Role for Astroglia-Derived BDNF and MSK1 in Homeostatic Synaptic Plasticity
by
Ulyana Lalo
1, 
Alexander Bogdanov
2,
Guy W. J. Moss
3,
Bruno G. Frenguelli
1 and
Yuriy Pankratov
1,* 1
School of Life Sciences, University of Warwick, Gibbet Hill Campus, Coventry CV4 7AL, UK
2
Institute for Chemistry and Biology, Immanuel Kant Baltic Federal University 2 Universitetskaya str., Kaliningrad 236040, Russia
3
Department of Neuroscience, Physiology and Pharmacology, University College London, London WC1E 6BT, UK
*
Author to whom correspondence should be addressed.
Neuroglia 2018, 1(2), 381-394; https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020026
Submission received: 5 November 2018
/
Accepted: 12 November 2018
/
Published: 22 November 2018
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Homeostatic scaling of synaptic strength in response to environmental stimuli may underlie the beneficial effects of an active lifestyle on brain function. Our previous results highlighted a key role for brain-derived neurotrophic factor (BDNF) and mitogen- and stress-activated protein kinase 1 (MSK1) in experience-related homeostatic synaptic plasticity. Astroglia have recently been shown to serve as an important source of BDNF. To elucidate a role for astroglia-derived BDNF, we explored homeostatic synaptic plasticity in transgenic mice with an impairment in the BDNF/MSK1 pathway (MSK1 kinase dead knock-in (KD) mice) and impairment of glial exocytosis (dnSNARE mice). We observed that prolonged tonic activation of astrocytes caused BDNF-dependent upregulation of excitatory synaptic currents accompanied by enlargement of synaptic boutons. We found that exposure to environmental enrichment (EE) and caloric restriction (CR) strongly upregulated excitatory but downregulated inhibitory synaptic currents in old wild-type mice, thus counterbalancing the impact of ageing on synaptic transmission. In parallel, EE and CR enhanced astrocytic Ca2+-signalling. Importantly, we observed a significant deficit in the effects of EE and CR on synaptic transmission in the MSK1 KD and dnSNARE mice. Combined, our results strongly support the importance of astrocytic exocytosis of BDNF for the beneficial effects of EE and CR on synaptic transmission and plasticity in the ageing brain.
1. Introduction
The ability of neurons to autonomously scale their synaptic strength in response to hyper- or hypoexcitability of their neighbours is instrumental for brain adaptation to environmental challenges during development, adulthood, and ageing [1,2,3]. Such forms of adaptive changes in synaptic strength are generally termed homeostatic synaptic plasticity [2]. The responsiveness of synapses and neuronal networks to environmental stimuli has been linked to the potential beneficial effects of an active lifestyle on the ageing brain [4,5,6,7,8].
Brain-derived neurotrophic factor (BDNF) has emerged as an important mediator of synaptic adaptation to both activity deprivation and environmental enrichment [1,9,10]. Increased production of BDNF has been observed in rodents in response to environmental enrichment [1,3], which, via the BDNF tropomyosin receptor kinase B (TrkB receptor), promotes dendritic growth, and increased spine density [11].
The role of BDNF in synaptic adaptation relies on a number of proteins, including the cytoskeletal-associated protein Arc/Arg3.1 and mitogen- and stress-activated protein kinase 1 (MSK1). In particular, our previous data have shown a quintessential role for MSK1 in BDNF-mediated homeostatic synaptic scaling in vitro, and the effects of environmental enrichment (EE) on synaptic transmission in hippocampus in vivo [9]. Since the release of BDNF can be increased by various environmental stimuli, such as physical activity or a change in diet [9,12,13], the BDNF/MSK1 pathway might play an important role in the beneficial effects of both physical activity and caloric restriction (CR) on synaptic function, in particular in the ageing brain.
Recent data has highlighted an important role for astroglia as a source of BDNF [14,15]. It has been recently demonstrated that astrocytes can release BDNF via Ca2+-dependent exocytosis [14]. In addition, astrocytes, which mediate neurovascular coupling and orchestrate metabolic support of neurons [16,17,18], are strategically positioned to mediate the effects of enhanced physical activity and CR. Our recent results show that EE and CR can both enhance astroglial Ca2+-signalling [19]. Thus, we hypothesise that astroglia-derived BDNF could be instrumental for homeostatic synaptic scaling and experience-dependent synaptic plasticity.
To verify this hypothesis, we explored the effects of tonic activation of astrocytes in situ and in vivo (via environmental stimuli) on synaptic scaling in transgenic mice with an impairment of BDNF/MSK1 signalling (MSK1 kinase dead knock-in mice) and an impairment of glial exocytosis (dnSNARE mice).
2. Materials and Methods
All animal work was carried out in accordance with UK legislation and “3R” strategy; research did not involve non-human primates. This project was approved by the University of Warwick Animal Welfare and Ethical Review Body (AWERB), approval number G13-19, and regulated under the auspices of the UK Home Office Animals (Scientific Procedures) Act licenses P1D8E11D6 and I3EBF4DB9. Experiments were performed in astrocytes and neurons of the hippocampus and somatosensory cortex of dn-SNARE transgenic mice [19,20,21,22], their wild-type littermates (WT), and transgenic mice with MSK1 kinase dead knock-in (MSK1 KD mice) [9]; the MSK1 KD mice had the same genetic background as dnSNARE mice (C57/Bl6). We used mice of two age groups, 6–12 (average 7.8) weeks and 9–15 (average 12.7) months. We compared animals kept under standard housing conditions (SH) vs. animals exposed to the EE from birth [9], including ad libitum access to the running wheel, or kept on mild CR diet (food intake individually regulated to maintain the body weight loss of 10–15%) for 4 to 6 weeks.
