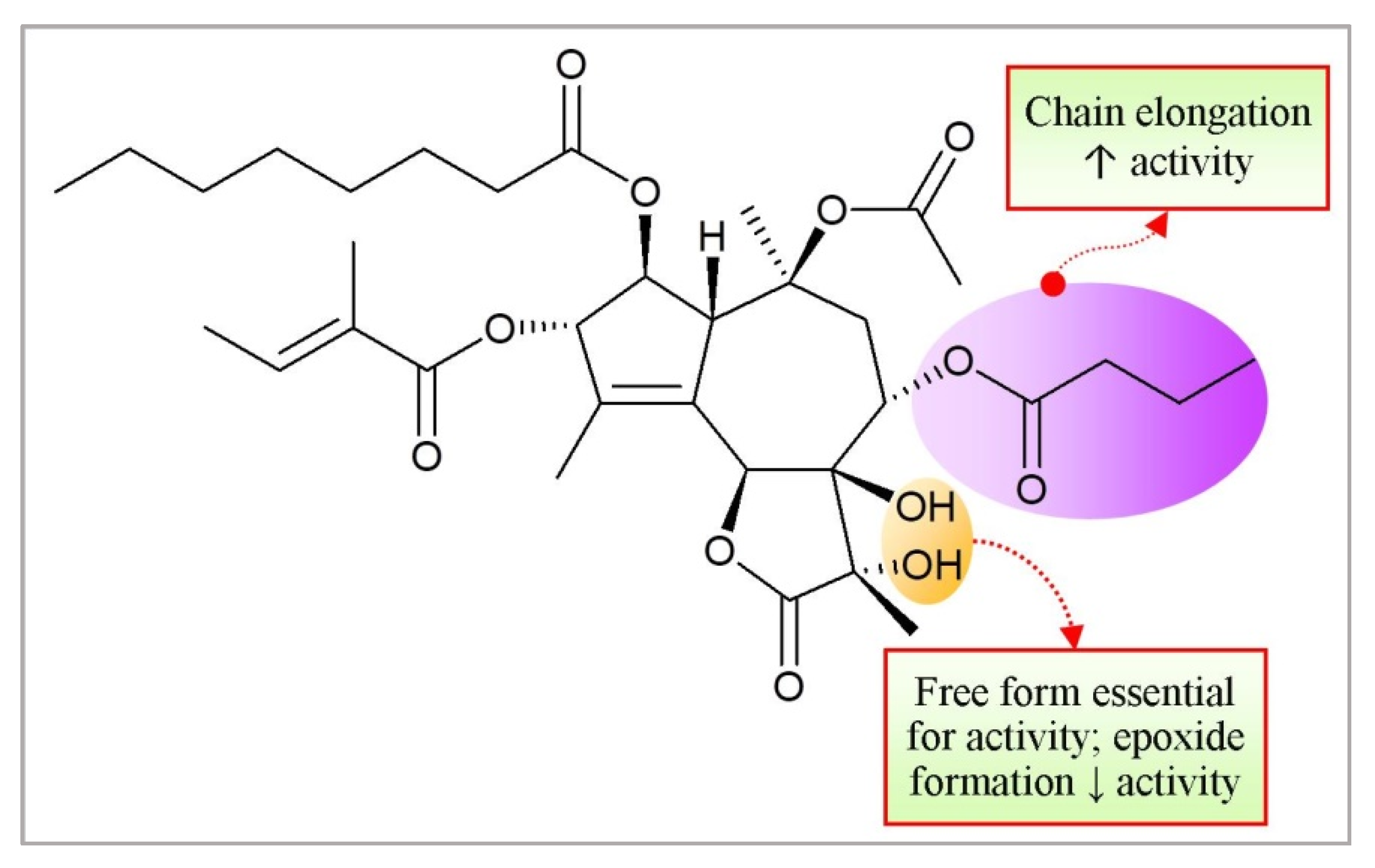
3.2. Taxanes
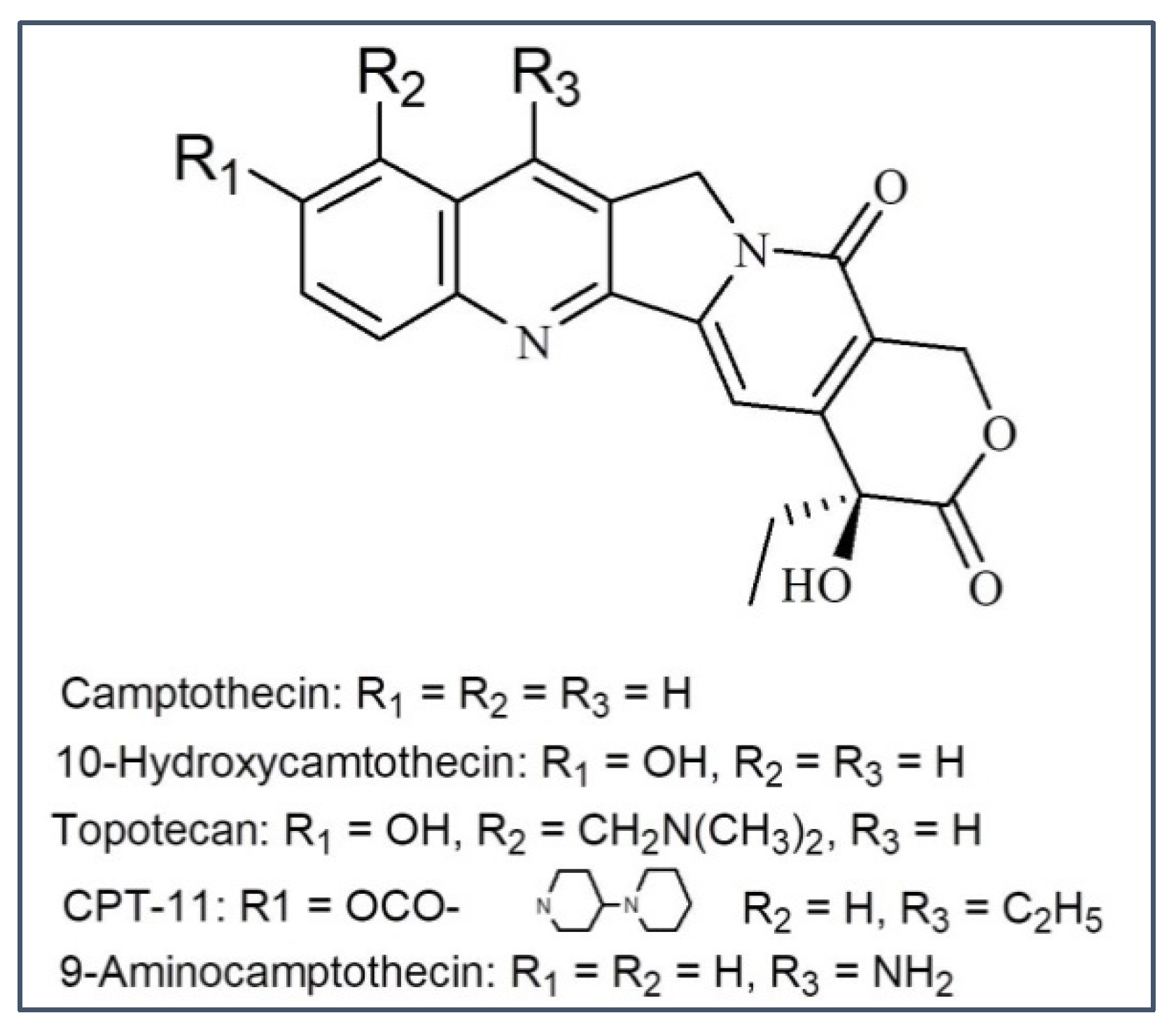
3.3. Camptothecin
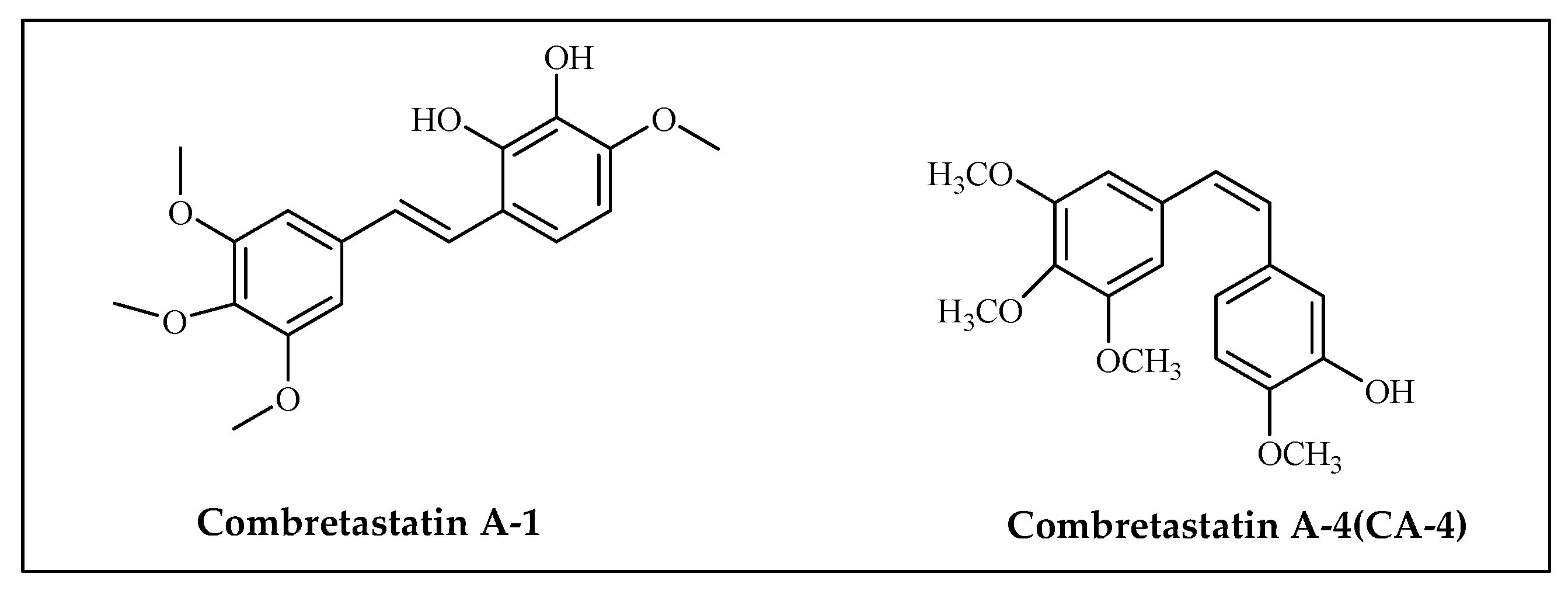
3.4. Combretastatin
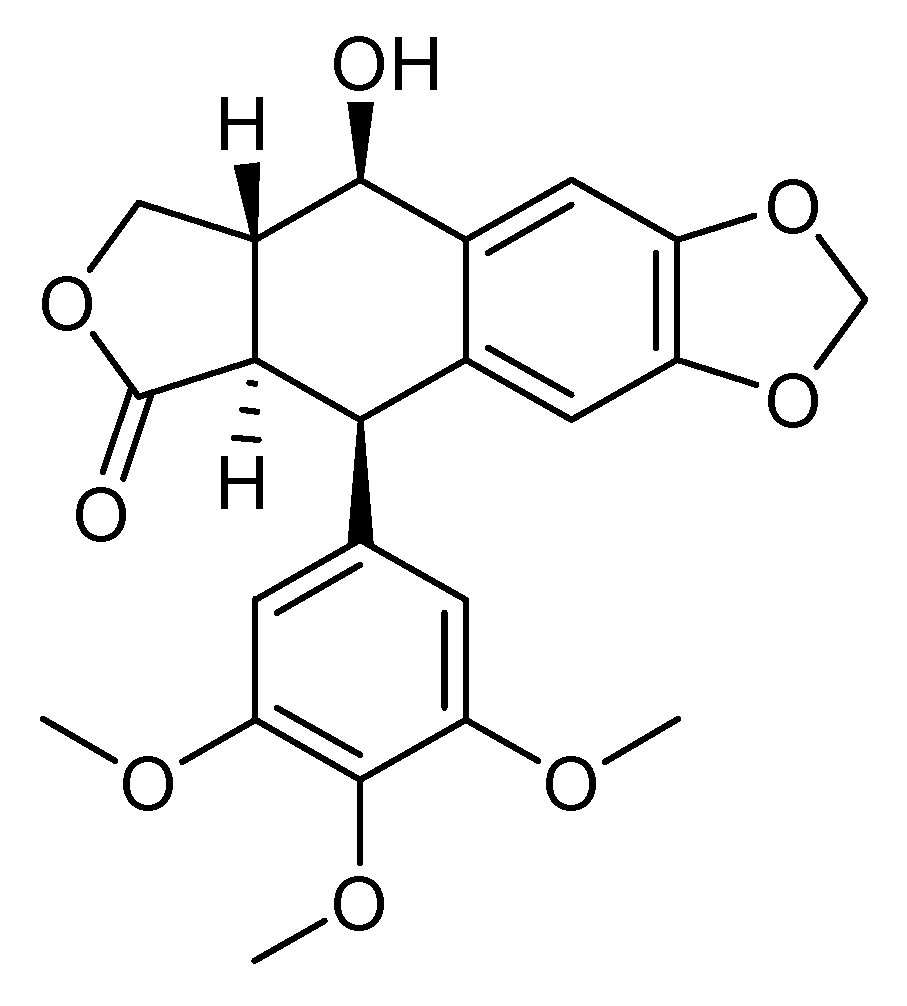
3.5. Podophyllotoxin
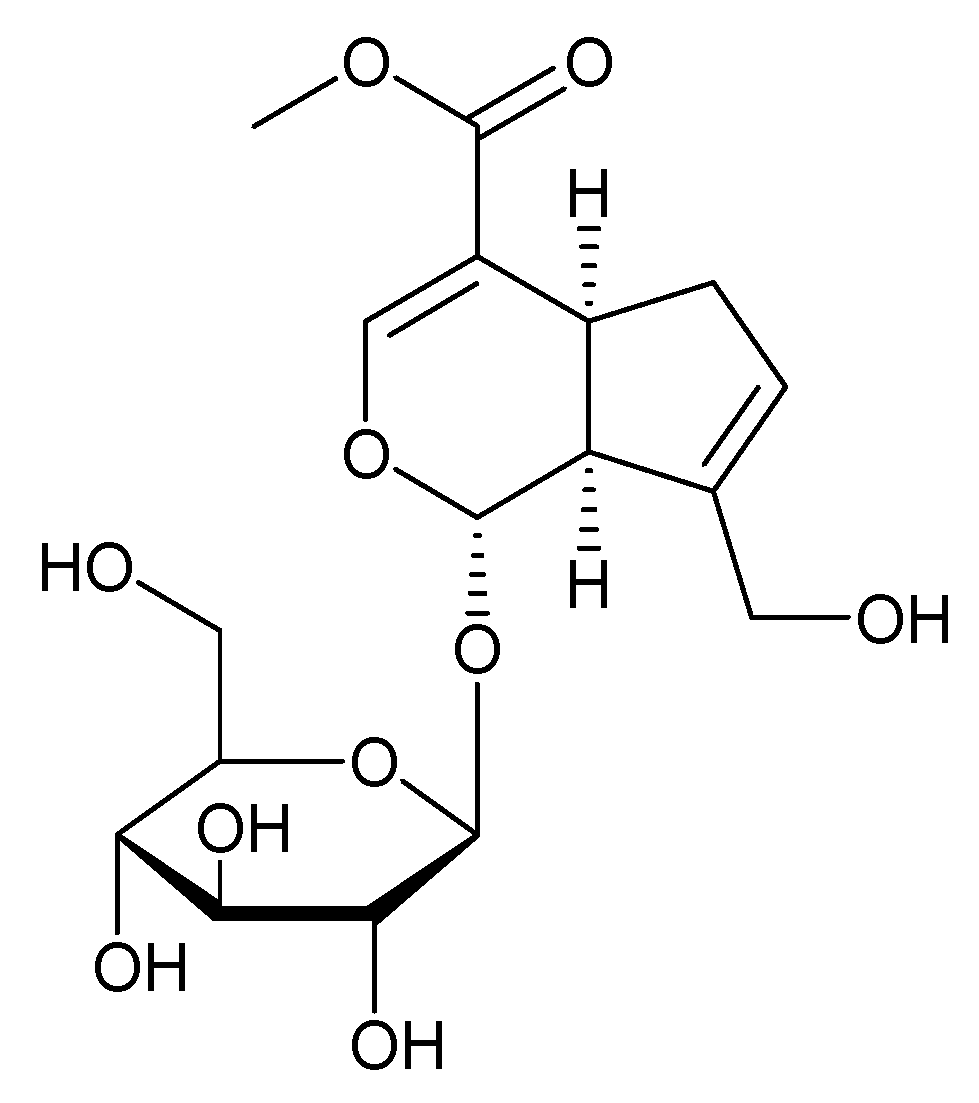
3.6. Geniposide and Their Derivatives
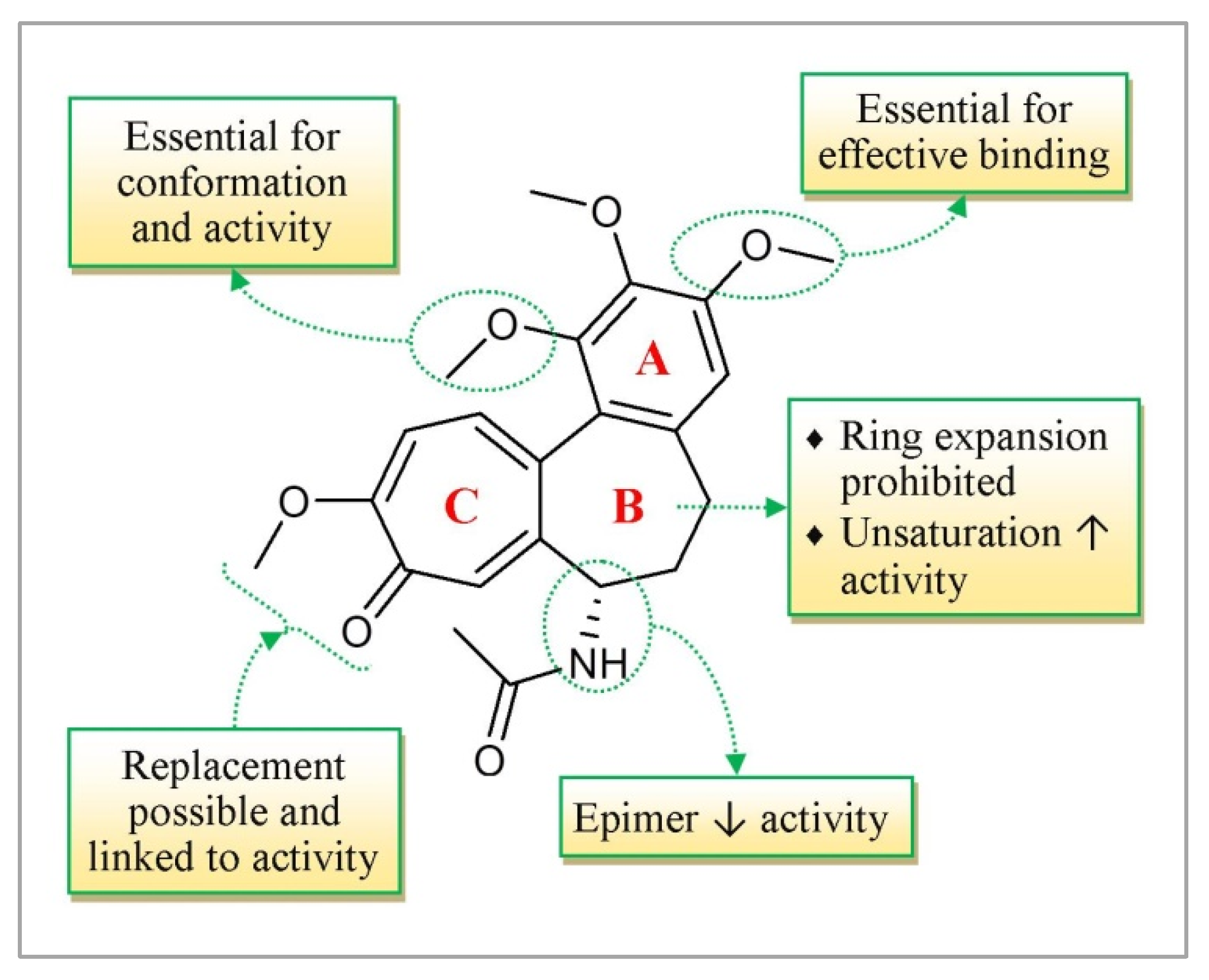
3.7. Colchicine
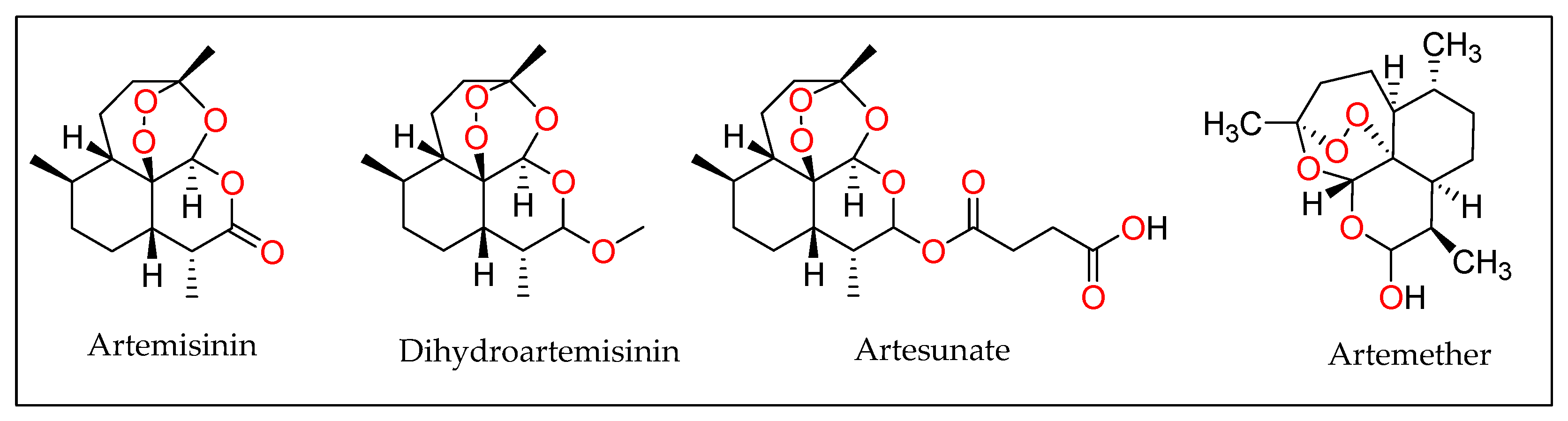
3.8. Artesunate
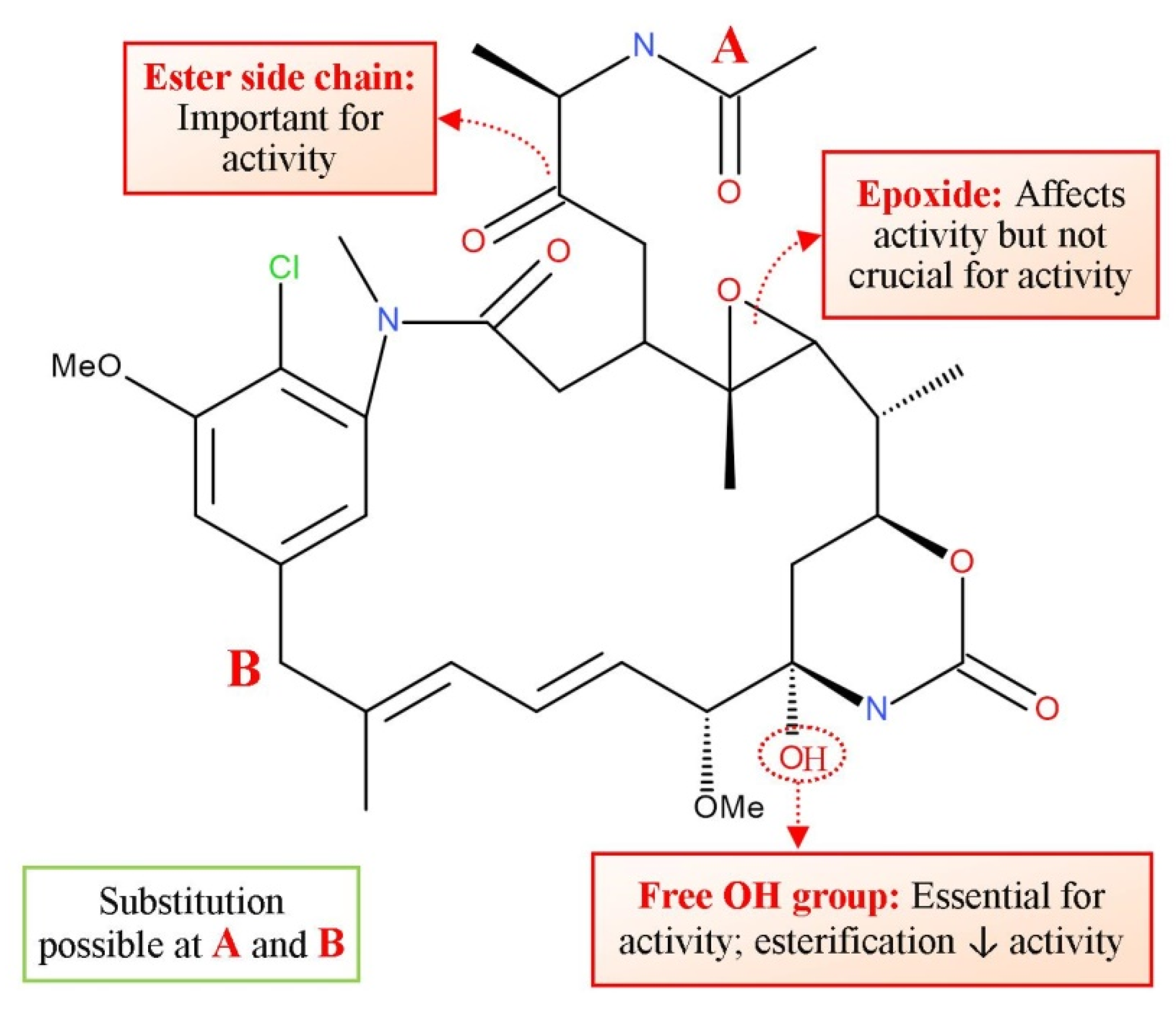
3.9. Homoharrigtonine
3.10. Salvicine
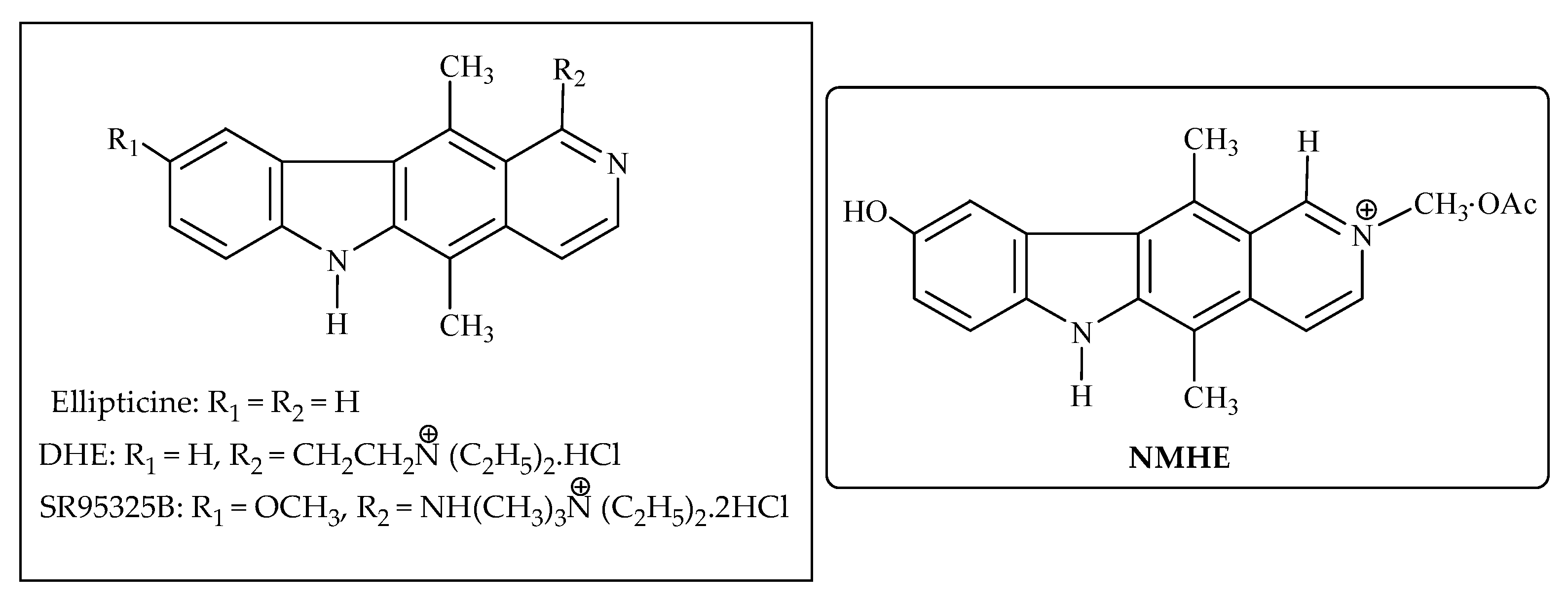
3.11. Ellipticine
3.12. Roscovitine
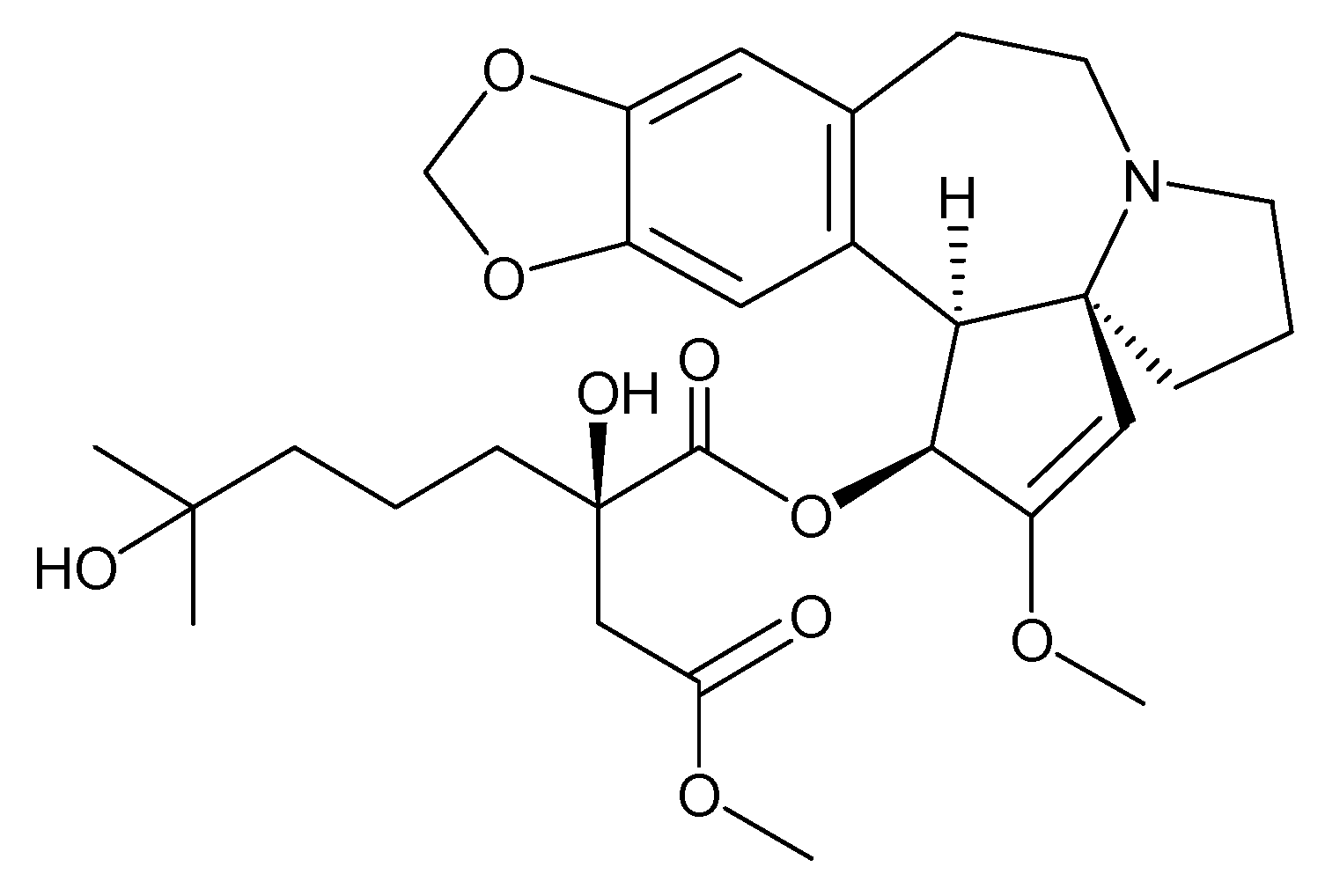
3.13. Maytansin
3.14. Thapsigargin
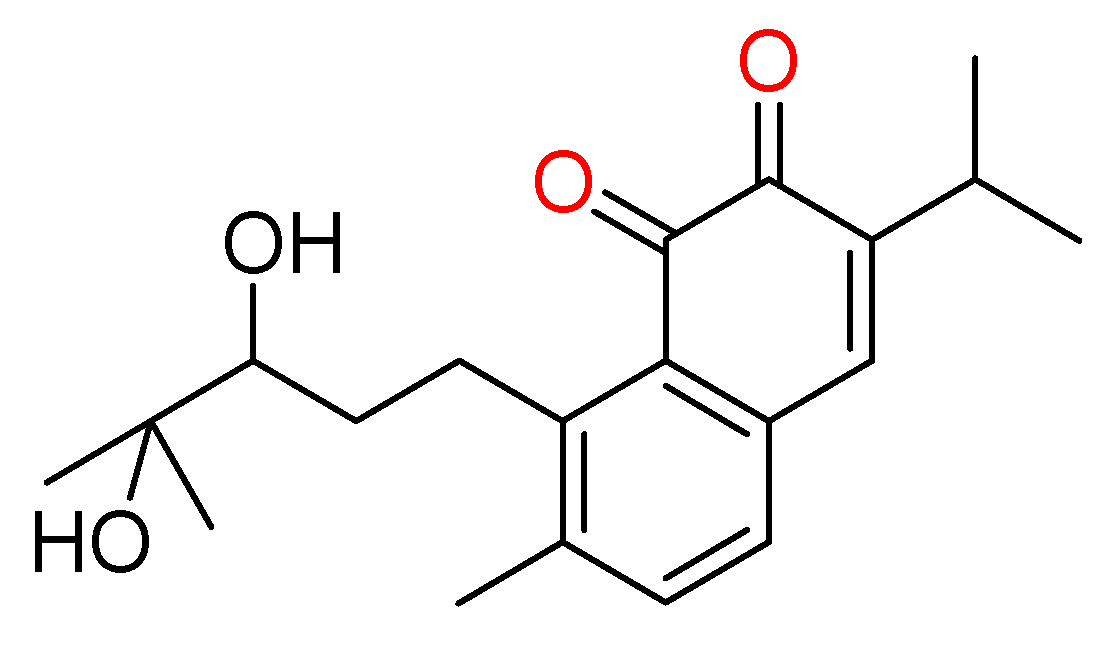
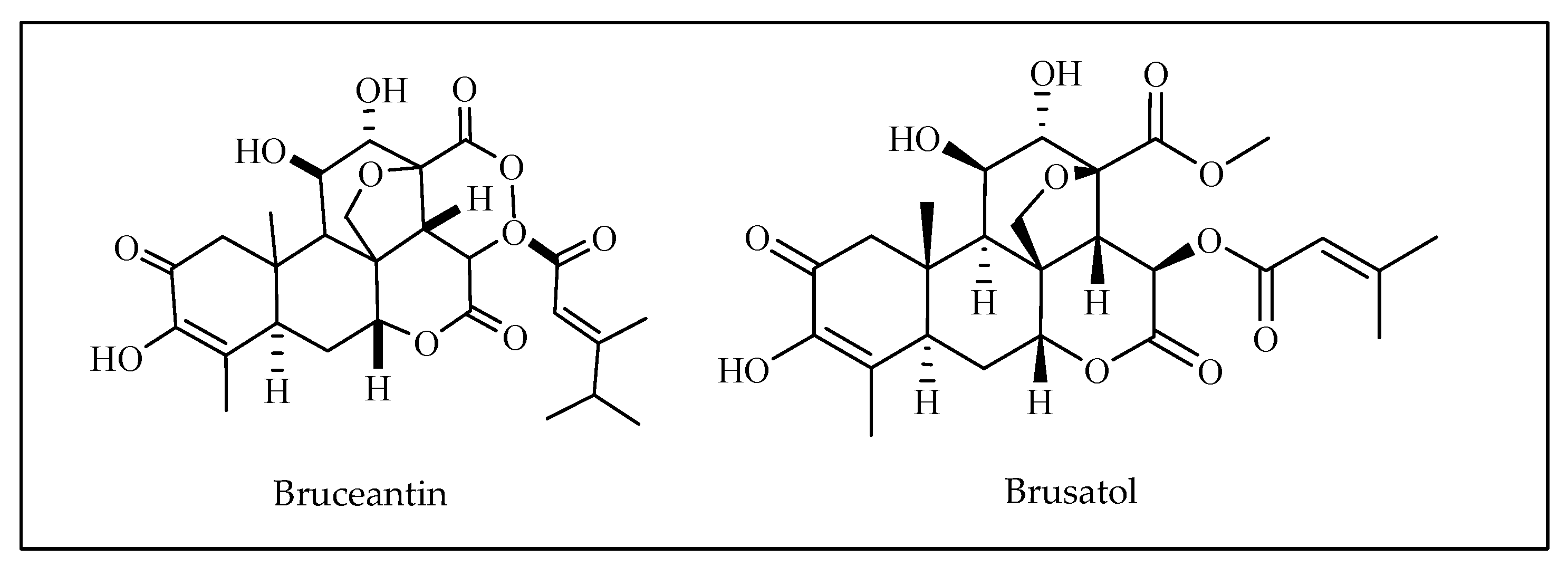
3.15. Bruceantin
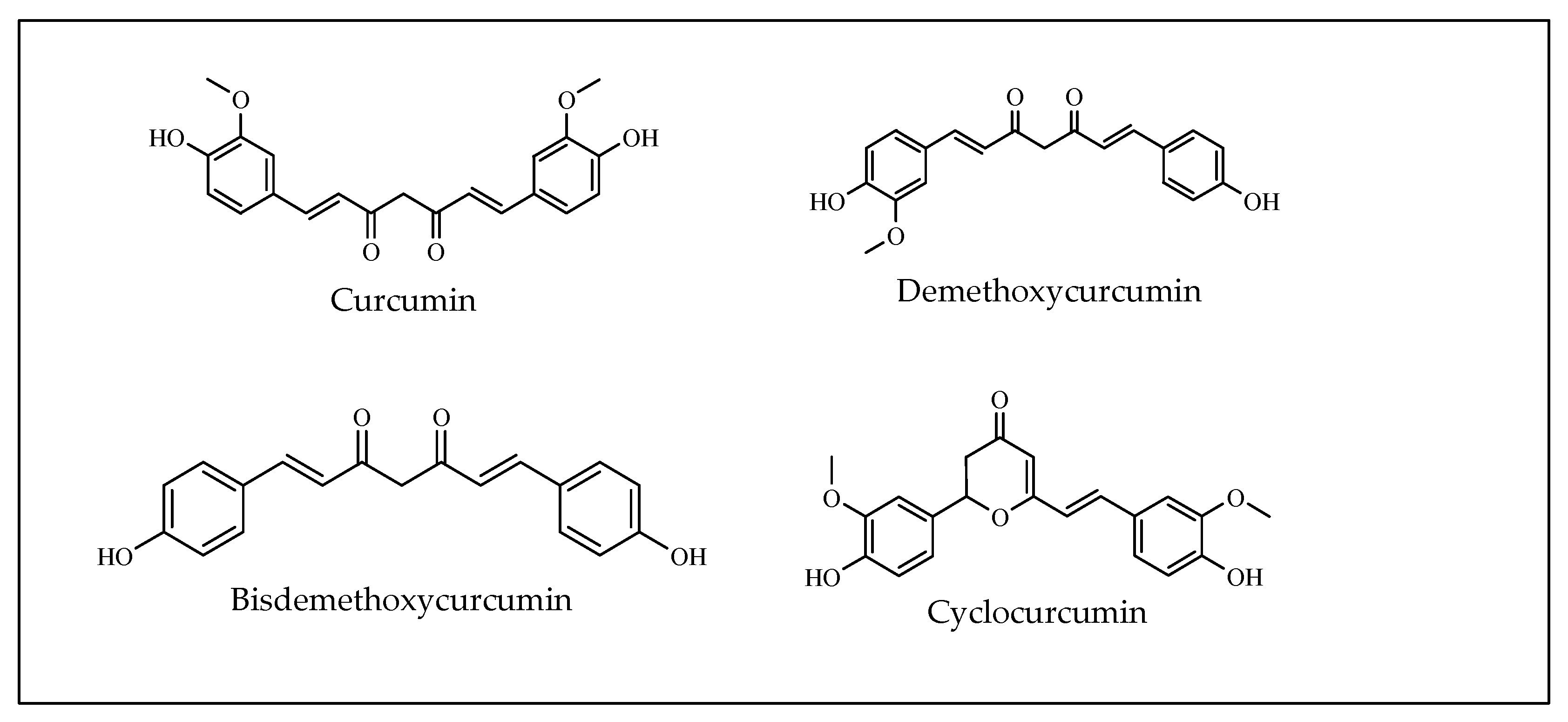
3.16. Curcumin
4. Current Drug Therapy vs. Bioactive Phytochemicals: Limitations and Challenges
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prance, G.T. Floristic inventory of the tropics: Where do we stand? Ann. Mo. Bot. Gard. 1977, 64, 659–684. [Google Scholar] [CrossRef]
- Balandrin, M.F.; Klocke, J.A.; Wurtele, E.S.; Bollinger, W.H. Natural Plant Chemicals: Sources of Industrial and Medicinal Materials. Science 1985, 228, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Ochwang’I, D.O.; Kimwele, C.N.; Oduma, J.A.; Gathumbi, P.K.; Mbaria, J.M.; Kiama, S.G. Medicinal plants used in treatment and management of cancer in Kakamega County, Kenya. J. Ethnopharmacol. 2014, 151, 1040–1055. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, K.; Biswas, B.; Raja, I.M.; Fukase, K. A Review of Cytotoxic Plants of the Indian Subcontinent and a Broad-Spectrum Analysis of Their Bioactive Compounds. Molecules 2020, 25, 1904. [Google Scholar] [CrossRef]
- Mukherjee, B. Nanotherapeutics-for-the-Treatment-of-Hepatocellular-Carcinoma. Nanother. Treat. Hepatocell. Carcinoma 2022, 29, 219–247. [Google Scholar]
- Mazumder, K.; Tanaka, K.; Fukase, K. Cytotoxic Activity of Ursolic Acid Derivatives Obtained by Isolation and Oxidative Derivatization. Molecules 2013, 18, 8929–8944. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [Green Version]
- Bailly, C.; Quetin-Leclercq, J.; Stevigny, C. Cytotoxic and Antitumor Potentialities of Aporphinoid Alkaloids. Curr. Med. Chem. Agents 2005, 5, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Butler, T.; Maravent, S.; Boisselle, J.; Valdes, J.; Fellner, C. A review of 2014 cancer drug approvals, with a look at 2015 and beyond. Pharm. Ther. 2015, 40, 191–205. [Google Scholar]
- Chekem, L.; Wierucki, S. Extraction de l’artémisinine et synthèse de ses dérivés artésunate et artéméther. Méd. Trop. 2006, 66, 602. [Google Scholar] [CrossRef]
- Khazir, J.; Mir, B.A.; Pilcher, L.; Riley, D.L. Role of plants in anticancer drug discovery. Phytochem. Lett. 2014, 7, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Bennouna, J.; Delord, J.-P.; Campone, M.; Nguyen, L. Vinflunine: A New Microtubule Inhibitor Agent. Clin. Cancer Res. 2008, 14, 1625–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ab Schutz, F.; Bellmunt, J.; Rosenberg, J.E.; Choueiri, T.K. Vinflunine: Drug safety evaluation of this novel synthetic vinca alkaloid. Expert Opin. Drug Saf. 2011, 10, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Himes, R.H. Interactions of the catharanthus (Vinca) alkaloids with tubulin and microtubules. Pharmacol. Ther. 1991, 51, 257–267. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting Microtubules by Natural Agents for Cancer Therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Falkson, G.; Burger, W. A Phase II Trial of Vindesine in Hepatocellular Cancer. Oncology 1995, 52, 86–87. [Google Scholar] [CrossRef]
- Wang, H.-K.; Lee, K.-H. Plant-derived anticancer agents and their analogs currently in clinical use or in clinical trials. Bot. Bull. Acad. Sin. Taipei 1997, 38, 225–236. [Google Scholar]
- Bretti, S.; Bonardi, G.M.; Celano, A.; Comandone, A.; Casadio, C.; Marchisio, U.; Forconi, G.; Manzoni, S.; Bumma, C. Non small cell lung cancer treatment with carboplatin and vindesine: A phase II study. Anticancer Res. 1992, 12, 1459–1461. [Google Scholar]
- Crawford, J. Update: Vinorelbine (navelbine) in non-small cell lung cancer. In Proceedings of the Seminars in Oncology, Paris, France, 1 August 1996; pp. 2–7. [Google Scholar]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Moudi, M.; Go, R.; Yien, C.Y.S.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231. [Google Scholar]
- Almagro, L.; Fernández-Pérez, F.; Pedreño, M.A. Indole Alkaloids from Catharanthus roseus: Bioproduction and Their Effect on Human Health. Molecules 2015, 20, 2973–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichota, A.; Gwozdzinski, K. Anticancer Activity of Natural Compounds from Plant and Marine Environment. Int. J. Mol. Sci. 2018, 19, 3533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, M.A. Mechanism of Action of Antitumor Drugs that Interact with Microtubules and Tubulin. Curr. Med. Chem. Agents 2012, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Singer, W.D.; Jordan, M.A.; Wilson, L.; Himes, R.H. Binding of vinblastine to stabilized microtubules. Mol. Pharmacol. 1989, 36, 366–370. [Google Scholar] [PubMed]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Paintrand, M.-R.; Pignot, I. Navelbine: An Ultrastructural Study of Its Effects. QJM 1983, 32, 115–124. [Google Scholar] [CrossRef]
- Meininger, V.; Binet, S.; Chaineau, E.; Fellous, A. In situ response to vinka alkaloids by microtubules in cultured post-implanted mouse embryos. Biol. Cell 1990, 68, 21–29. [Google Scholar] [CrossRef]
- Binet, S.; Chaineau, E.; Fellous, A.; Lataste, H.; Krikorian, A.; Couzinier, J.-P.; Meininger, V. Immunofluorescence study of the action of navelbine, vincristine and vinblastine on mitotic and axonal microtubules. Int. J. Cancer 1990, 46, 262–266. [Google Scholar] [CrossRef]
