An Efficient Workflow for Screening and Stabilizing CRISPR/Cas9-Mediated Mutant Lines in Bombyx mori



Abstract
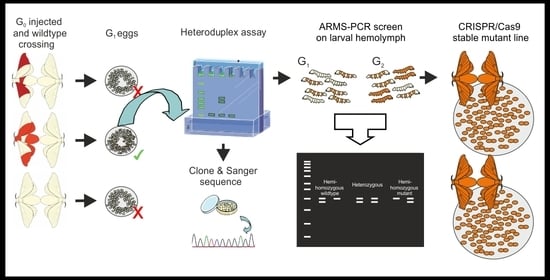
:
1. Introduction
2. Experimental Design
2.1. Initial Identification of Mutations
2.2. Mutation Sequence Determination
2.3. Hemolymph Sampling and Screening with ARMS-PCR
2.4. Materials
- Ice and ice box
- Masking tape
- Lab tissue
- Forceps (PharmaPlast, Thessaloniki, Greece; Cat. no.: 161)
- 15 mL sterile tubes (SARSTEDT, Nümbrecht, Germany; Cat. no.: 62.554.502)
- 50 mL sterile tubes (SARSTEDT, Nümbrecht, Germany; Cat. no.: 62.547.254)
- 1.5 mL sterile microtubes (SARSTEDT, Nümbrecht, Germany; Cat. no.: 72.706.200)
- μStripPro 0.2 mL PCR tubes (SARSTEDT, Nümbrecht, Germany; Cat. no.: 72.985.99)
- Hypodermic needle (32G) (BioTekne SRL, Bologna, Italy; Cat. no.: AM32G)
- Pipette tips (P10), (SARSTEDT, Nümbrecht, Germany; Cat. no.: 70.3010)
- Pipette tips (P200), (SARSTEDT, Nümbrecht, Germany; Cat. no.: 70.3030.020)
- Pipette tips (P1000), (SARSTEDT, Nümbrecht, Germany; Cat. no.: 70.3050.20)
- Sterile vented Petri dishes, 94 mm (Roll S.A.S., Sacco, Italy; Cat. no.: 182480)
- 70% ethanol (VWR, Milan, Italy; Cat. no.: 20821.310)
- TAE buffer (50×: Tris free base: 242 g, disodium EDTA 18.61 g, glacial acetic acid 57.1 mL, double-distilled H2O to 1 L)
- TBE buffer (5×: 104 g tris base 27.5 g, boric acid 40 mL, 0.5 M EDTA, 700 mL double-distilled H2O, pH to 8.0, adjust to 1 L)
- LB Broth Miller (Merck Life Science, Milano, Italy; Cat. no.: L3522)
- Phusion Green Hot Start II High Fidelity PCR Master Mix (ThermoFisher, Waltham, MA, USA, Cat. no.: F566L)
- DreamTaq Green PCR Master Mix (2X) (ThermoFisher, Waltham, MA, USA, Cat. no.: K1082)
- Agarose (EuroClone, Milano, Italy; Cat. no.: EMR920500)
- Ethidium bromide (Merck Life Science, Milano, Italy; Cat. no.: E1510)
- 1 Kb Plus DNA Ladder (ThermoFisher, Waltham, MA, USA, Cat. no.: 10787018)
- Acrylamide/Bis-acrylamide 30% (29:1) (Merck, Darmstadt, Germany; Cat. no.: A3574)
- Ammonium persulphate (APS; Merck Life Science, Milano, Italy; Cat. no.: A3678)
- N, N, N′, N′-Tetramethylethylenediamine (TEMED; Merck Life Science, Milano, Italy; Cat. no.: T9281)
- StrataClone PCR Cloning Kit (Agilent Technologies, Santa Clara, CA, USA; Cat. no.: 240205)
- Ampicillin (Merck Life Science, Milano, Italy; Cat. no.: 59349).
2.5. Equipment
- Micropipettes (P10, P200, P1000)
- 1.5 mL Microtube racks
- Gel Doc™ XR+ (Bio-Rad, Hercules, CA, USA; Cat. no.: 1708182)
- NanoDrop 2000c (Thermo Scientific, Waltham, MA, USA; Cat. no.: ND-2000C)
- Thermal Cycler Veriti™ (Thermo Scientific, Waltham, MA, USA; Cat. no.: 4375786)
- Vert Slab (Amersham Bioscience, San Francisco, CA, USA).