2.1. Slice and Cell Preparation
Mice were anaesthetized by halothane and then decapitated, in accordance with UK legislation. Brains were removed rapidly after decapitation and placed into ice-cold physiological saline containing (mM): NaCl 130, KCl 3, CaCl2 0.5, MgCl2 2.5, NaH2PO4 1, NaHCO3 25, glucose 15, pH of 7.4 gassed with 95% O2–5% CO2. Transverse slices (260 μm) were cut at 4 °C and then placed in physiological saline containing (mM): NaCl 130, KCl 3, CaCl2 2.5, MgCl2 1, NaH2PO4 1, NaHCO3 22, glucose 15, pH of 7.4 gassed with 95% O2–5% CO2 and kept for 1.5 to 5 h prior to cell isolation and recording.
Astrocytes were identified by their morphology under differential contrast observation, green fluorescent protein (GFP) fluorescence (astrocytes from dn-SNARE mice) or staining with sulforhodamine 101 (astrocytes from WT and MSK1 KD mice). After recording, the identification of astrocytes was confirmed via their functional properties (high potassium conductance, low input resistance, and strong activity of glutamate transporters) as described previously [20,21,22,23]. To facilitate high-quality whole-cell recordings in the brain tissue slices of old mice, tissue slices were treated with a vibrating glass ball to remove the upper layer of dead cells and expose healthy neurons [24].
Hippocampal cultures were prepared from WT and MKS1 KD mice as described previously [9]. Briefly, hippocampi were dissected, recovered by enzymatic digestion with trypsin and dissociated through a Gilson pipette. For immunocytochemistry and for miniature excitatory postsynaptic currents (mEPSC) recordings, cells were plated at a density of approximately 100,000 per dish onto 22-mm glass coverslips coated with poly-l-lysine. Cultures were maintained at 37 °C in neurobasal medium (Gibco, Themo Fisher Scientific, UK) containing 1% l-glutamine (Gibco), 1% penicillin–streptomycin (Gibco) and 2% B27 supplement (Gibco) in a 95% O2/5% CO2-humidified incubator. Neurons were used between 12 to 16 days in vitro.
2.2. Electrophysiological Recordings
Whole-cell voltage-clamp recordings from cortical neurones and astrocytes were made with patch pipettes (4–5 MΩ) filled with intracellular solution (in mM): 110 CsCl, 10 NaCl, 10 HEPES, 5 MgATP, 1 d-Serine, 0.1 EGTA, pH 7.35. Currents were monitored using an MultiClamp 700B patch-clamp amplifier (Axon Instruments, Union City, CA, USA) filtered at 2 kHz and digitized at 4 kHz. Experiments were controlled by a Digidata1440A data acquisition board (Axon Instruments) and WinWCP software (Strathclyde University, Edinburgh, UK); data were analysed by custom lab software. Liquid junction potentials were compensated with the patch-clamp amplifier. The series and input resistances were 5–7 MΩ and 700–1200 MΩ, respectively; both series and input resistance varied by less than 20% in the cells accepted for analysis.
2.3. High-Resolution Scanning of Synaptic Boutons
Synaptic morphology was imaged in the neuronal cultures using the super-resolution hopping probe ion conductance microscopy, an advanced version of scanning ion conductance microscopy (SICM) [25]. The identification of synaptic boutons and SICM 3D topographical imaging were carried out using a custom-modified SICM scanner ICNano (Ionoscope, London, UK), as described previously [25]. Briefly, the sample was positioned in the X-Y directions with a nanopositioning stage (Physik Instrumente, Karlsruhe, Germany) and the scanning pipette was positioned in Z-direction with a piezoelectric actuator (PI). The fine-tipped scanning nanopipettes were pulled from borosilicate glass (outer/internal diameter 1/0.5 mm) with a horizontal laser puller P-2000 (Sutter Instruments, Novato, CA); pipette resistance was in the range of 80 to 100 MΩ corresponding to the estimated tip diameter of 90–120 nm. Nanopipettes were held in voltage-clamp mode with an Axopatch 200B patch-clamp amplifier (Axon Instruments); the amplifier head-stage was mounted on the z-scanning head. The amplifier output signal was monitored by the SICM electronics, which simultaneously controlled sample and pipette positioning.
The scan system was mounted onto an inverted microscope Nikon TE2000-U (Nikon Instruments, Kingston, UK) equipped with epifluorescence illumination. The sample was preloaded with the fluorescent synaptic marker FM1-43 (3 μM, 15 min loading followed by 15 min washout). FM1-43 fluorescence was imaged 100X 1.3 numerical aperture (NA) oil immersion objective and an EM-CCD camera (Andor iXon3, Andor Technology, Belfast, UK). Synaptic boutons were identified by matching the tentative boutons in topography and FM1-43 fluorescence [25]. The raw SICM data were processed using Gwyddion 5.0 microscopy analysis software (Czech Metrology Institute, Brno, Czech Republic) [26]. After constructing a 3D topographical image of identified synaptic varicosities, their maximal span in X-Y-Z dimensions (Δx, Δy and Δz) was determined and the effective size was calculated as the square root of (Δx2 + Δy2 + Δz2) quotient. The synaptic varicosity volume was estimated as the volume of an ellipsoid of Δx, Δy and Δz dimensions.
2.4. Multiphoton Fluorescent Ca2+-Imaging in Astrocytes
To monitor the cytoplasmic free Ca2+concentraton ([Ca2+]i) in situ, astrocytes of neocortical slices were loaded via 30 min incubation with 1 µM of Rhod-2AM (dnSNARE mice) or Oregon Green Bapta-2AM and sulphorhodamine 101 (wild-type and MSK1 KD mice) at 33 °C. Two-photon images of neurons and astrocytes were acquired at 5 Hz frame rate using a Zeiss LSM-7MP multiphoton microscope (Carl Zeiss, Jena, Germany) coupled to a MaiTai (SpectraPhysics, Santa Clara CA, USA) pulsing laser; experiments were controlled by ZEN LSM software (Carl Zeiss). Images were further analysed offline using ZEN LSM (Carl Zeiss) and ImageJ 1.52 (NIH) software [27]. The [Ca2+]i levels were expressed as ΔF/F ratio averaged over a region of interest (ROI). For analysis of spontaneous Ca2+-transients in astrocytes, three ROIs located over dendrites and one ROI located over the soma were chosen. Overall Ca2+-response to receptors agonists or synaptic stimulation was quantified using an ROI covering the whole cell image.