- Johnson, I.S.; Wright, H.F.; Svoboda, G.H.; Vlantis, J. Antitumor principles derived from Vinca rosea Linn, I. Vincaleukoblastine and leurosine. Cancer Res. 1960, 20, 1016–1022. [Google Scholar]
- Silvestri, R. New Prospects for Vinblastine Analogues as Anticancer Agents. J. Med. Chem. 2013, 56, 625–627. [Google Scholar] [CrossRef]
- World Health Organization. Meeting Report: WHO Technical Consultation: Nutrition-Related Health Products and the World Health Organization Model List of Essential Medicines–Practical Considerations And Feasibility: Geneva, Switzerland, 20–21 September 2018; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Maswadeh, H.; Demetzos, C.; Daliani, I.; Kyrikou, I.; Mavromoustakos, T.; Tsortos, A.; Nounesis, G. A molecular basis explanation of the dynamic and thermal effects of vinblastine sulfate upon dipalmitoylphosphatidylcholine bilayer membranes. Biochim. Biophys. Acta Biomembr. 2002, 1567, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Kyrikou, I.; Daliani, I.; Mavromoustakos, T.; Maswadeh, H.; Demetzos, C.; Hatziantoniou, S.; Giatrellis, S.; Nounesis, G. The modulation of thermal properties of vinblastine by cholesterol in membrane bilayers. Biochim. Biophys. Acta Biomembr. 2004, 1661, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandamudi, S.; Campbell, R.B. The drug loading, cytotoxicty and tumor vascular targeting characteristics of magnetite in magnetic drug targeting. Biomaterials 2007, 28, 4673–4683. [Google Scholar] [CrossRef] [PubMed]
- Dyke, R.; Nelson, R.; Brade, W. Vindesine a short review of preclinical and first clinical data. Cancer Chemother. Pharmacol. 1979, 2, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K.; Donehower, R.C. The clinical pharmacology and use of antimicrotubule agents in cancer chemotherapeutics. Pharmacol. Ther. 1991, 52, 35–84. [Google Scholar] [CrossRef]
- Bonfil, R.; Russo, D.M.; Binda, M.; Delgado, F.M.; Vincenti, M. Higher antitumor activity of vinflunine than vinorelbine against an orthotopic murine model of transitional cell carcinoma of the bladder. Urol. Oncol. Semin. Orig. Investig. 2002, 7, 159–166. [Google Scholar] [CrossRef]
- Bellmunt, J.; Théodore, C.; Demkov, T.; Komyakov, B.; Sengelov, L.; Daugaard, G.; Caty, A.; Carles, J.; Jagiello-Gruszfeld, A.; Karyakin, O.; et al. Phase III Trial of Vinflunine Plus Best Supportive Care Compared with Best Supportive Care Alone After a Platinum-Containing Regimen in Patients with Advanced Transitional Cell Carcinoma of the Urothelial Tract. J. Clin. Oncol. 2009, 27, 4454–4461. [Google Scholar] [CrossRef]
- Valle, A.C.V.; de Carvalho, A.C.; Andrade, R.V. Viscum Album-Literature Review. Int. J. Sci. Res. 2021, 10, 63–71. [Google Scholar]
- Orrù, S.; Scaloni, A.; Giannattasio, M.; Urech, K.; Pucci, P.; Schaller, G. Amino acid sequence, SS bridge arrangement and distribution in plant tissues of thionins from Viscum album. Biol. Chem. 1997, 378, 989–996. [Google Scholar] [CrossRef]
- Nazaruk, J.; Orlikowski, P. Phytochemical profile and therapeutic potential of Viscum album L. Nat. Prod. Res. 2015, 30, 373–385. [Google Scholar] [CrossRef]
- Loef, M.; Walach, H. Quality of life in cancer patients treated with mistletoe: A systematic review and meta-analysis. BMC Complement. Med. Ther. 2020, 20, 227. [Google Scholar] [CrossRef] [PubMed]
- Ioana, V.S.; Socaciu, C. The biological activity of European mistletoe (Viscum album) extracts and their pharmaceutical impact. Bull. USAMV-CN 2008, 63, 217–222. [Google Scholar]
- Lim, Y.C.; Rajabalaya, R.; Lee, S.H.F.; Tennakoon, K.U.; Le, Q.-V.; Idris, A.; Zulkipli, I.N.; Keasberry, N.; David, S.R. Parasitic Mistletoes of the Genera Scurrula and Viscum: From Bench to Bedside. Molecules 2016, 21, 1048. [Google Scholar] [CrossRef] [PubMed]
- Kienle, G.S.; Glockmann, A.; Schink, M.; Kiene, H. Viscum album L. extracts in breast and gynaecological cancers: A systematic review of clinical and preclinical research. J. Exp. Clin. Cancer Res. 2009, 28, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urech, K.; Schaller, G.; Ziska, P.; Giannattasio, M. Comparative study on the cytotoxic effect of viscotoxin and mistletoe lectin on tumour cells in culture. Phytother. Res. 1995, 9, 49–55. [Google Scholar] [CrossRef]
- Wrotek, S.; Skawiński, R.; Kozak, W. Immunostimulatory properties of mistletoe extracts and their application in oncology. Postepy Hig. Med. Dosw. 2014, 68, 1216–1224. [Google Scholar] [CrossRef]
- Stein, G.M.; Schaller, G.; Pfüller, U.; Schietzel, M.; Büssing, A. Thionins from Viscum album L: Influence of the viscotoxins on the activation of granulocytes. Anticancer Res. 1999, 19, 1037–1042. [Google Scholar]
- Tabiasco, J.; Pont, F.; Fournie, J.-J.; Vercellone, A. Mistletoe viscotoxins increase natural killer cell-mediated cytotoxicity. J. Biol. Inorg. Chem. 2002, 269, 2591–2600. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Holton, R.A.; Somoza, C.; Kim, H.B.; Liang, F.; Biediger, R.J.; Boatman, P.D.; Shindo, M.; Smith, C.C.; Kim, S. First total synthesis of taxol. 1. Functionalization of the B ring. J. Am. Chem. Soc. 1994, 116, 1597–1598. [Google Scholar] [CrossRef]
- Isah, T. Natural Sources of Taxol. Br. J. Pharm. Res. 2015, 6, 214–227. [Google Scholar] [CrossRef]
- Mody, M.D.; Gill, H.S.; Saba, N.F. The evolving and future role of taxanes in squamous cell carcinomas of the head and neck: A review. JAMA Otolaryngol. Head Neck Surg. 2016, 142, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.E.; Wani, M.C.; Cook, C.; Palmer, K.H.; McPhail, A.A.; Sim, G. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminata1, 2. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar] [CrossRef]