3. Procedure
3.1. Initial Identification of Mutations
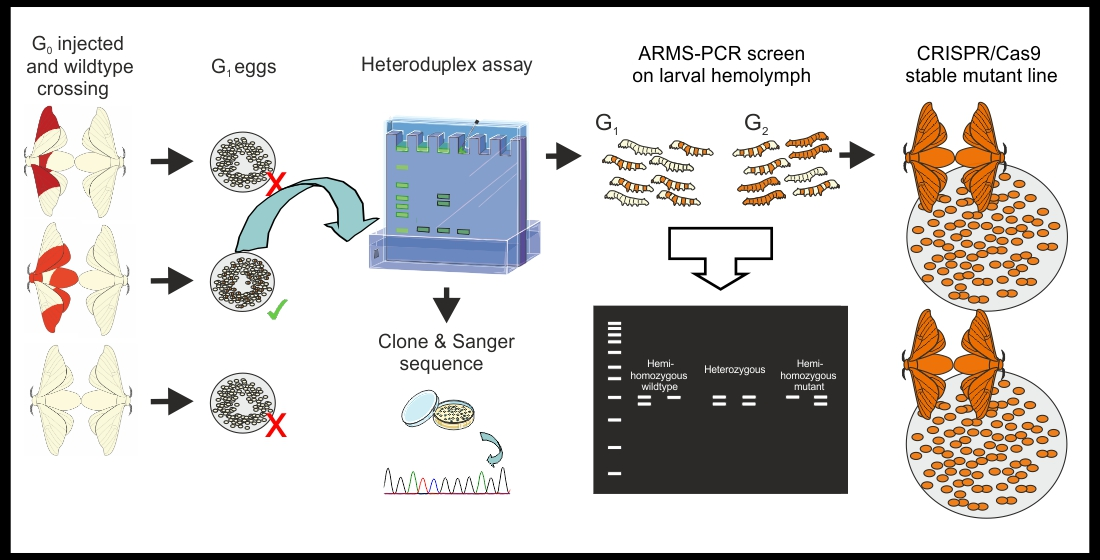
3.1.1. Genomic DNA Extraction from G1 Embryos, and PCR. Time for Completion: DNA Extraction-90 min. Per 24 Egg Samples. PCR and Agarose Gel Electrophoresis-4 h (Figure 2)
- Allow the G1 eggs to develop for three days, collect five eggs per egg batches, place each egg into a microtube. Label microtubes with the unique egg batch code, and replicate number (see Note 1).
- Crush the eggs with a pipette tip and perform DNA extraction and purification using the chosen DNA purification kit following the manufacturer’s instructions. Quantify DNA with a NanoDrop and prepare 20 ng/μL working solutions for each sample.
![Mps 04 00004 i001]() CRITICAL STEP Also perform DNA extraction on wildtype tissue from the same genetic background. This DNA will be used to obtain amplicons required for the mixed heteroduplex assay.
CRITICAL STEP Also perform DNA extraction on wildtype tissue from the same genetic background. This DNA will be used to obtain amplicons required for the mixed heteroduplex assay.![Mps 04 00004 i001]() CRITICAL STEP Use high fidelity polymerase when performing initial molecular screening. Calculate a sufficient PCR master mix volume for all your samples in 20 μL PCR reactions, include negative and positive controls. Calculate the required volume of wildtype amplicon (for heteroduplex formation, a minimum of 10 μL of wildtype amplicon per sample: e.g., 10 μL × 12 samples = 120 μL).
CRITICAL STEP Use high fidelity polymerase when performing initial molecular screening. Calculate a sufficient PCR master mix volume for all your samples in 20 μL PCR reactions, include negative and positive controls. Calculate the required volume of wildtype amplicon (for heteroduplex formation, a minimum of 10 μL of wildtype amplicon per sample: e.g., 10 μL × 12 samples = 120 μL).- Prepare a PCR master mix (see primer sequences in Figure 1 and Table 1), for 480 μL add reagents as in Table 2. Label and add 19 μL of PCR master mix to each PCR tube. Add 1 μL of the 20 ng/μL genomic DNA (gDNA) samples to each tube. Add 20 ng of wildtype gDNA to the positive control and wildtype PCR reactions, and 1 μL of H2O to the negative control. Briefly vortex and centrifuge. Run the PCR; 98 °C–2 min, [98 °C–20 s, 62 °C–30 s, 72 °C 20 s] × 30, 72 °C 2–min and hold at 4 °C.
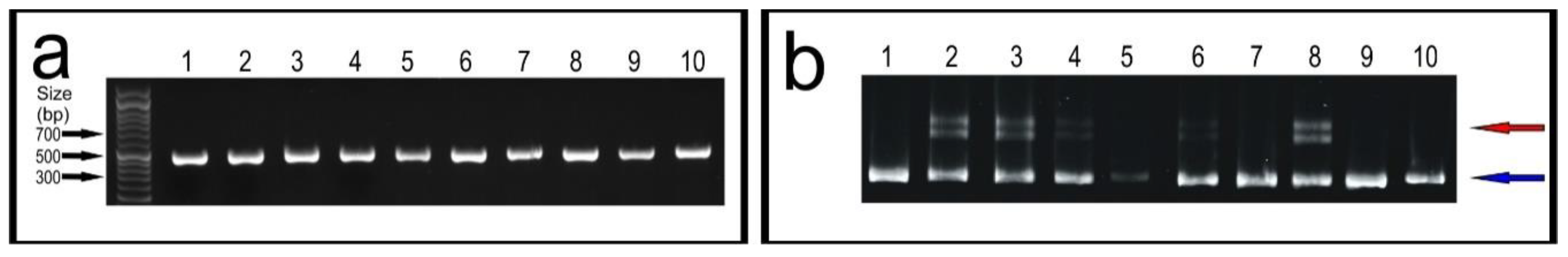
- Load 5 μL of the PCR reactions on a 1.2% agarose gel (TAE buffer) stained with ethidium bromide. Run the gel for 30 min. at 100 V and image on a GelDoc XR+, to confirm all the PCR reactions have been successful.

3.1.2. Heteroduplex Assay and Polyacrylamide Gel Electrophoresis. Time for Completion: Preparation for Heteroduplex Formation-90 min. PAGE Gel Preparation–3 h; Preparing, Loading and Running the PAGE Gel–4 h
- Prepare and label a PCR tube for each egg sample as well as positive and negative controls. To each tube, add 5 μL of the egg sample PCR amplicon.