2.5. Data Analysis
All data are presented as mean ± standard deviation (SD) and the statistical significance of differences between data groups was tested by two-tailed unpaired t-test, unless indicated otherwise. For all cases of statistical significance reported, the statistical power of the test was between 0.8 and 0.9.
The spontaneous transmembrane currents recorded in neurons were analysed offline using methods described previously [20,21]. The amplitude distributions of spontaneous and evoked currents were analysed with the aid of probability density functions and likelihood maximization techniques; all histograms shown were calculated as probability density functions. The amplitude distributions were fitted with either multiquantal binomial model or bimodal function consisting of two Gaussians with variable peak location, width and amplitude. Parameters of models were fit using likelihood maximization routine.
3. Results
3.1. Astroglia-Induced Homeostatic Synaptic Scaling in Cultured Neurons
A conventional approach to test molecular mechanisms of homeostatic synaptic plasticity is exposure of primary neuronal cultures to conditions of chronic inhibition or enhancement of neuronal firing, e.g., 24 h-long incubation with tetrodotoxin or picrotoxin [9,10]. To probe the specific role of astrocytes, we modified this approach and selectively enhanced astroglial Ca2+-signalling with TFLLR, an agonist of PAR-1 receptors [20]. The efficiency and specificity of PAR-1 receptor-mediated activation of astrocytes and lack of such action in neurons have been verified previously [20].
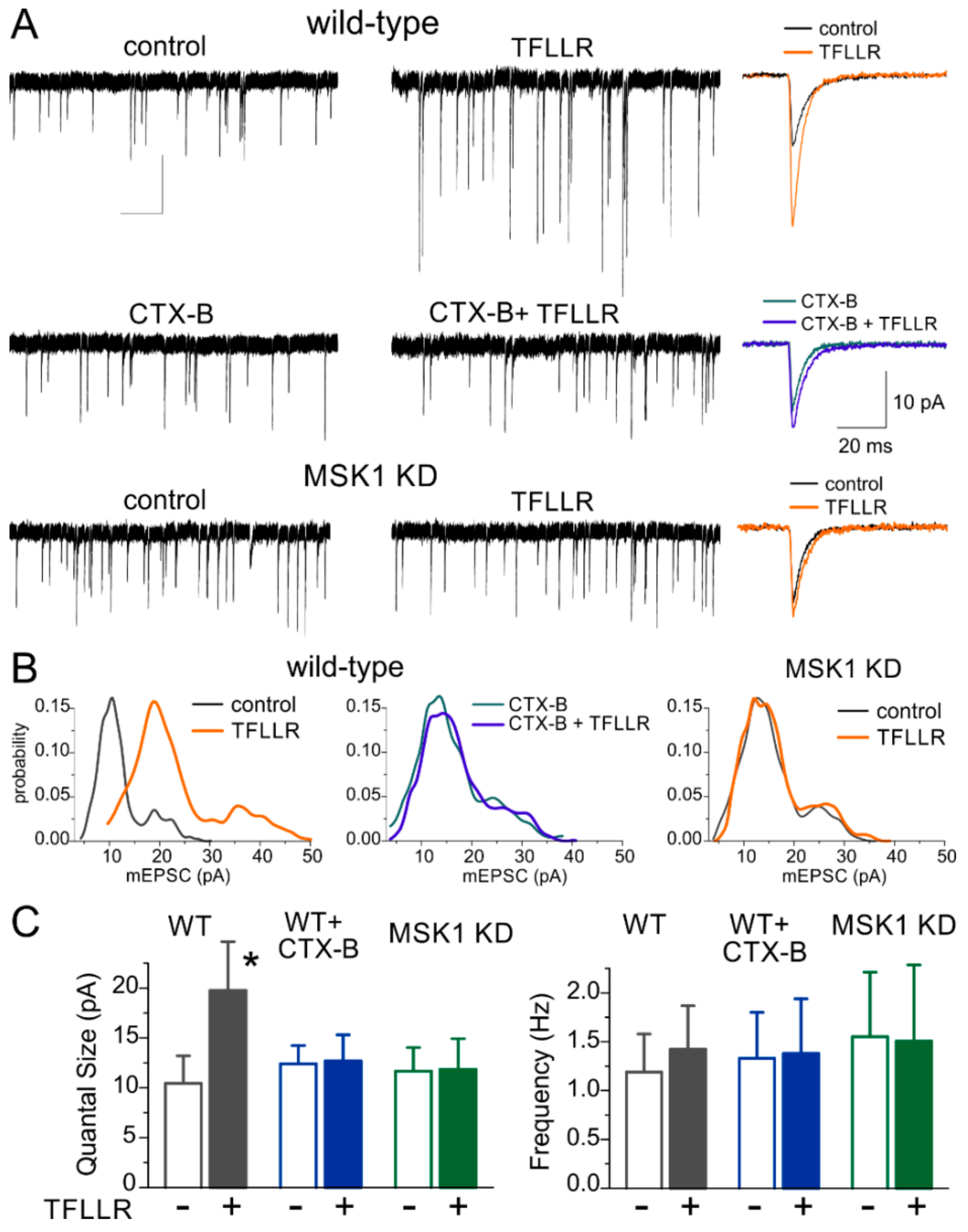
The 24 h-long incubation with TFLLR (3 µM) caused a considerable increase in the amplitude of mEPSCs in wild-type hippocampal pyramidal neurons (Figure 1A) suggesting that enhancement of astrocytic signalling can induce synaptic scaling. The average mEPSC amplitude increased from 14.01 ± 2.77 pA (n = 11) to 26.7 ± 6.55 pA (n = 9, p < 0.01).