- Patankar, N.; Waterhouse, D. Nano-particulate drug delivery systems for camptothecins. Cancer Ther. 2012, 8, 90–104. [Google Scholar]
- Hertzberg, R.P.; Caranfa, M.J.; Hecht, S.M. On the mechanism of topoisomerase I inhibition by camptothecin: Evidence for binding to an enzyme-DNA complex. Biochemistry 1989, 28, 4629–4638. [Google Scholar] [CrossRef]
- Hsiang, Y.H.; Lihou, M.G.; Liu, L. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989, 49, 4629–4638. [Google Scholar]
- Jaxel, C.; Kohn, K.W.; Wani, M.C.; Wall, M.E.; Pommier, Y. Structure-activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: Evidence for a specific receptor site and a relation to antitumor activity. Cancer Res. 1989, 49, 1465–1469. [Google Scholar]
- Kingsbury, W.D.; Boehm, J.C.; Jakas, D.R.; Holden, K.G.; Hecht, S.M.; Gallagher, G.; Caranfa, M.J.; McCabe, F.L.; Faucette, L.F.; Johnson, R.K.; et al. Synthesis of water-soluble (aminoalkyl)camptothecin analogs: Inhibition of topoisomerase I and antitumor activity. J. Med. Chem. 1991, 34, 98–107. [Google Scholar] [CrossRef]
- Wang, H.-K.; Liu, S.-Y.; Taylor, G.; Lee, K.-H. Synthesis of novel water-soluble 7-(aminoacylhydrazono)-formyl camptothecins with potent inhibition of DNA topoisomerase I. Bioorg. Med. Chem. 1994, 2, 1397–1402. [Google Scholar] [CrossRef]
- Wang, H.-K.; Lin, S.-Y.; McPhail, A.T.; Lee, K.-H. The synthesis of 5-substituted camptothecins as potential inhibitors of DNA topoisomerase I. Bioorg. Med. Chem. Lett. 1995, 5, 77–82. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Wahl, M.L.; Flowers, J.L.; Sen, B.; Colvin, M.; Dewhirst, M.W.; Manikumar, G.; Wani, M.C. Camptothecin analogs with enhanced activity against human breast cancer cells. II. Impact of the tumor pH gradient. Cancer Chemother. Pharmacol. 2005, 57, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Zunino, F.; Dallavalle, S.; Laccabue, D.; Beretta, G.; Merlini, L.; Pratesi, G. Current Status and Perspectives in the Development of Camptothecins. Curr. Pharm. Des. 2002, 8, 2505–2520. [Google Scholar] [CrossRef] [PubMed]
- Stefański, T.; Mikstacka, R.; Kurczab, R.; Dutkiewicz, Z.; Kucińska, M.; Murias, M.; Zielińska-Przyjemska, M.; Cichocki, M.; Teubert, A.; Kaczmarek, M.; et al. Design, synthesis, and biological evaluation of novel combretastatin A-4 thio derivatives as microtubule targeting agents. Eur. J. Med. Chem. 2018, 144, 797–816. [Google Scholar] [CrossRef]
- Lawrence, N.J.; Hepworth, L.A.; Rennison, D.; McGown, A.T.; Hadfield, J.A. Synthesis and anticancer activity of fluorinated analogues of combretastatin A-4. J. Fluor. Chem. 2003, 123, 101–108. [Google Scholar] [CrossRef]
- Maya, A.B.; Pérez-Melero, C.; Mateo, C.; Alonso, D.; Fernández, J.L.; Gajate, C.; Mollinedo, F.; Peláez, R.; Caballero, E.; Medarde, M. Further Naphthylcombretastatins. An Investigation on the Role of the Naphthalene Moiety. J. Med. Chem. 2004, 48, 556–568. [Google Scholar] [CrossRef]
- Pettit, G.R.; Rhodes, M.R.; Herald, D.L.; Hamel, E.; Schmidt, J.M.; Pettit, R.K. Antineoplastic agents. 445. Synthesis and evaluation of structural modifications of (Z)-and (E)-combretastatin A-4. J. Med. Chem. 2005, 48, 4087–4099. [Google Scholar] [CrossRef]
- Gaukroger, K.; Hadfield, J.A.; Lawrence, N.J.; Nolan, S.; McGown, A.T. Structural requirements for the interaction of combretastatins with tubulin: How important is the trimethoxy unit? Org. Biomol. Chem. 2003, 1, 3033–3037. [Google Scholar] [CrossRef]
- Hang, J.-Y.; Yang, M.-F.; Chang, C.-Y.; Chen, C.-M.; Kuo, C.-C.; Liou, J.-P. 2-Amino and 2‘-Aminocombretastatin Derivatives as Potent Antimitotic Agents. J. Med. Chem. 2006, 49, 6412–6415. [Google Scholar] [CrossRef]
- Canel, C.; Moraes, R.M.; Dayan, F.E.; Ferreira, D. Podophyllotoxin. Phytochemistry 2000, 54, 115–120. [Google Scholar] [CrossRef]
- Hande, K.R. Topoisomerase II inhibitors. Update Cancer Ther. 2008, 3, 13–26. [Google Scholar] [CrossRef]
- Rao, R.D.; Krishnan, S.; Fitch, T.R.; Schomberg, P.J.; DiNapoli, R.P.; Nordstrom, K.; Scheithauer, B.; O’Fallon, J.R.; Maurer, M.J.; Buckner, J.C. Phase II trial of carmustine, cisplatin, and oral etoposide chemotherapy before radiotherapy for grade 3 astrocytoma (anaplastic astrocytoma): Results of North Central Cancer Treatment Group trial 98-72-51. Int. J. Radiat. Oncol. 2005, 61, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, H.J.; Lee, J.W.; Park, J.D.; Park, K.D.; Shin, H.Y.; Ahn, H.S. Irinotecan, vincristine, cisplatin, cyclophosphamide, and etoposide for refractory or relapsed medulloblastoma/PNET in pediatric patients. Child’s Nerv. Syst. 2013, 29, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadi, M.; Sharifi, M.; Behmanesh, M.; Moyano, E.; Bonfill, M.; Cusido, R.M.; Palazon, J. Podophyllotoxin: Current approaches to its biotechnological production and future challenges. Eng. Life Sci. 2010, 10, 281–292. [Google Scholar] [CrossRef]
- Pan, L.; Chai, H.-B.; Kinghorn, A.D. Discovery of new anticancer agents from higher plants. Front. Biosci. 2012, 4, 142. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Yang, L.; Tian, X. Podophyllotoxin: Current Perspectives. Curr. Bioact. Compd. 2007, 3, 37–66. [Google Scholar] [CrossRef]
- Zhang, X.; Rakesh, K.; Shantharam, C.; Manukumar, H.; Asiri, A.; Marwani, H.; Qin, H.-L. Podophyllotoxin derivatives as an excellent anticancer aspirant for future chemotherapy: A key current imminent needs. Bioorg. Med. Chem. 2018, 26, 340–355. [Google Scholar] [CrossRef]
- Xiao, X.-G.; Wang, S.; Xia, S.; Zou, M.; Li, Y.; Wei, Y.; Mei, Q.; Chen, Y. Retrospective study of irinotecan/cisplatin followed by etoposide/cisplatin or the reverse sequence in extensive-stage small cell lung cancer. OncoTargets Ther. 2015, 8, 2209–2214. [Google Scholar] [CrossRef] [Green Version]