![Mps 04 00004 i001]() CRITICAL STEP Add 5 μL of wildtype PCR amplicon to each PCR sample, vortex and centrifuge the mixtures. Place the samples in a thermocycler and perform a temperature denaturing ramp-down; 98 °C for 3 min. and decrease the temperature 1 °C every 20 s. Hold the ramp-down at 25 °C. If mutations are present in a G1 sample, heteroduplexes will have formed. Store the samples at 4 °C until use or at −20 °C for longer storage. Keep in mind that samples from hemizygous females bearing a per mutation will not produce heteroduplexes without the addition of the wildtype amplicon.
CRITICAL STEP Add 5 μL of wildtype PCR amplicon to each PCR sample, vortex and centrifuge the mixtures. Place the samples in a thermocycler and perform a temperature denaturing ramp-down; 98 °C for 3 min. and decrease the temperature 1 °C every 20 s. Hold the ramp-down at 25 °C. If mutations are present in a G1 sample, heteroduplexes will have formed. Store the samples at 4 °C until use or at −20 °C for longer storage. Keep in mind that samples from hemizygous females bearing a per mutation will not produce heteroduplexes without the addition of the wildtype amplicon.- Under a chemical hood prepare a 15% non-denaturing PAGE gel. For a 30 mL gel, add; 18 Ω H2O (22 mL), Acrylamide/Bis 29:1% (9 mL) and 5× TBE (3.5 mL), to a 50 mL tube. Close the lid tightly and gently invert the solution several times. Under the fume hood, loosen the lid and allow the mix to de-gas for 15 min. To cast the gel, add; 400 μL of 10% APS, and 40 μL of TEMED. Close the tube and gently invert the solution several times. Cast the gel and allow polymerizing for a minimum of 2 h.
![Mps 04 00004 i002]() PAUSE STEP PAGE gels can be stored for a few days at 4 °C–after the gel solidifies, dampen lab tissue with 1× TBE buffer and cover the top and bottom of the gel plates, seal the gel in a plastic bag. Do not store PAGE gels for more than a few days.