To confirm that the observed synaptic scaling was BDNF-dependent, we incubated wild-type cultured neurones in TFLLR with Cyclotraxin-B (CTX-B, 10 nM), a selective and potent TrkB receptor inhibitor which has been shown to block the actions of BDNF [28]. Addition of the TrkB receptor inhibitor blocked the TFFLR-induced enhancement of mEPSCs (Figure 1A). Furthermore, the glia-induced synaptic scaling was impaired in cell cultures derived from MSK1 KD mice (Figure 1B,C) indicating the crucial role for BDNF-MSK1 pathway in this phenomenon.
Analysis of amplitude distributions of mEPSCs (Figure 1B) showed that the increase in the average amplitude occurred due to an increase in the unitary quantal size of synaptic response (manifested as the main peak of the amplitude distribution). The incubation with TFLLR alone caused a robust increase in the quantal size of mEPSCs recorded in wild type neurons, whereas there was no significant change in mEPSC frequency (Figure 1B,C). These results imply that glia-induced BDNF-dependent homeostatic synaptic scaling occurs mainly via postsynaptic mechanisms, similar to previous reports on various forms of homeostatic synaptic plasticity.
3.2. Astroglia-Induced Homeostatic Changes in Synaptic Morphology
An enhancement of strength of excitatory synapses at postsynaptic locus can occur via two principle pathways: an increase in efficacy of neurotransmitter receptors (i.e., permeability and open time of glutamate receptor-associated ion channels) or an increase in their surface expression. These functional alterations are often accompanied by changes in synaptic morphology, in particular an increase in the size of dendritic spines. Brain-derived neurotrophic factor has been previously implicated in the regulation of synaptic morphology via induction of Arc/Arg3.1 gene [9,29,30].
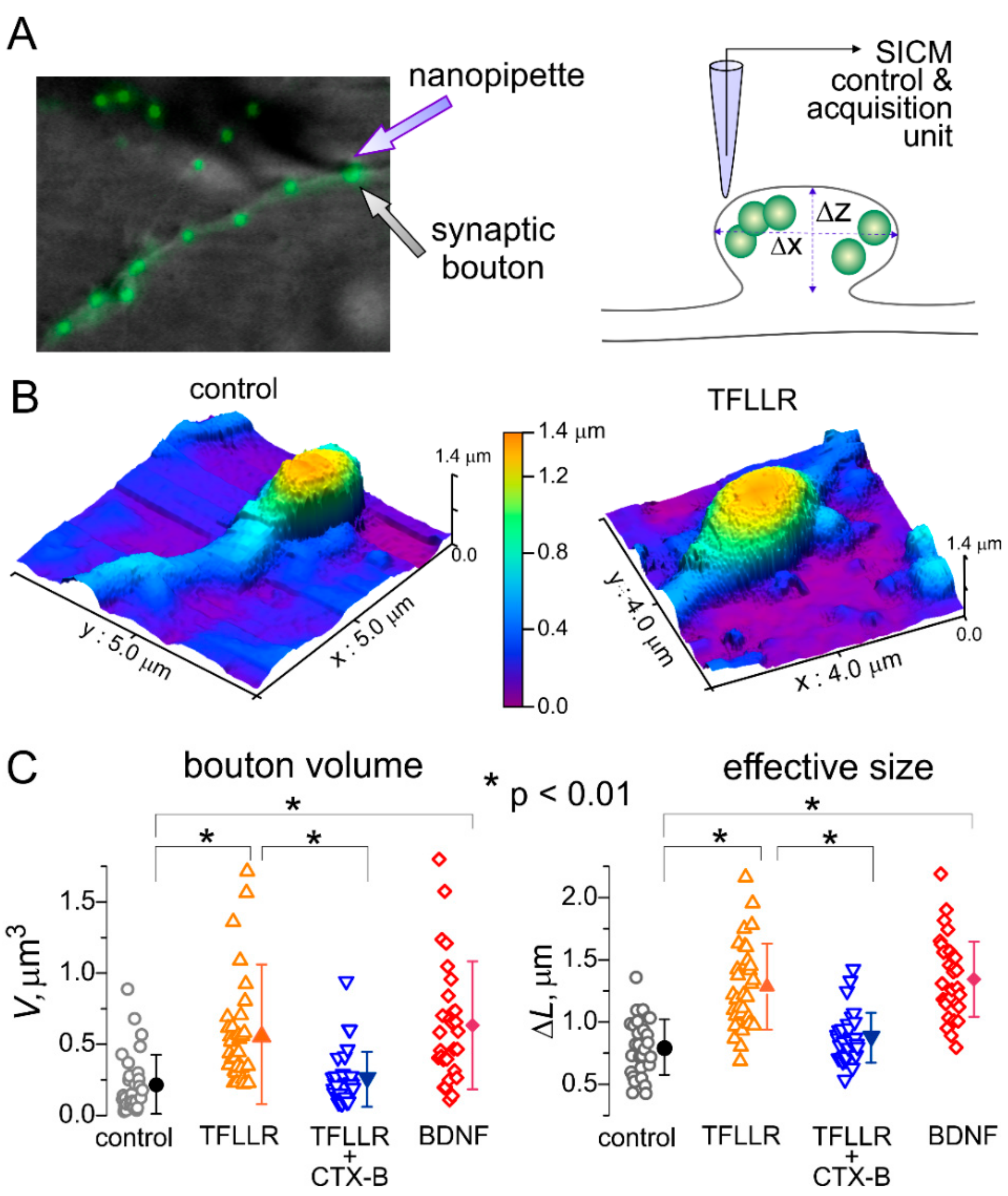
To elucidate whether this cascade is involved in the TFFLR-induced synaptic scaling, we assessed the alterations in the size and shape of postsynaptic sites using SICM. This technique is based on decrease in the ionic current passing through a glass microelectrode in close proximity of cells (or any other obstacles). The SICM method enables the imaging of live cells at nanoscale resolution and adequately maps the shape and size of small subcellular structures, such as dendrites, axons and synaptic boutons [25]. This technique has been successfully used before in the structure–function studies of synaptic networks of cultured hippocampal neurons and mechano-sensory stereocilia of cochlear hair cells [25,31].