- Shan, M.; Yu, S.; Yan, H.; Guo, S.; Xiao, W.; Wang, Z.; Zhang, L.; Ding, A.; Wu, Q.; Li, S.F.Y. A Review on the Phytochemistry, Pharmacology, Pharmacokinetics and Toxicology of Geniposide, a Natural Product. Molecules 2017, 22, 1689. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Du, S.; Lu, Y.; Rao, X. [Studies on O/W partition coefficient and absorption kinetics of geniposide in fructus gardeniae extract in rat intestine]. Zhongguo Zhongyao Zazhi = China, J. Chin. Mater. Med. 2009, 34, 1840–1844. [Google Scholar]
- Li, N.; Li, L.; Wu, H.; Zhou, H. Antioxidative Property and Molecular Mechanisms Underlying Geniposide-Mediated Therapeutic Effects in Diabetes Mellitus and Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.-Y.; Wang, G.-F.; Liu, Z.-Q.; Rao, J.-J.; Lü, L.; Xu, W.; Wu, S.-G.; Zhang, J.-J. Effect of geniposide, a hypoglycemic glucoside, on hepatic regulating enzymes in diabetic mice induced by a high-fat diet and streptozotocin. Acta Pharmacol. Sin. 2009, 30, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Li, G.; Hölscher, C.; Li, L. Neuroprotective effects of geniposide on Alzheimer’s disease pathology. Rev. Neurosci. 2015, 26, 371–383. [Google Scholar] [CrossRef]
- Zhang, A.; Chang, D.; Zhang, Z.; Li, F.; Li, W.; Wang, X.; Li, Y.; Hua, Q. In Vitro Selection of DNA Aptamers that Binds Geniposide. Molecules 2017, 22, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Zhang, X.; Jin, G.; Shi, Z.; Sun, W.; Chen, F. Geniposide reduces development of streptozotocin-induced diabetic nephropathy via regulating nuclear factor-kappa B signaling pathways. Fundam. Clin. Pharmacol. 2017, 31, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-S.; Zhang, T.; Yu, S.-C.; Ding, Y.; Zhang, L.-Y.; Qiu, C.; Jin, D. Transformation of Geniposide into Genipin by Immobilized β-Glucosidase in a Two-Phase Aqueous-Organic System. Molecules 2011, 16, 4295–4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habtemariam, S.; Lentini, G. Plant-Derived Anticancer Agents: Lessons from the Pharmacology of Geniposide and Its Aglycone, Genipin. Biomedicines 2018, 6, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yao, J.; Luo, Y.; Han, Y.; Wang, Z.; Du, L. P38 MAP kinase mediates apoptosis after genipin treatment in non-small-cell lung cancer H1299 cells via a mitochondrial apoptotic cascade. J. Pharmacol. Sci. 2013, 121, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, E.N.; Patnaik, J.; Mullins, M.R.; Lemasters, J.J. Free Tubulin Modulates Mitochondrial Membrane Potential in Cancer Cells. Cancer Res. 2010, 70, 10192–10201. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.-Y.; Wu, C.-C.; Chuang, Y.-H.; Chuang, W.-L. Anti-cancer mechanisms of clinically acceptable colchicine concentrations on hepatocellular carcinoma. Life Sci. 2013, 93, 323–328. [Google Scholar] [CrossRef]
- Zemer, D.; Revach, M.; Pras, M.; Modan, B.; Schor, S.; Sohar, E.; Gafni, J. A Controlled Trial of Colchicine in Preventing Attacks of Familial Mediterranean Fever. N. Engl. J. Med. 1974, 291, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.R.; Peeper, D.S. Metastasis mechanisms. Biochim. Biophys. Acta Rev. Cancer 2009, 1796, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.D.; Ashby, W.J.; Lewis, J.D.; Zijlstra, A. Targeting tumor cell motility to prevent metastasis. Adv. Drug Deliv. Rev. 2011, 63, 568–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugio, K.; Maruyama, M.; Tsurufuji, S.; Sharma, P.N.; Brossi, A. Separation of tubulin-binding and anti-inflammatory activity in colchicine analogs and congeners. Life Sci. 1987, 40, 35–39. [Google Scholar] [CrossRef]
- Zweig, M.H.; Chignell, C.F. Interaction of some colchicine analogs, vinblastine and podophyllotoxin with rat brain microtubule protein. Biochem. Pharmacol. 1973, 22, 2141–2150. [Google Scholar] [CrossRef]
- Tatematsu, H.; Kilkuskie, R.E.; Corrigan, A.J.; Bodner, A.J.; Lee, K.-H. Anti-Aids Agents, 3. Inhibitory Effects of Colchicine Derivatives on HIV Replication in H9 Lymphocyte Cells. J. Nat. Prod. 1991, 54, 632–637. [Google Scholar] [CrossRef]
- Tu, Y. Artemisinin-A Gift from Traditional Chinese Medicine to the World (Nobel Lecture). Angew. Chem. Int. Ed. 2016, 55, 10210–10226. [Google Scholar] [CrossRef]
- Rutteman, G.R.; Erich, S.A.; Mol, J.A.; Spee, B.; Grinwis, G.; Fleckenstein, L.; London, A.C.; Efferth, T. Safety and efficacy field study of artesunate for dogs with non-resectable tumours. Anticancer Res. 2013, 33, 1819–1827. [Google Scholar]
- Chaturvedi, D.; Goswami, A.; Saikia, P.P.; Barua, N.C.; Rao, P.G. Artemisinin and its derivatives: A novel class of anti-malarial and anti-cancer agents. Chem. Soc. Rev. 2009, 39, 435–454. [Google Scholar] [CrossRef]
- Itoh, Y.; Brossi, A.; Hamel, E.; Lin, C.M. Colchicine Models: Synthesis and Binding to Tubulin of Tertamethoxybiphenyls. Helv. Chim. Acta 1988, 71, 1199–1209. [Google Scholar] [CrossRef]
- Shi, Q.; Verdier-Pinard, P.; Brossi, A.; Hamel, E.; McPhail, A.T.; Lee, K.-H. Antitumor Agents. 172. Synthesis and Biological Evaluation of Novel Deacetamidothiocolchicin-7-ols and Ester Analogs as Antitubulin Agents. J. Med. Chem. 1997, 40, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Levy, V.; Zohar, S.; Bardin, C.; Vekhoff, A.; Chaoui, D.; Rio, B.; Legrand, O.; Sentenac, S.; Rousselot, P.; Raffoux, E.; et al. A phase I dose-finding and pharmacokinetic study of subcutaneous semisynthetic homoharringtonine (ssHHT) in patients with advanced acute myeloid leukaemia. Br. J. Cancer 2006, 95, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Fresno, M.; Jimenez, A.; Vazquez, D. Inhibition of Translation in Eukaryotic Systems by Harringtonine. J. Biol. Inorg. Chem. 1977, 72, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.T. Harringtonine, an inhibitor of initiation of protein biosynthesis. Mol. Pharmacol. 1975, 11, 511–519. [Google Scholar] [PubMed]