PAUSE STEP PAGE gels can be stored for a few days at 4 °C–after the gel solidifies, dampen lab tissue with 1× TBE buffer and cover the top and bottom of the gel plates, seal the gel in a plastic bag. Do not store PAGE gels for more than a few days. - Prepare the PAGE gel in 1× TBE running buffer and load 7.5 μL of the samples from the temperature ramp-down (N.B. Phusion Green Master Mix contains loading dye). Load an appropriate DNA ladder, negative PCR control, and a positive control (wildtype only PCR amplicon). Run the gel at 50 V for 1 h, then increase to 200 V for 2.5 h. After the run, remove the gel from the plates, wash it three times with distilled water, submerge the gel in 75–100 mL of distilled water and add 5 μL of ethidium bromide (10 mg/mL), gently shake for 15 min. Image the gel on a Gel Doc™ XR+.

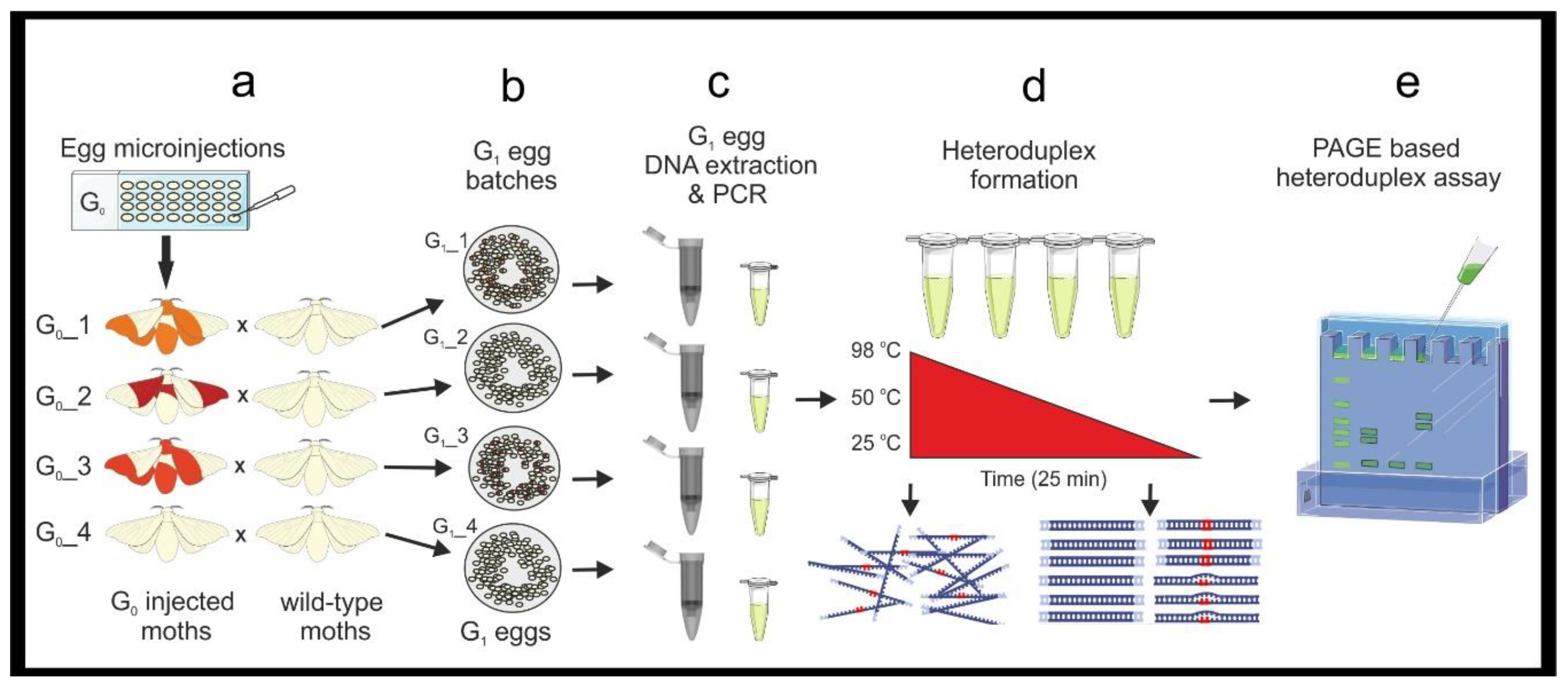
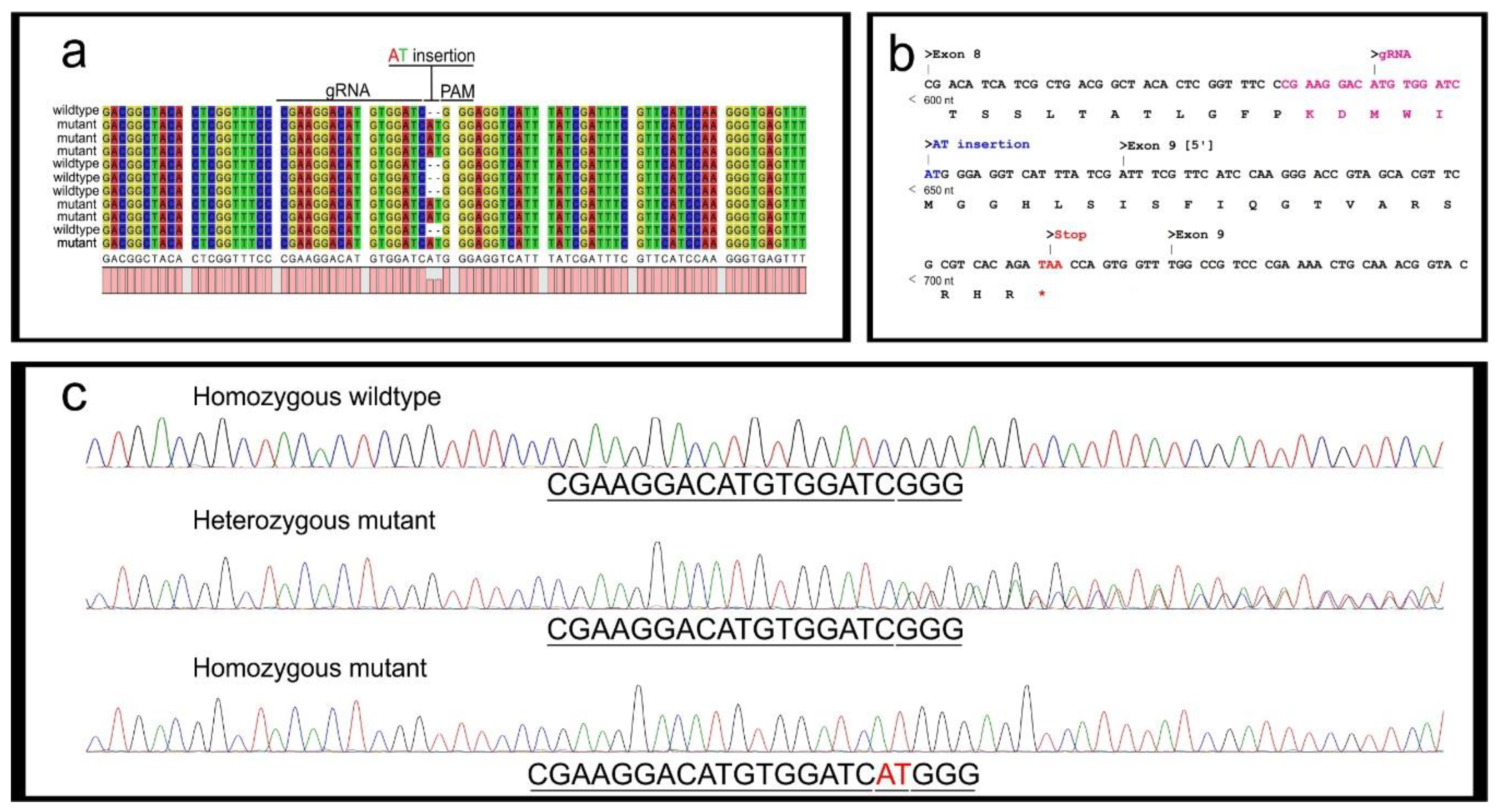
3.2. Mutant Sequence Determination
- Perform PCR using standard Taq polymerase and the gDNA previously extracted from the known mutated G1 egg samples (see point 2 in Section 3.1.1). Prepare 480 μL of DreamTaq master mix as in Table 3 (see primer sequences in Figure 1 and Table 1). Vortex briefly and centrifuge.
- Add 19 μL of the master mix to a PCR tube for each sample and add 1 μL of G1 embryonic gDNA (20 ng/μL), prepare a positive control with wildtype gDNA (20 ng) and a negative control with 1 μL of H2O. Briefly vortex and centrifuge the mixtures. Perform PCR (95 °C for 3 min, [95 °C for 30 s–51 °C for 30 s–72 °C for 45 s] × 25, 72 °C 10 min.) and store the PCR products at 4 °C. Run 5 μL of each on a 1.2% agarose gel (as in point 6 in Section 3.1.1) to confirm all samples have amplified the fragment.