To test for the effects of astroglia-derived BDNF on synaptic morphology, we examined live neurons from wild-type mice hippocampal cultures incubated with TFLLR alone, TFLLR with the inhibitor of TrkB receptors, or BDNF (Figure 2A). To identify functional synapses, neurons were labelled with FM1-43, an activity-dependent marker of synaptic vesicles, prior to SICM imaging (Figure 2A). Whenever a FM1-43 signal was observed, varicosities could be mapped at the SICM images; with the size and shape of varicosities consistent with the geometry expected of synaptic boutons. We targeted fluorescently-labelled varicosities located on distal dendrites to increase the probability of encountering excitatory synapses. Upon 3D-mapping of identified synaptic boutons (Figure 2B), their volume and effective size were evaluated as described in Methods (Figure 2C).
Consistent with our observations of astroglia-induced increase in synaptic strength, 24 h-incubation with TFLLR caused significant, up to 60%, increase in the average size of synapses (from 0.798 ± 0.223 µm to 1.248 ± 0.345 µm, p < 0.01). Correspondingly, the average volume of synaptic boutons showed >2.5-fold increase, from 0.218 ± 0.208 µm3 to 0.569 ± 0.489 µm3 (n = 31, p < 0.01).
The effect of TFLLR was effectively antagonized by Cyclotraxin-B (average size increased by just 10%) and reproduced by incubation with BDNF (Figure 3C).
Combined with data on the changes in mEPSC amplitude (Figure 1), these results demonstrate that activation of astrocytes can engage the BDNF-dependent molecular cascade, which is instrumental for homeostatic regulation of synaptic strength.
3.3. Astrocytes Participate in Homeostatic Plasticity In Vivo
BDNF-mediated homeostatic synaptic scaling has been implicated in the positive effects of environmental enrichment on synaptic transmission [9]. There is also evidence that an increase in BDNF can underlie the beneficial effects of a low-calorie diet on brain function [12,32]. Previously, we demonstrated that environmental enrichment and caloric restriction can enhance Ca2+-signalling in neocortical astrocytes [19]. Hence, one might predict that an increase in astroglial Ca2+-dependent BDNF release plays a key role in the beneficial effects of EE and CR on synaptic transmission in the aging brain. To explore the role of this signalling mechanism in a behavioural model of experience-dependent synaptic plasticity, we examined the effect of environmental enrichment and caloric restriction on excitatory and inhibitory synaptic transmission in the neocortex.
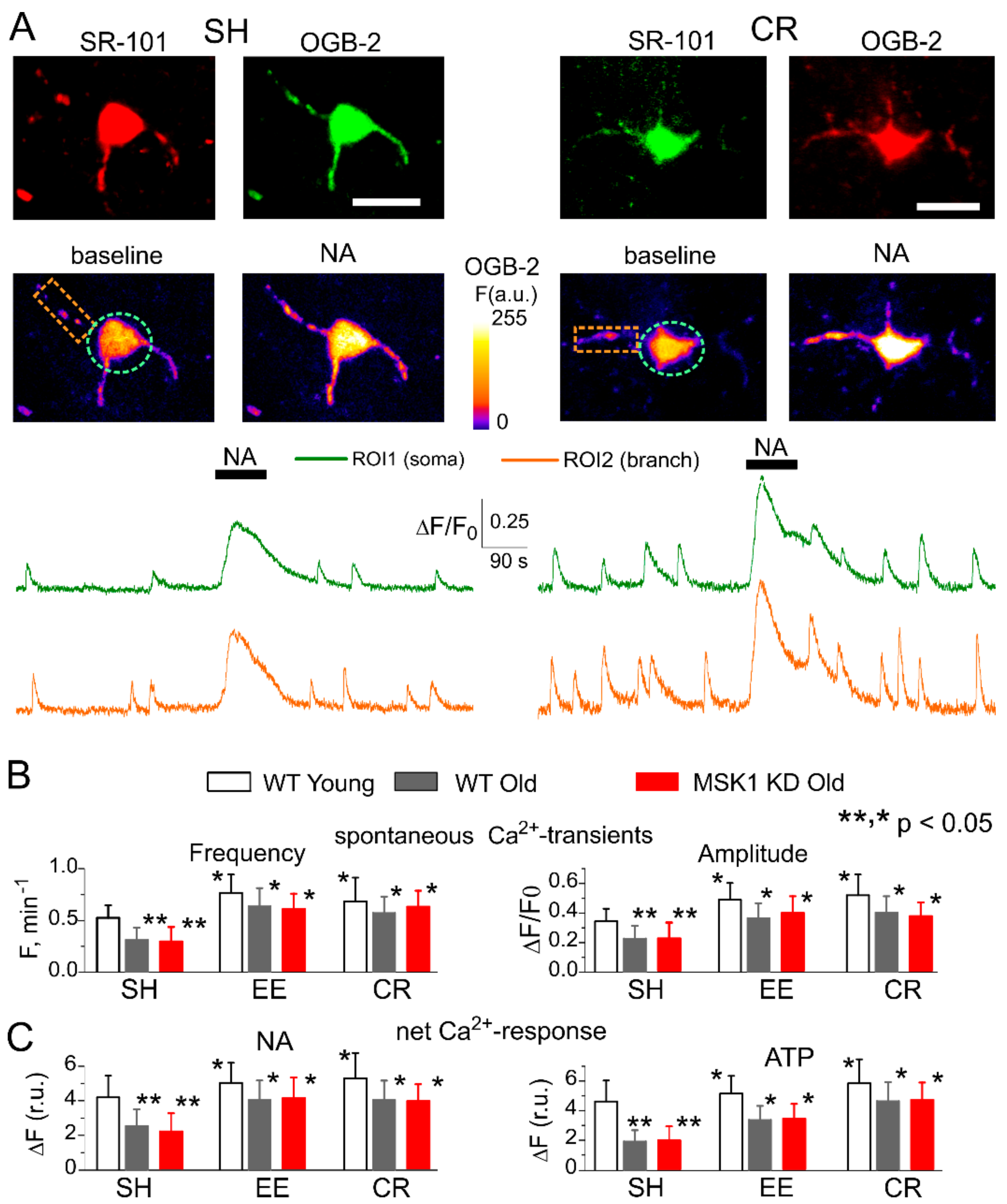
First, we verified that EE and CR can enhance Ca2+-signalling in neocortical astrocytes of MSK1 KD mice. Astroglial Ca2+-signalling was monitored using multiphoton fluorescence microscopy as described previously [20,22,23]. We measured spontaneous cytosolic Ca2+ transients in the branches and soma of neocortical astrocytes of 6–12 week-old (young adults) and 9–15 months old (old) mice (Figure 3). There was no significant difference in Ca2+-signalling between neocortical astrocytes of old wild-type and MSK1 KD mice (Figure 3). Similar to our previous reports, the amplitude and frequency of spontaneous astrocytic Ca2+-transients in old WT and MSK1 KD mice raised in standard housing (SH) were significantly lower than those measured in the young SH WT mice. Most importantly, EE and CR significantly increased spontaneous astroglial Ca2+-signalling in the old WT and MSK1 KD mice to a similar extent (Figure 3A,B).