- Gürel, G.; Blaha, G.; Moore, P.B.; Steitz, T.A. U2504 Determines the Species Specificity of the A-Site Cleft Antibiotics: The Structures of Tiamulin, Homoharringtonine, and Bruceantin Bound to the Ribosome. J. Mol. Biol. 2009, 389, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isah, T. Anticancer alkaloids from trees: Development into drugs. Pharmacogn. Rev. 2016, 10, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.-S.; Ding, J.; Tang, Q.-M.; Li, M.; Zhao, M.; Lu, L.-J.; Chen, L.-J.; Yuan, S.-T. Synthesis and antitumour activity of novel diterpenequinone salvicine and the analogs. Bioorg. Med. Chem. Lett. 1999, 9, 2731–2736. [Google Scholar] [CrossRef]
- Meng, L.-H.; Zhang, J.-S.; Ding, J. Salvicine, a novel DNA topoisomerase II inhibitor, exerting its effects by trapping enzyme-DNA cleavage complexes. Biochem. Pharmacol. 2001, 62, 733–741. [Google Scholar] [CrossRef]
- Qing, C.; Jiang, C.; Zhang, J.-S.; Ding, J. Induction of apoptosis in human leukemia K-562 and gastric carcinoma SGC-7901 cells by salvicine, a novel anticancer compound. Anti-Cancer Drugs 2001, 12, 51–56. [Google Scholar] [CrossRef]
- Qing, C.; Zhang, J.; Ding, J. In vitro cytotoxicity of salvicine, a novel diterpenoid quinone. Zhongguo Yao Li Xue Bao = Acta Pharmacol. Sin. 1999, 20, 297–302. [Google Scholar]
- Goodwin, S.; Smith, A.; Horning, E. Alkaloids of Ochrosia elliptica Labill. J. Am. Chem. Soc. 1959, 81, 1903–1908. [Google Scholar] [CrossRef]
- Lin, Y.M.; Juichi, M.; Wu, R.Y.; Lee, K.H. Antitumor agents. LXIX. Alkaloids of Ochrosia acuminata. Planta Med. 1985, 51, 545–546. [Google Scholar] [CrossRef] [PubMed]
- Froelich-Ammon, S.J.; Patchan, M.W.; Osheroff, N.; Thompson, R.B. Topoisomerase II Binds to Ellipticine in the Absence or Presence of DNA: Characterization of enzyme∙ drug interactions by fluorescence spectroscopy. J. Biol. Chem. 1995, 270, 14998–15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, Y.-C.; Kuo, P.-L.; Hsu, Y.-L.; Cho, C.-Y.; Lin, C.-C. Ellipticine induces apoptosis through p53-dependent pathway in human hepatocellular carcinoma HepG2 cells. Life Sci. 2006, 78, 2550–2557. [Google Scholar] [CrossRef]
- Kuo, P.-L.; Hsu, Y.-L.; Chang, C.-H.; Lin, C.-C. The mechanism of ellipticine-induced apoptosis and cell cycle arrest in human breast MCF-7 cancer cells. Cancer Lett. 2005, 223, 293–301. [Google Scholar] [CrossRef]
- Rouëssé, J.; Spielmann, M.; Turpin, F.; Le Chevalier, T.; Azab, M.; Mondésir, J. Phase II study of elliptinium acetate salvage treatment of advanced breast cancer. Eur. J. Cancer 1993, 29, 856–859. [Google Scholar] [CrossRef]
- Stiborová, M.; Frei, E. Ellipticines as DNA-targeted chemotherapeutics. Curr. Med. Chem. 2014, 21, 575–591. [Google Scholar] [CrossRef]
- Rouëssé, J.; Huertas, D.; Sancho-Garnier, H.; Le Chevalier, T.; Amiel, J.; Brulé, G.; Tursz, T.; Mondésir, J. 2 N methyl 9 hydroxy-ellipticine in treatment of metastatic breast cancers (author’s transl). Bull. Cancer 1981, 68, 437–441. [Google Scholar]
- Kattan, J.; Durand, M.; Droz, J.-P.; Mahjoubi, M.; Marino, J.-P.; Azab, M. Phase I Study of Retelliptine Dihydrochloride (SR 95325 B) Using a Single Two-Hour Intravenous Infusion Schedule. Am. J. Clin. Oncol. 1994, 17, 242–245. [Google Scholar] [CrossRef]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef]
- Popowycz, F.; Fournet, G.; Schneider, C.; Bettayeb, K.; Ferandin, Y.; Lamigeon, C.; Tirado, O.M.; Mateo-Lozano, S.; Notario, V.; Colas, P.; et al. Pyrazolo[1,5-a]-1,3,5-triazine as a Purine Bioisostere: Access to Potent Cyclin-Dependent Kinase Inhibitor (R)-Roscovitine Analogue. J. Med. Chem. 2009, 52, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oumata, N.; Bettayeb, K.; Ferandin, Y.; Demange, L.; Lopez-Giral, A.; Goddard, M.-L.; Myrianthopoulos, V.; Mikros, E.; Flajolet, M.; Greengard, P.; et al. Roscovitine-Derived, Dual-Specificity Inhibitors of Cyclin-Dependent Kinases and Casein Kinases 1. J. Med. Chem. 2008, 51, 5229–5242. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Sallam, H.; Ferandin, Y.; Popowycz, F.; Fournet, G.; Hassan, M.; Echalier, A.; Bernard, P.; Endicott, J.; Joseph, B.; et al. N-&-N, a new class of cell death-inducing kinase inhibitors derived from the purine roscovitine. Mol. Cancer Ther. 2008, 7, 2713–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacCallum, D.E.; Melville, J.; Frame, S.; Watt, K.; Anderson, S.; Gianella-Borradori, A.; Lane, D.P.; Green, S.R. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II–dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005, 65, 5399–5407. [Google Scholar] [CrossRef] [Green Version]
- Yarotskyy, V.; Elmslie, K. Roscovitine, a cyclin-dependent kinase inhibitor, affects several gating mechanisms to inhibit cardiac L-type (Ca (V) 1.2) calcium channels. Br. J. Pharmacol. 2007, 152, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhang, T.; Jiang, T.; Zhang, R.; Teng, Z.-H.; Li, C.; Gu, Z.-P.; Mei, Q. Wortmannin potentiates roscovitine-induced growth inhibition in human solid tumor cells by repressing PI3K/Akt pathway. Cancer Lett. 2009, 286, 232–239. [Google Scholar] [CrossRef]
- Goodyear, S.; Sharma, M. Roscovitine regulates invasive breast cancer cell (MDA-MB231) proliferation and survival through cell cycle regulatory protein cdk5. Exp. Mol. Pathol. 2007, 82, 25–32. [Google Scholar] [CrossRef]
- Ljungman, M.; Paulsen, M.T. The cyclin-dependent kinase inhibitor roscovitine inhibits RNA synthesis and triggers nuclear accumulation of p53 that is unmodified at Ser15 and Lys382. Mol. Pharmacol. 2001, 60, 785–789. [Google Scholar]