- Dilute the PCR products 1:10 in NF H2O. Ligate the PCR amplicons into the StrataClone PCR Cloning vector, following the manufacturer’s protocol. Perform one ligation reaction for each sample.
![Mps 04 00004 i002]() PAUSE STEP Ligated plasmids can be stored as a reaction mixture at −20 °C before transformation.
PAUSE STEP Ligated plasmids can be stored as a reaction mixture at −20 °C before transformation.- Transform competent cells following the provided protocol and incubate ON at 37 °C.
- Select at least five colonies from each ON plate. Aseptically prepare five 5 mL-aliquots of sterile LB broth with ampicillin (100 μg/mL) in 15 mL tubes. Using a sterile pipette tip inoculate each 5 mL LB aliquot with one colony. Incubate at 37 °C with vigorous shaking ON.
- Isolate the plasmids using the PureYield™ Plasmid Miniprep System (Promega, Dane County, WI, USA) following the manufacture’s protocol. Elute the DNA in NF H2O (required for Sanger sequencing). Use a NanoDrop to check the quality and quantity of the samples.
- Prepare the plasmid DNA for Sanger sequencing following the service provider’s guidelines.
- A protocol for computational analysis of Sanger sequences is available in Supplementary Methods A.
- Design ARMS-PCR primers for the appropriate mutation/s identified by Sanger sequencing (Table 1).
![Mps 04 00004 i001]() CRITICAL STEP Test the ARMS-PCR primers using wildtype gDNA. Mutant (M)-ARMS-PCR primers must not generate an amplicon using wildtype gDNA.
CRITICAL STEP Test the ARMS-PCR primers using wildtype gDNA. Mutant (M)-ARMS-PCR primers must not generate an amplicon using wildtype gDNA.
3.3. Hemolymph Sampling and Screening with ARMS-PCR
3.3.1. Hemolymph Sampling and Larvae Rearing. Time for Completion: Hemolymph Sampling-2–3 h Per 50 Worms. DNA Extraction–90 min. Per 24 Samples. Larvae Rearing–6–7 Days for the 5th Instar and 12–14 Days for Pupation and Adult Emergence
- Rear G1 egg batches bearing the mutation until the 1st–2nd day of the 5th larval instar.
- For each positive G1 batch, process at least 30–50 larvae.
- Sterilize the workspace with 70% ethanol. For each larva, label one 1.5 mL microtube and one sterile Petri dish with the same code. Repeat for each larva, in advance. Place the labeled microtubes on ice, on the workbench near the labeled Petri dishes.
- On the workbench, prepare; a small beaker with 50 mL of 70% ethanol, lab tissue, a microtube rack, a P20 micropipette, sterile tips, hypodermic needles, and waste containers for general lab waste, and hypodermic needles.
- Using forceps, select a larva and carefully submerge it into the 70% ethanol beaker for 3 s. Gently blot the larva dry on lab tissue and let air dry for a few seconds. While air drying, open a labeled microtube and take its paired Petri dish.
- Pick up and gently fold the worm head-to-tail, exposing the dermis between the body folds (Figure 6a). Hold the larva over the microtube and use a hypodermic needle to gently pierce the dermis without deeply entering the body cavity (Figure 6b), a drop of hemolymph will pool at the wound (Figure 6c).
![Mps 04 00004 i001]() CRITICAL STEP Collect the hemolymph drop using a P20 micropipette (Figure 6d) and place into the microtube. Do not squeeze the larvae or attempt to withdraw additional hemolymph from the wound with the micropipette. Store the hemolymph on ice.
CRITICAL STEP Collect the hemolymph drop using a P20 micropipette (Figure 6d) and place into the microtube. Do not squeeze the larvae or attempt to withdraw additional hemolymph from the wound with the micropipette. Store the hemolymph on ice.- Place the larva into the appropriately labeled Petri dish. Continue sampling remaining larvae. Allow the sampled larvae heal for a minimum of 20 min. After healing, feed larvae in the Petri dishes.
![Mps 04 00004 i002]() PAUSE STEP Hemolymph samples can be frozen at −20 °C until use.
PAUSE STEP Hemolymph samples can be frozen at −20 °C until use.- To rear sampled G1 larvae: everyday, remove the Petri dish lid and place upside down. Using forceps, transfer the larva to the lid, empty the frass and dry food from the dish, if necessary wipe the dish with ethanol soaked lab tissue before transferring the larva back with fresh food.
- After 6–7 days the larvae will prepare to spin their cocoon. Empty and clean the dish, add a folded square of lab tissue to facilitate cocoon spinning. After spinning begins do not interfere with the larvae for 4 days, then the pupae can be cut from the cocoons and sexed (Figure 6e).
3.3.2. DNA Extraction from Hemolymph and Screening with ARMS-PCR
- Perform gDNA extraction on 20 μL of hemolymph per sample using the PureYield Plasmid Miniprep System, following the manufacturer’s protocol. Using a NanoDrop, determine the samples quality and quantity. Prepare 20 ng/μL working gDNA solutions for each sample.