We also assessed the responses of astrocytes to exogenous activation of NA and ATP receptors since these neurotransmitters are released during enhanced neuronal and physical activity [21,33,34] and potentially can mediate a link between environmental enrichment and the function of astrocytes. There is also growing evidence of the importance of α1AR and P2Y receptors for astrocytic Ca2+-signalling and glia–neuron interactions [21,22,33,34]. EE and CR had a moderate effect on the amplitudes of astrocytic responses to NA (3 µM) and ATP (30 µM) in the young mice, but caused a significant enhancement of astroglial responses at the older age, both in the WT and MSK1 KD mice (Figure 3C).
These results have verified that Ca2+-signalling in astrocytes, and especially the effects of EE and CR, are not affected by MSK1 KD knock-in. We have previously demonstrated that astroglial Ca2+-signalling is not altered in the dnSNARE mice either [19,20,23]. Hence, if any difference in the impact of EE and CR on synaptic transmission in the dnSNARE and MSK1 KD mice were observed, they should be attributed not to the deficit in astroglial Ca2+-elevation, but rather to the impairment of downstream signalling cascades, namely astroglial exocytosis and BDNF-mediated regulation of neuronal synaptic strength.
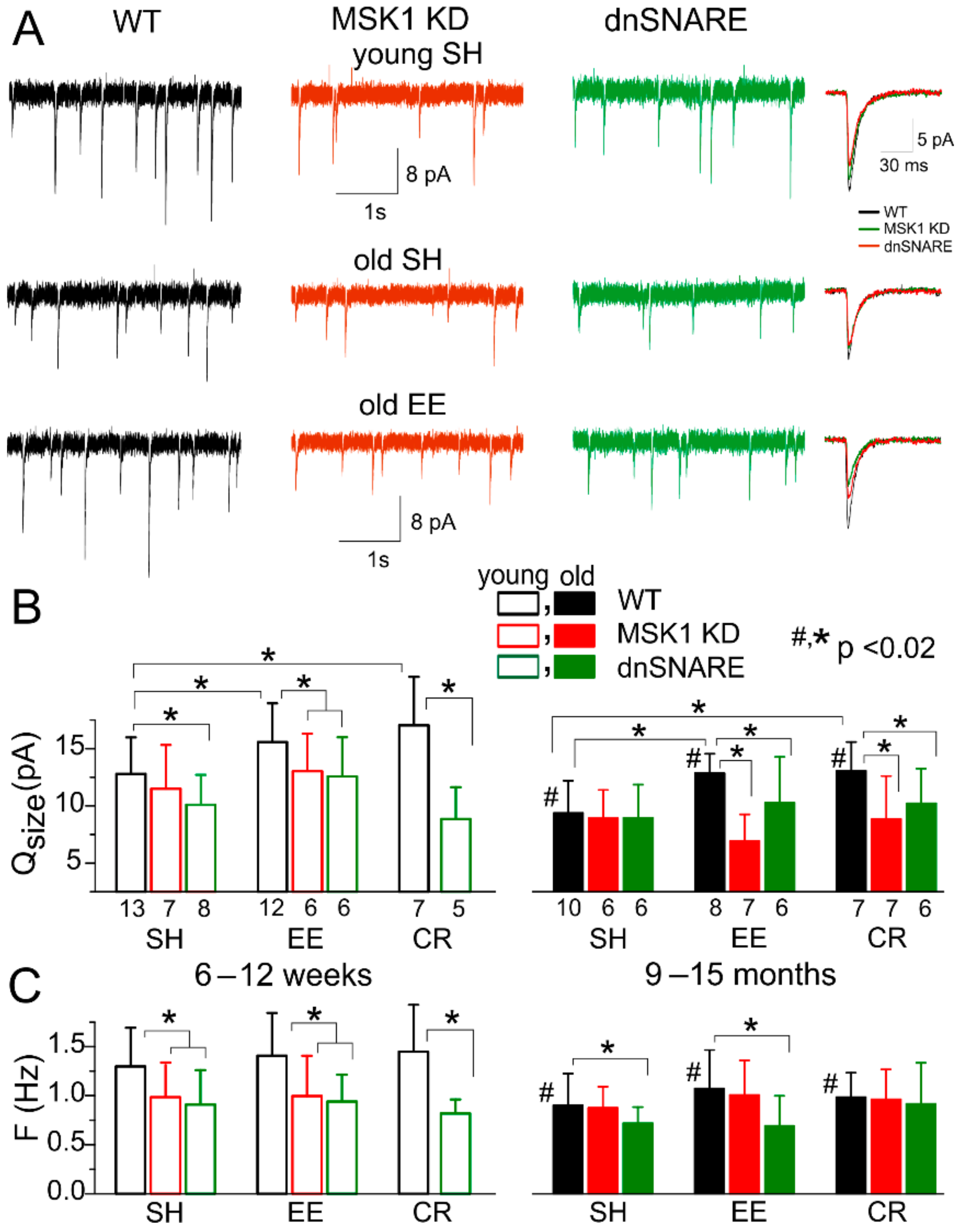
To test this, we recorded AMPA receptor-mediated mEPSCs in neocortical pyramidal neurons in the presence of the GABAA receptor antagonist picrotoxin (100 µM), and the P2 Receptor antagonist PPADS (10 µM). Exposure of wild-type mice to EE from birth to 6–12 weeks increased the average amplitude of mEPSCs to 18.6 ± 6.04 pA (n = 12), as compared to 21.3 ± 5.97 (n = 13) in the SH mice. The difference between the EE and SH mice was even larger for the older mice (Figure 4A,B), which might be related to the age-related decline in the amplitude of excitatory synaptic currents. In the older mice, the mEPSC amplitude increased from 10.86 ± 3.55 pA (SH, n = 10) to 14.34 ± 3.47 pA (EE, n = 8).