- Corey, E.; Weigel, L.O.; Chamberlin, A.R.; Cho, H.; Hua, D.H. Total synthesis of maytansine. J. Am. Chem. Soc. 1980, 102, 6613–6615. [Google Scholar] [CrossRef]
- Bhattacharyya, B.; Wolff, J. Maytansine binding to the vinblastine sites of tubulin. FEBS Lett. 1977, 75, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Cassady, J.M.; Chan, K.K.; Floss, H.G.; Leistner, E. Recent Developments in the Maytansinoid Antitumor Agents. Chem. Pharm. Bull. 2004, 52, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.B.; Lin, C.M.; Hamel, E. Maytansine inhibits nucleotide binding at the exchangeable site of tubulin. Biochem. Biophys. Res. Commun. 1985, 128, 1239–1246. [Google Scholar] [CrossRef]
- Takahashi, M.; Iwasaki, S.; Kobayashi, H.; Okuda, S.; Murai, T.; Sato, Y. Rhizoxin binding to tubulin at the maytansine-binding site. Biochim. Biophys. Acta-Gen. Subj. 1987, 926, 215–223. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Komoda, Y.; Branfman, A.R.; Dailey, R.G., Jr.; Zimmerly, V.A. Tumor inhibitors. 96. Novel maytansinoids. Structural interrelations and requirements for antileukemic activity. J. Am. Chem. Soc. 1974, 96, 3706–3708. [Google Scholar] [CrossRef] [PubMed]
- Kirschning, A.; Harmrolfs, K.; Knobloch, T. The chemistry and biology of the maytansinoid antitumor agents. Comptes Rendus. Chim. 2008, 11, 1523–1543. [Google Scholar] [CrossRef]
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolpert-Defilippes, M.K.; Bono, V.H., Jr.; Dion, R.L.; Johns, D.G. Initial studies on maytansine-induced metaphase arrest in 11210 mürine leukemia cells. Biochem. Pharmacol. 1975, 24, 1735–1738. [Google Scholar] [CrossRef]
- Remillard, S.; Rebhun, L.I.; Howie, G.A.; Kupchan, S.M. Antimitotic Activity of the Potent Tumor Inhibitor Maytansine. Science 1975, 189, 1002–1005. [Google Scholar] [CrossRef]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor Delivery and In Vivo Processing of Disulfide-Linked and Thioether-Linked Antibody−Maytansinoid Conjugates. Bioconjug. Chem. 2009, 21, 84–92. [Google Scholar] [CrossRef]
- Lopus, M.; Oroudjev, E.; Wilson, L.; Wilhelm, S.; Widdison, W.; Chari, R.; Jordan, M.A. Maytansine and Cellular Metabolites of Antibody-Maytansinoid Conjugates Strongly Suppress Microtubule Dynamics by Binding to Microtubules. Mol. Cancer Ther. 2010, 9, 2689–2699. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Dong, Y.-H.; Wang, D.-F.; Liu, Z.-P. Tubulin Maytansine Site Binding Ligands and their Applications as MTAs and ADCs for Cancer Therapy. Curr. Med. Chem. 2020, 27, 4567–4576. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Bajaj, S.O. Promising anticancer drug thapsigargin: A perspective toward the total synthesis. Synth. Commun. 2017, 48, 1–13. [Google Scholar] [CrossRef]
- Thi Quynh Doan, N.; Brogger Christensen, S. Thapsigargin, origin, chemistry, structure-activity relationships and prodrug development. Curr. Pharm. Des. 2015, 21, 5501–5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Søhoel, H.; Jensen, A.-M.L.; Møller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Olsen, C.E.; Christensen, S.B. Natural products as starting materials for development of second-generation SERCA inhibitors targeted towards prostate cancer cells. Bioorg. Med. Chem. 2006, 14, 2810–2815. [Google Scholar] [CrossRef] [PubMed]
- Skytte, D.M.; Møller, J.V.; Liu, H.; Nielsen, H.O.; Svenningsen, L.E.; Jensen, C.M.; Olsen, C.E.; Christensen, S.B. Elucidation of the topography of the thapsigargin binding site in the sarco-endoplasmic calcium ATPase. Bioorg. Med. Chem. 2010, 18, 5634–5646. [Google Scholar] [CrossRef]
- Winther, A.-M.L.; Liu, H.; Sonntag, Y.; Olesen, C.; le Maire, M.; Soehoel, H.; Olsen, C.-E.; Christensen, S.B.; Nissen, P.; Møller, J.V. Critical Roles of Hydrophobicity and Orientation of Side Chains for Inactivation of Sarcoplasmic Reticulum Ca2+-ATPase with Thapsigargin and Thapsigargin Analogs. J. Biol. Chem. 2010, 285, 28883–28892. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; De Raeymaecker, J.; Hovgaard, J.B.; Smaardijk, S.; Vandecaetsbeek, I.; Wuytack, F.; Møller, J.V.; Eggermont, J.; De Maeyer, M.; Christensen, S.B. Structure/activity relationship of thapsigargin inhibition on the purified Golgi/secretory pathway Ca2+/Mn2+-transport ATPase (SPCA1a). J. Biol. Chem. 2017, 292, 6938–6951. [Google Scholar] [CrossRef] [Green Version]
- Canová, N.K.; Kmoníčková, E.; Martínek, J.; Zídek, Z.; Farghali, H. Thapsigargin, a selective inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases, modulates nitric oxide production and cell death of primary rat hepatocytes in culture. Cell Biol. Toxicol. 2007, 23, 337–354. [Google Scholar] [CrossRef]
- Deniaud, A.; el Dein, O.S.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2007, 27, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Wong, P.-M.; Gammoh, N.; Jiang, X. Distinct Autophagosomal-Lysosomal Fusion Mechanism Revealed by Thapsigargin-Induced Autophagy Arrest. Mol. Cell 2011, 42, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Cuendet, M.; Pezzuto, J.M. Antitumor Activity of Bruceantin: An Old Drug with New Promise. J. Nat. Prod. 2003, 67, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.-K.; Zhang, Z.-J.; Dong, S.-H.; Wang, Y.-X.; Song, S.-J.; Huang, X.-X. Quassinoids: Phytochemistry and antitumor prospect. Phytochemistry 2021, 187, 112769. [Google Scholar] [CrossRef] [PubMed]