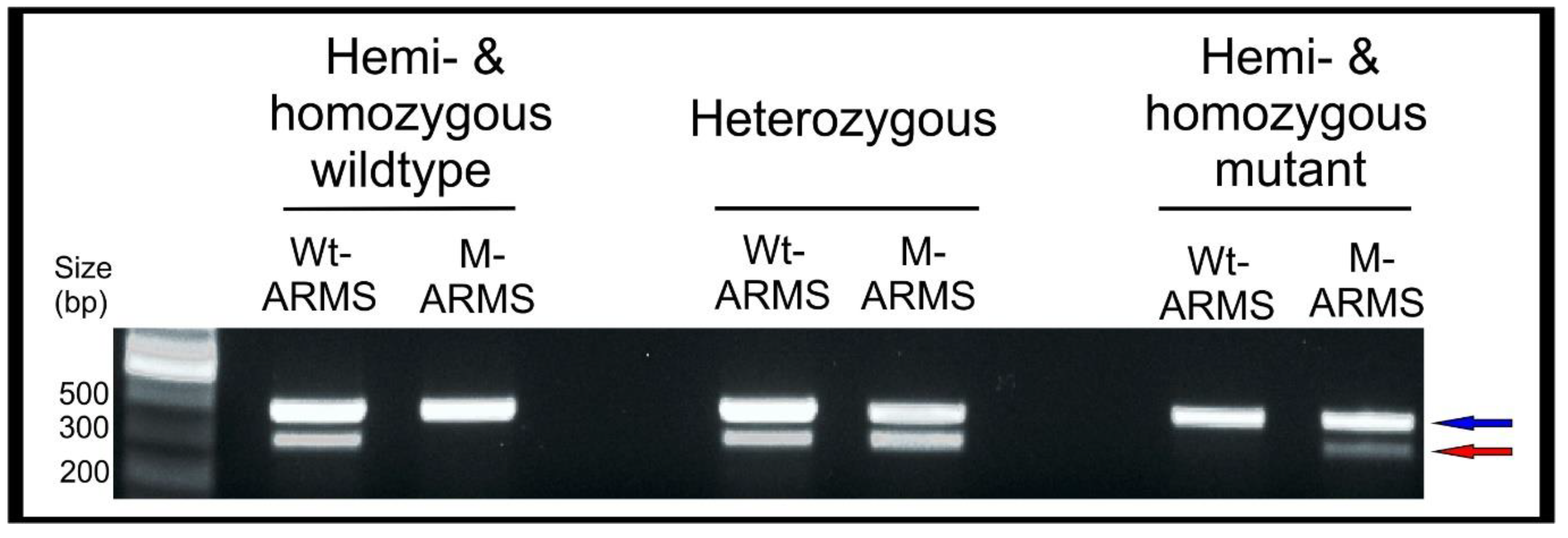
![Mps 04 00004 i001]() CRITICAL STEP High fidelity polymerase cannot be used for ARMS-PCR. Prepare PCR master mixes for both wildtype (WT) and mutant (M) ARMS-PCR reactions as in Table 4.
CRITICAL STEP High fidelity polymerase cannot be used for ARMS-PCR. Prepare PCR master mixes for both wildtype (WT) and mutant (M) ARMS-PCR reactions as in Table 4.- For each gDNA sample, prepare two PCR tubes with 9 μL of the WT-ARMS –PCR and M-ARMS-PCR master mixes. Add 1 μL (20 ng) of gDNA to both reactions. Repeat for each individual gDNA sample. Include a negative control, a positive control is not required as each reaction contains internal positive control primers. Vortex briefly and centrifuge the mixtures. Run the PCR (95 °C for 3 min, [95 °C for 30 s–51 °C for 30 s–72 °C for 45 s] × 34, 72 °C for 10 min.) and store the PCR products at 4 °C or use immediately.
- Prepare a 1.2% agarose gel with 1 × TAE buffer and stain with ethidium bromide. Load a DNA ladder (5 μL of GeneRuler 1 kb) into the first well. Always be consistent with sample loading: load 5 μL of the WT-ARMS-PCR reaction followed by 5 μL of the M-ARMS-PCR reaction for an individual hemolymph gDNA sample in order to easily compare the two reactions. Run the gel (30 min at 100 V) and image on a GelDoc XR+.
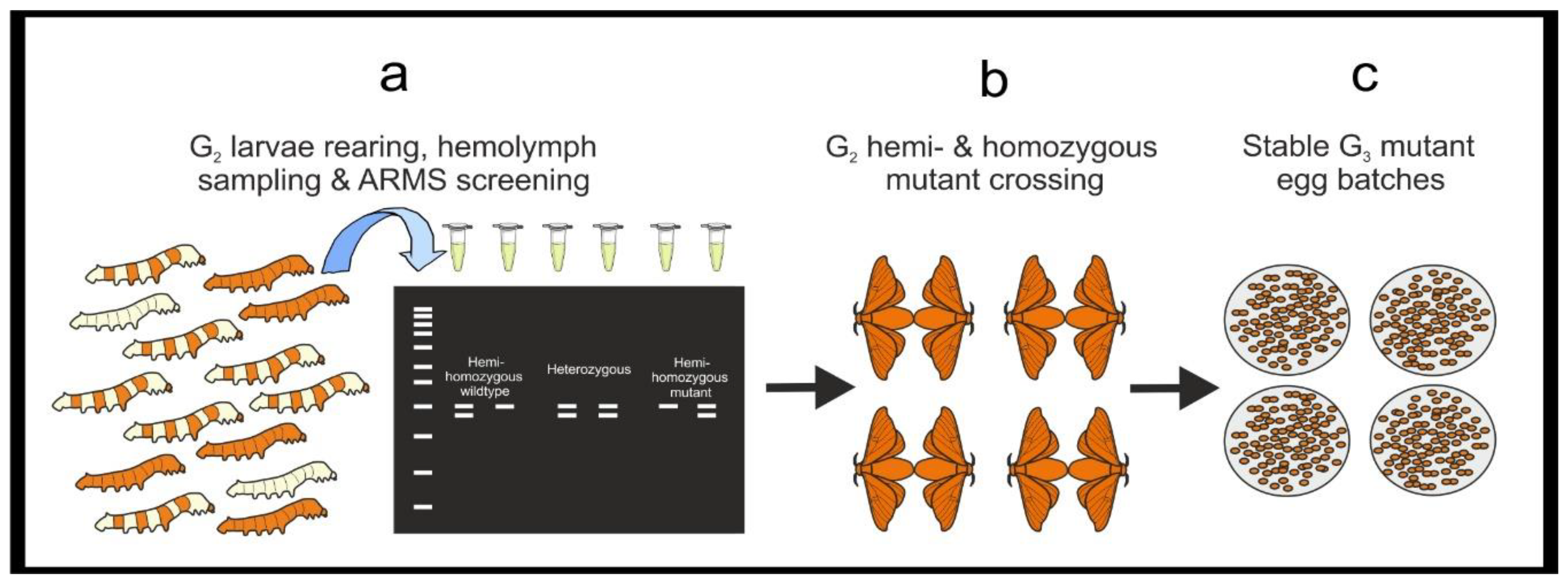
- Maintain G1 larvae showing a positive amplification M-ARMS reaction and discard those showing only a positive amplification WT-ARMS reaction. Rear larvae until the adult stage and in-breed G1 moths bearing mutation to generate the G2 egg batches. Notes for breeding B. mori moths can be found in Supplementary Methods B.