Exposure to CR had similar positive effect on mEPSCs (Figure 4B). The observed effects of both EE and CR were, most likely, of postsynaptic origin since they were accompanied by a significant increase in quantal size, whereas the frequency of mEPSCs did not undergo considerable changes (Figure 4C).
In stark contrast to the WT mice, mEPSCs recorded from MSK1 KD mice undergo the same age-related decline, but did not exhibit the EE- or CR-induced increase in amplitude (Figure 4A,B). In line with data obtained in the MSK1 KD mice, the dnSNARE mice showed only modest EE- and CR-induced increase in the average mEPSCs amplitude. The difference in the quantal size of mEPSCs in the WT and dnSNARE mice was statistically significant only for the EE in the younger age (p < 0.05). The incomplete inhibition of EE-induced plasticity in the young dnSNARE mice could be attributed to the mosaic (50–60% of cells) expression of dnSNARE and therefore incomplete loss of glial exocytosis. In addition, neuronal release of BDNF cannot be excluded.
The dependence of EE- and CR-induced upscaling of excitatory synaptic currents on the MSK1 signalling pathway and astrocytic exocytosis closely agrees with our results obtained in cell cultures (Figure 1 and Figure 2). Combined together, our in vitro and ex vivo data strongly support the crucial importance of astroglial release of BDNF for homeostatic plasticity of excitatory synaptic transmission.
Finally, we explored the experience-dependent plasticity of inhibitory synaptic signalling. We recorded GABA receptor-mediated miniature inhibitory synaptic currents (mIPSCs) at a membrane potential of −80 mV in the presence of glutamate and P2X receptor antagonists (DNQX and PPADS, respectively). The pattern of age- and environment-related alterations in the GABAergic synaptic currents was different to the changes exhibited by mEPSCs. Firstly, both the quantal size and frequency of mIPSCs undergo dramatic increase in older WT and the MSK1 KD mice (Figure 5). In addition, GABAergic currents were upregulated in the dnSNARE mice of both age groups as compared to their WT-littermates (Figure 5A,B); this result is in line with our previous observations [19]. Secondly, exposure of the wild-type mice to EE and CR efficiently downregulated inhibitory synaptic signalling, especially in the neurons of older mice (Figure 5A,B). In contrast to mEPSCs, the EE- and CR-induced downscaling of mIPSCs was absent in the dnSNARE but not in the MSK1 KD mice. This might be explained by participation of other gliotransmitters, in particular ATP [19], in astroglial-driven modulation of GABA receptors.
Combined, our data strongly support the importance of astrocytic exocytosis and BDNF/MSK1-mediated signalling for the beneficial effects of EE and CR on synaptic transmission in the ageing brain.
4. Discussion
Our experiments in cultured neurons demonstrated that long-term enhancement of astrocytic Ca2+-signalling can lead to neuronal synaptic scaling that engages the same BDNF/MSK1 cascade that has been shown to underlie homeostatic synaptic plasticity induced by alterations in neuronal firing [9,10]. The astrocyte-induced alterations in synaptic strength (Figure 1) and morphology (Figure 2) depend on the activity of BDNF TrkB receptors and MSK1. Combined with recently reported data unequivocally showing the ability of astrocytes to release BDNF via exocytosis [14], our results strongly suggest that astrocyte-derived BDNF can modulate homeostatic synaptic scaling.
Our ex vivo data also highlighted the importance of astroglial-derived BDNF and the BDNF/MSK1 pathway for experience-dependent homeostatic synaptic plasticity. Firstly, we observed that exposure of mice to EE and CR can induce an increase in glutamatergic (Figure 4), and a decrease in GABAergic synaptic transmission (Figure 5). The impact of EE and CR was significantly reduced in dnSNARE mice, indicating the critical importance of astrocytic exocytosis, which is very likely the main pathway of BDNF release [14]. Secondly, the effects of EE and CR on synaptic transmission and plasticity differed significantly in the wild-type and MSK1 KD mice (Figure 4 and Figure 5), where one of the main pathways of BDNF-mediated homeostatic plasticity was impaired [9]. Finally, the exposure to EE and CR led to significant upregulation of Ca2+-signalling in astrocytes, which did not differ in the WT and transgenic mice. The most likely explanation for all our observations of changes in glutamatergic mEPSCs would be the enhancement of Ca2+-dependent release of BDNF from astrocytes which, in turn, upregulates the strength of excitatory synapses via MSK1-dependent cascade.