- Crocker, K.E.; Pei, Y.; Robertson, E.S.; Winkler, J.D. Synthesis of a novel bruceantin analog via intramolecular etherification. Can. J. Chem. 2020, 98, 270–272. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vartanian, S.; Ma, T.P.; Lee, J.; Haverty, P.M.; Kirkpatrick, D.S.; Yu, K.; Stokoe, D. Application of Mass Spectrometry Profiling to Establish Brusatol as an Inhibitor of Global Protein Synthesis. Mol. Cell. Proteom. 2016, 15, 1220–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata-Greenwood, E.; Cuendet, M.; Sher, D.; Gustin, D.; Stock, W.; Pezzuto, J.M. Brusatol-mediated induction of leukemic cell differentiation and G1 arrest is associated with down-regulation of c-myc. Leukemia 2002, 16, 2275–2284. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.-T.; Kim, C.W.; Kim, H.G.; Lee, J.-S.; Park, H.J. Brusatol-Mediated Inhibition of c-Myc Increases HIF-1α Degradation and Causes Cell Death in Colorectal Cancer under Hypoxia. Theranostics 2017, 7, 3415–3431. [Google Scholar] [CrossRef]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef] [Green Version]
- Rajasekhar Reddy, A.; Dinesh, P.; Prabhakar, A.S.; Umasankar, K.; Shireesha, B.; Bhagavan Raju, M. A comprehensive review on SAR of curcumin. Mini Rev. Med. Chem. 2013, 13, 1769–1777. [Google Scholar] [CrossRef]
- Ahsan, H.; Parveen, N.; Khan, N.U.; Hadi, S. Pro-oxidant, anti-oxidant and cleavage activities on DNA of curcumin and its derivatives demethoxycurcumin and bisdemethoxycurcumin. Chem. Interact. 1999, 121, 161–175. [Google Scholar] [CrossRef]
- Bachar, S.C.; Mazumder, K.; Bachar, R.; Aktar, A.; Al Mahtab, M. A Review of Medicinal Plants with Antiviral Activity Available in Bangladesh and Mechanistic Insight into Their Bioactive Metabolites on SARS-CoV-2, HIV and HBV. Front. Pharmacol. 2021, 12, 732891. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, K.; Nabila, A.; Aktar, A.; Farahnaky, A. Bioactive Variability and In Vitro and In Vivo Antioxidant Activity of Unprocessed and Processed Flour of Nine Cultivars of Australian lupin Species: A Comprehensive Substantiation. Antioxidants 2020, 9, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alalaiwe, A.; Fang, J.-Y.; Lee, H.-J.; Chiu, C.-H.; Hsu, C.-Y. The Demethoxy Derivatives of Curcumin Exhibit Greater Differentiation Suppression in 3T3-L1 Adipocytes Than Curcumin: A Mechanistic Study of Adipogenesis and Molecular Docking. Biomolecules 2021, 11, 1025. [Google Scholar] [CrossRef] [PubMed]
- Noorafshan, A.; Ashkani-Esfahani, S. A Review of Therapeutic Effects of Curcumin. Curr. Pharm. Des. 2013, 19, 2032–2046. [Google Scholar] [CrossRef]
- Gupta, A.; Khan, S.; Manzoor, M.; Yadav, A.; Sharma, G.; Anand, R.; Gupta, S. Anticancer Curcumin: Natural Analogues and Structure-Activity Relationship. Stud. Nat. Prod. Chem. 2017, 54, 355–401. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kumar, A.; Bharti, A.C. Anticancer potential of curcumin: Preclinical and clinical studies. Anticancer Res. 2003, 23, 363–398. [Google Scholar]
- Sharma, R.A.; Ireson, C.R.; Verschoyle, R.D.; Hill, K.A.; Williams, M.L.; Leuratti, C.; Manson, M.M.; Marnett, L.J.; Steward, W.P.; Gescher, A. Effects of dietary curcumin on glutathione S-transferase and malondialdehyde-DNA adducts in rat liver and colon mucosa: Relationship with drug levels. Clin. Cancer Res. 2001, 7, 1452–1458. [Google Scholar]
- Cheng, A.L.; Hsu, C.-H.; Lin, J.K.; Hsu, M.M.; Ho, Y.-F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.-R.; Ming-Shiang, W.; et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001, 21, 2895–2900. [Google Scholar]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II Trial of Curcumin in Patients with Advanced Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef] [Green Version]
- Gediya, L.K.; Njar, V.C. Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2009, 4, 1099–1111. [Google Scholar] [CrossRef]
- Gyanani, V.; Haley, J.C.; Goswami, R. Challenges of Current Anticancer Treatment Approaches with Focus on Liposomal Drug Delivery Systems. Pharmaceuticals 2021, 14, 835. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Tumor Microenvironment and the Response to Anticancer Therapy. Cancer Biol. Ther. 2002, 1, 453–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Colombo, A.; Fedeli, L.; Pavesi, A.; Quaroni, S.; Saracchi, M.; Ventrella, G. Isolation of endophytic fungi and actinomycetes taxane producers. Ann. Microbiol. 2000, 50, 3–14. [Google Scholar]
- Aung, T.N.; Qu, Z.; Kortschak, R.D.; Adelson, D.L. Understanding the Effectiveness of Natural Compound Mixtures in Cancer through Their Molecular Mode of Action. Int. J. Mol. Sci. 2017, 18, 656. [Google Scholar] [CrossRef]
- Cao, B.; Chen, H.; Gao, Y.; Niu, C.; Zhang, Y.; Li, L. CIP-36, a novel topoisomerase II-targeting agent, induces the apoptosis of multidrug-resistant cancer cells in vitro. Int. J. Mol. Med. 2014, 35, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Oliveira, P.; Otero, P.; Pereira, A.; Chamorro, F.; Carpena, M.; Echave, J.; Fraga-Corral, M.; Simal-Gandara, J.; Prieto, M. Status and Challenges of Plant-Anticancer Compounds in Cancer Treatment. Pharmaceuticals 2021, 14, 157. [Google Scholar] [CrossRef]
- Li, Q.-Y.; Zu, Y.-G.; Shi, R.-Z.; Yao, L.-P. Review Camptothecin: Current Perspectives. Curr. Med. Chem. 2006, 13, 2021–2039. [Google Scholar] [CrossRef]
- Liu, P.; Qin, Y.; Wu, L.; Yang, S.; Li, N.; Wang, H.; Xu, H.; Sun, K.; Zhang, S.; Han, X.; et al. A phase I clinical trial assessing the safety and tolerability of combretastatin A4 phosphate injections. Anti-Cancer Drugs 2014, 25, 462–471. [Google Scholar] [CrossRef]
- Dhillon, N.; Wolff, R.A.; Abbruzzese, J.L.; Hong, D.S.; Camacho, L.H.; Li, L.; Braiteh, F.S.; Kurzrock, R. Phase II clinical trial of curcumin in patients with advanced pancreatic cancer. J. Clin. Oncol. 2006, 24, 14151. [Google Scholar] [CrossRef]
- Luo, C.Y.; Tang, J.Y.; Wang, Y.P. Homoharringtonine: A New Treatment Option for Myeloid Leukemia. Hematology 2004, 9, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Issell, B. The podophyllotoxin derivatives VP16-213 and VM26. Cancer Chemother. Pharmacol. 1982, 7, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Sparano, J.A. Taxanes for Breast Cancer: An Evidence-Based Review of Randomized Phase II and Phase III Trials. Clin. Breast Cancer 2000, 1, 32–40. [Google Scholar] [CrossRef]