- Rear G2 larvae until the 5th instar and perform DNA extraction from hemolymph and ARMS-PCR as from Section 3.3.1.
- Select hemi- and homozygous mutant G2 larvae and rear until adult stage. Mate them to generate a G3 mutant stable line.
4. Expected Results
4.1. Initial Identification of Mutations
4.2. Mutant Sequence Determination
4.3. Screening with ARMS-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meng, X.; Zhu, F.; Chen, K. Silkworm: A promising model organism in life science. J. Insect Sci. 2017, 17, 97. [Google Scholar] [CrossRef]
- Willis, J.H.; Wilkins, A.S.; Goldsmith, M.R. A Brief History of Lepidoptera as Model Systems. Molecular Model Systems in the Lepidoptera; Goldsmith, M.R., Wilkins, A.S., Eds.; Cambridge University Press: Cambridge, UK, 1995; pp. 1–20. [Google Scholar] [CrossRef]
- Xu, H.; O’Brochta, D.A. Advanced technologies for genetically manipulating the silkworm Bombyx mori, a model Lepidopteran insect. Proc. R. Soc. B 2015, 282, 20150487. [Google Scholar] [CrossRef] [Green Version]
- Tamura, T.; Thibert, C.; Royer, C.; Kanda, T.; Eappen, A.; Kamba, M.; Shirk, P. Germline transformation of the silkworm Bombyx mori L. using a piggyBac transposon-derived vector. Nat. Biotechnol. 2000, 18, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Takasu, Y.; Kobayashi, I.; Beumer, K.; Uchino, K.; Sezutsu, H.; Sajwan, S.; Carroll, D.; Tamura, T.; Zurovec, M. Targeted mutagenesis in the silkworm Bombyx mori using zinc finger nuclease mRNA injection. Insect Biochem. Mol. Biol. 2010, 40, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhang, S.; Wang, F.; Liu, Y.; Liu, Y.; Xu, H.; Xia, Q. Highly efficient and specific genome editing in silkworm using custom TALENs. PLoS ONE 2012, 7, e45035. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Reed, R.D. A practical guide to CRISPR/Cas9 genome editing in Lepidoptera. In Diversity and Evolution of Butterfly Wing Patterns; Sekimura, H.T., Nijhout, F., Eds.; Springer Nature: Singapore, 2017; pp. 155–172. [Google Scholar] [CrossRef]
- Blayney, J.; Foster, E.M.; Jagielowicz, M.; Kreuzer, M.; Morotti, M.; Reglinski, K.; Xiao, J.H.; Hublitz, P. Unexpectedly high levels of inverted re-insertions using paired sgRNAs for genomic deletions. Methods Protoc. 2020, 3, 53. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Z.; Xu, J.; Zeng, B.; Ling, L.; You, L.; Chen, Y.; Huang, Y.; Tan, A. The CRISPR/Cas system mediates efficient genome engineering in Bombyx mori. Cell Res. 2013, 1414–1416. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Chang, J.; Wang, X.; Liu, Y.; Zhang, J.; Lu, W.; Gao, J.; Shi, R.; Zhao, P.; Xia, Q. CRISPR/Cas9 mediated multiplex genome editing and heritable mutagenesis of BmKu70 in Bombyx mori. Sci. Rep. 2014, 4489. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Xin, H.; Roy, B.; Dai, J.; Miao, Y.; Gao, G. Heritable genome editing with CRISPR/Cas9 in the silkworm, Bombyx mori. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Zhang, Z.; Aslam, A.F.; Liu, X.; Li, M.; Huang, Y.; Tan, A. Functional analysis of Bombyx Wnt1 during embryogenesis using the CRISPR/Cas9 system. J. Insect Physiol. 2015, 79, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, X.; Li, X.; Pu, Q.; Luo, C.; Xu, L.; Peng, X.; Liu, S. Genetic manipulation of microRNAs in the silk gland of silkworm, Bombyx mori. Biol. Proced. Online 2019, 21, 16. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Xu, X.; Li, X.; Zhang, Y.; Huang, Y.; Li, K.; Xu, J. CRISPR disruption of Bmovo resulted in the failure of emergence and affected the wing and gonad development in the silkworm Bombyx mori. Insects 2019, 10, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.W.; Jeong, C.Y.; Yu, J.H.; Kim, S.B.; Kang, S.K.; Kim, S.W.; Kim, N.S.; Kim, K.Y.; Park, J.W. Bombyx mori kynurenine 3-monooxygenase gene editing and insect molecular breeding using the clustered regularly interspaced short palindromic repeat/CRISPR associated protein 9 system. Biotechnol. Prog. 2020, e3054. [Google Scholar] [CrossRef] [PubMed]