- Furuse, K.; Kubota, K.; Kawahara, M.; Ogawara, M.; Kinuwaki, E.; Motomiya, M.; Nishiwaki, Y.; Niitani, H.; Sakuma, A. A Phase II study of vinorelbine, a new derivative of vinca alkaloid, for previously untreated advanced non-small cell lung cancer. Lung Cancer 1994, 11, 385–391. [Google Scholar] [CrossRef]
- Ghosh, S.; Dutta, S.; Sarkar, A.; Kundu, M.; Sil, P.C. Targeted delivery of curcumin in breast cancer cells via hyaluronic acid modified mesoporous silica nanoparticle to enhance anticancer efficiency. Colloids Surf. B Biointerfaces 2021, 197, 111404. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VAs | Cancers | Common Side Effects |
|---|---|---|
| VBL | Leukemia, non-Hodgkin’s and Hodgkin’s lymphoma, breast cancers, nephroblastoma, Ewing’s sarcoma, small cell lung cancer, testicular carcinoma, and germ cell tumors | Toxicity to white blood cells, nausea, vomiting, constipation, dyspnea, chest or tumor pain, wheezing, fever, antidiuretic hormone secretion, and weight loss |
| VCR | Philadelphia chromosome-negative acute lymphoblastic leukemia, B-cell lymphoma, metastatic melanoma, breast cancer, glioma, colorectal cancer, non-Hodgkin’s and Hodgkin’s lymphoma, neuroblastoma, rhabdomyosarcoma, multipole myeloma, and Wilm’s tumor | Important peripheral neuropathy, nausea, vomiting, diarrhea, bloating, stomach/abdominal cramps, mouth sore, dizziness, headache, hair loss, constipation, loss of appetite, and weight loss |
| VDS | Pediatric solid tumors, malignant melanoma, blast crisis of chronic myeloid leukemia, acute lymphocytic leukemia, metastatic colorectal cancer, breast cancer, and renal and esophageal carcinomas | Leukopenia, thrombocytopenia, fatigue, constipation, sore mouth, difficulty swallowing paralytic ileus, loss of sensation, nerve pain, diarrhea, convulsions, depression, and weight loss |
| VRL | The wide antitumor spectrum of activity, such as advanced breast cancer, advanced metastatic non-small cell lung cancer, and rhabdomyosarcoma | Neuropathy, nausea or vomiting, muscle weakness, constipation, diarrhea, anemia, and weight loss |
| VFN | Metastatic and advanced urothelial cancer after failure of platin containing therapy | Neuropathy, nausea or vomiting, muscle weakness, constipation, abdominal pain, vomiting or nausea, stomatitis, diarrhea, alopecia myalgia, fatigue, and weight loss |
| Class of Phytochemicals | Pharmacological Action | Clinical Trial | Type of Cancer | Molecular Targets | Ref. |
|---|---|---|---|---|---|
| Camptothecin Irinotecan Topotecan | Stabilizes topoisomerase I-DNA complex thereby preventing re-ligation of single strand breaks resulting in lethal double-stranded breaks in DNA. | Prospective phase I clinical trial | Ovarian, cervical, colorectal, and small cell lung cancer (SCLC) | Topoisomerase I | [180] |
| Combretastatin A4 | Inhibits polymerization of tubulin causing disruption of the tumor endothelial cells lining the tumor vasculature | Prospective phase I clinical trial | Polypoidal choroidal vasculopathy, anaplastic thyroid cancers | Tubulin | [181] |
| Curcumin | Multiple actions on mutagenesis, cell cycle regulation, apoptosis, oncogene expression and metastasis | Prospective phase II clinical trial | Patients with advanced pancreatic cancer, urinary bladder cancer, uterine cervical neoplasm, or intestinal metaplasia | - | [182] |
| Homoharringtonine | Binds to large ribosomal subunit, which affects chain elongation and prevents protein synthesis | Prospective phase I/II clinical trial | Chronic myeloid leukemia | Ribosomoal protein | [183] |
| Podophyllotoxin Etoposide Teniposide | Inhibits DNA synthesis by forming a complex with topoisomerase II and DNA. | Prospective phase I clinical trial | Osteosarcoma, NSCLC cervical, nasopharyngeal, colon, breast, prostate, and testicular cancer | Topoisomerase II | [184] |
| Taxanes Cabazitaxel Docetaxe Paclitaxel | Inhibit microtubule function resulting in cell cycle arrest and aberrant mitosis. | Phase II clinical trial | NSCLC, head and neck, breast, prostate, gastric adenocarcinoma | Tubulin | [185] |
| Vinca alkaloids Vinblastine Vincristine Vindesine Vinflunine Vinorelbine | Inhibit microtubule polymerization and assembly, leading to metaphase arrest and cell death. | Phase II clinical trial | NSCLC, breast, lung, leukemia, Hodgkin and non-Hodgkin lymphomas, testicular carcinoma, Kaposi’s sarcoma, and second-line transitional cell carcinoma of the urothelium (TCCU) | Tubulin | [186] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazumder, K.; Aktar, A.; Roy, P.; Biswas, B.; Hossain, M.E.; Sarkar, K.K.; Bachar, S.C.; Ahmed, F.; Monjur-Al-Hossain, A.S.M.; Fukase, K. A Review on Mechanistic Insight of Plant Derived Anticancer Bioactive Phytocompounds and Their Structure Activity Relationship. Molecules 2022, 27, 3036. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27093036
Mazumder K, Aktar A, Roy P, Biswas B, Hossain ME, Sarkar KK, Bachar SC, Ahmed F, Monjur-Al-Hossain ASM, Fukase K. A Review on Mechanistic Insight of Plant Derived Anticancer Bioactive Phytocompounds and Their Structure Activity Relationship. Molecules. 2022; 27(9):3036. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27093036
Chicago/Turabian StyleMazumder, Kishor, Asma Aktar, Priyanka Roy, Biswajit Biswas, Md. Emran Hossain, Kishore Kumar Sarkar, Sitesh Chandra Bachar, Firoj Ahmed, A. S. M. Monjur-Al-Hossain, and Koichi Fukase. 2022. "A Review on Mechanistic Insight of Plant Derived Anticancer Bioactive Phytocompounds and Their Structure Activity Relationship" Molecules 27, no. 9: 3036. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27093036