- Nartey, M.A.; Sun, X.; Qin, S.; Hou, C.X.; Li, M.W. CRISPR/Cas9-based knockout reveals that the clock gene timeless is indispensable for regulating circadian behavioral rhythms in Bombyx mori. Insect Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Q.; Wang, Y.; Li, J.; Gao, C. Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 2014, 9, 2395–2410. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Van Meir, E.G. A simple genotyping method to detect small CRISPR-Cas9 induced indels by agarose gel electrophoresis. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Xu, Y.; Yu, S.; Lu, L.; Ding, M.; Cheng, J.; Song, G.; Gao, X.; Yao, L.; Fan, D.; et al. An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci. Rep. 2014, 4, 1–8. [Google Scholar] [CrossRef]
- Newton, C.R.; Graham, A.; Heptinstall, L.E.; Powell, S.J.; Summers, C.; Kalsheker, N.; Smith, J.C.; Markham, A.F. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989, 17, 2503–2516. [Google Scholar] [CrossRef]
- Brady, D.; Saviane, A.; Romoli, O.; Tettamanti, G.; Sandrelli, F.; Cappellozza, S. Oral Infection in a Germ-Free Bombyx mori Model, Immunity in Insects; Sandrelli, F., Tettamanti, G., Eds.; Springer Nature: New York, NY, USA, 2020; pp. 217–231. [Google Scholar] [CrossRef]
- Cappellozza, L.; Cappellozza, S.; Saviane, A.; Sbrenna, G. Artificial diet rearing system for the silkworm Bombyx mori (Lepidoptera: Bombycidae): Effect of vitamin C deprivation on larval growth and cocoon production. Appl. Entomol. Zool 2005, 40, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, E.K.; Tao, C.; Mario, A.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Synthego Performance Analysis, ICE Analysis. Available online: https://ice.synthego.com/#/ (accessed on 14 December 2020).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Gene | Accession no. | Start Position | End Position | Fragment Length (bps) | Sequence (5′–3′) |
|---|---|---|---|---|---|---|
| per_FOR | period | LOC692522 | 10,240 | 10,259 | 482 | ACTGCATGACGGCAACTTT |
| per_REV | 10,702 | 10,722 | TGCCGTTCCTTTTGAAATTC | |||
| ARMS_WT_FOR | period | LOC692522 | 10,417 | 10,435 | 305 | GAAGGACATGTGGATCGG |
| ARMS_M_FOR | 10,417 | 10,437 | 307 | CCGAAGGACATGTGGATCAT | ||
| cyc_FOR | cycle | LOC692530 | 69,748 | 69,774 | 541 | CCTGAATAGTTACCAAATACATTTGA |
| cyc_REV | 70,265 | 70,286 | CGAATTTTGGTGGTCGTGTAT |
| Reagent | Concentration | Volume (μL) |
|---|---|---|
| Phusion Green Master Mix | 2× | 250 |
| per_FOR | 10 μM | 25 |
| per_REV | 10 μM | 25 |
| H2O | - | 180 |
| Reagent | Concentration | Volume (μL) |
|---|---|---|
| DreamTaq (2×) mix | 2× | 250 |
| per_FOR | 10 μM | 25 |
| per_REV | 10 μM | 25 |
| H2O | - | 180 |
| Reagent | Concentration | WT-ARMS-PCR vol. (μL) | M-ARMS-PCR vol. (μL) |
|---|---|---|---|
| DreamTaq Green | 2× | 250 | 250 |
| cyc_FOR | 10 μM | 25 | 25 |
| cyc_REV | 10 μM | 25 | 25 |
| per_REV | 10 μM | 25 | 25 |
| ARMS_WT_FOR | 10 μM | 25 | 0 |
| ARMS_M_FOR | 10 μM | 0 | 25 |
| H2O | - | 150 | 150 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brady, D.; Saviane, A.; Cappellozza, S.; Sandrelli, F. An Efficient Workflow for Screening and Stabilizing CRISPR/Cas9-Mediated Mutant Lines in Bombyx mori. Methods Protoc. 2021, 4, 4. https://0-doi-org.brum.beds.ac.uk/10.3390/mps4010004
Brady D, Saviane A, Cappellozza S, Sandrelli F. An Efficient Workflow for Screening and Stabilizing CRISPR/Cas9-Mediated Mutant Lines in Bombyx mori. Methods and Protocols. 2021; 4(1):4. https://0-doi-org.brum.beds.ac.uk/10.3390/mps4010004
Chicago/Turabian StyleBrady, Daniel, Alessio Saviane, Silvia Cappellozza, and Federica Sandrelli. 2021. "An Efficient Workflow for Screening and Stabilizing CRISPR/Cas9-Mediated Mutant Lines in Bombyx mori" Methods and Protocols 4, no. 1: 4. https://0-doi-org.brum.beds.ac.uk/10.3390/mps4010004