As for the homeostatic plasticity of GABAergic transmission, it was only weakly affected by the impairment of the BDNF/MSK1 pathway, but was critically dependent on astroglial exocytosis. These results highlight an important difference in astroglial-driven homeostatic regulation of excitatory and inhibitory synaptic transmission: the former is modulated mainly via the BDNF/MSK1 pathway whereas the latter is modulated, most likely, by other gliotransmitters. As our previous results suggest, the vesicular release of ATP can play an important role in astroglial-driven modulation of GABA receptors [20].
Our data highlighted interesting trends in age-related alterations in synaptic transmission: a decrease in the efficacy of excitatory synapses, and an increase in the efficacy of inhibitory synapses, but neither were accompanied by marked change in the frequency of synaptic events. These observations suggest that, at least at early stages of ageing, neurons retain a significant number of functional synapses, and cognitive decline can be associated with dysregulation, rather than complete loss of synapses. Furthermore, our present (Figure 3) and previous data [19,35,36] on age-related decline in astroglial Ca2+-signalling and the release of gliotransmitters suggest a very plausible reason of such dysregulation—impairment of glia-driven modulation of synaptic activity. This hypothesis is strongly supported by observation that manipulations which facilitate astroglial signalling, e.g., EE, CR, reversed the changes in excitatory and inhibitory synaptic signalling back to a “younger” state (Figure 4 and Figure 5). Also, the notion of impairment of glial regulation of balance between the excitatory and inhibitory signals onto principal neurons as a putative cause of age-related cognitive decline agrees with our previous observation that enhancement of astrocytic Ca2+-signalling, including EE- and CR-induced, can rescue long-term synaptic plasticity in old mice [19,35,36]. Nonetheless, this model of age-related alterations of synaptic function needs to be explored further, in particular in relation to neurodegenerative disorders.
To conclude, our results strongly support the physiological importance of astroglial exocytosis, in particular the release of BDNF, for communication between astrocytes and neurons and experience-related synaptic plasticity across a lifetime.
Author Contributions
Conceptualization, B.G.F. and Y.P.; Data Curation, U.L., G.W.J.M., and Y.P.; Formal Analysis, U.L., A.B., and Y.P.; Investigation, U.L. and A.B.; Methodology, G.W.J.M., B.G.F., and Y.P.; Resources, G.W.J.M. and B.G.F.; Supervision, Y.P.; Writing—original draft, U.L. and Y.P.
Funding
This work was supported by the BBSRC UK grants number BB/K009192/1 to Y.P. and BB/F021445/1 to B.G.F. and Y.P. and BB/D01817X/1 to G.W.J.M.
Acknowledgments
The authors thank Professor J. Simon Arthur (University of Dundee) for developing and providing MSK1 KD mice.
Conflicts of Interest
Authors declare no conflicts of interests
References
- Baroncelli, L.; Braschi, C.; Spolidoro, M.; Begenisic, T.; Sale, A.; Maffei, L. Nurturing brain plasticity: Impact of environmental enrichment. Cell Death Differ. 2010, 17, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Turrigiano, G.G. Strength through diversity. Neuron 2008, 60, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Nithianantharajah, J.; Hannan, A.J. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat. Rev. Neurosci. 2006, 7, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Hillman, C.H.; Erickson, K.I.; Kramer, A.F. Be smart, exercise your heart: Exercise effects on brain and cognition. Nat. Rev. Neurosci. 2008, 9, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Mercken, E.M.; Carboneau, B.A.; Krzysik-Walker, S.M.; de Cabo, R. Of mice and men: The benefits of caloric restriction, exercise, and mimetics. Ageing Res. Rev. 2012, 11, 390–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merzenich, M.M.; Van Vleet, T.M.; Nahum, M. Brain plasticity-based therapeutics. Front. Hum. Neurosci. 2014, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Nithianantharajah, J.; Hannan, A.J. The neurobiology of brain and cognitive reserve: Mental and physical activity as modulators of brain disorders. Prog. Neurobiol. 2009, 89, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H. Exercise and the brain: Something to chew on. Trends Neurosci. 2009, 32, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.A.; Hunter, C.J.; Palygin, O.; Wauters, S.C.; Martin, K.J.; McKenzie, C.; McKelvey, K.; Morris, R.G.; Pankratov, Y.; Arthur, J.S.; et al. MSK1 regulates homeostatic and experience-dependent synaptic plasticity. J. Neurosci. 2012, 32, 13039–13051. [Google Scholar] [CrossRef] [PubMed]
- Turrigiano, G.G.; Leslie, K.R.; Desai, N.S.; Rutherford, L.C.; Nelson, S.B. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 1998, 391, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Cowansage, K.K.; LeDoux, J.E.; Monfils, M.H. Brain-derived neurotrophic factor: A dynamic gatekeeper of neural plasticity. Curr. Mol. Pharmacol. 2010, 3, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Griffioen, K.J.; Wan, R.; Mattson, M.P. Brain-derived neurotrophic factor as a regulator of systemic and brain energy metabolism and cardiovascular health. Ann. N. Y. Acad. Sci. 2012, 1264, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.H.; Yu, J.T.; Tan, L. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease: Risk, Mechanisms, and Therapy. Mol. Neurobiol. 2015, 52, 1477–1493. [Google Scholar] [CrossRef] [PubMed]
- Stenovec, M.; Lasic, E.; Bozic, M.; Bobnar, S.T.; Stout, R.F., Jr.; Grubisic, V.; Parpura, V.; Zorec, R. Ketamine Inhibits ATP-Evoked Exocytotic Release of Brain-Derived Neurotrophic Factor from Vesicles in Cultured Rat Astrocytes. Mol. Neurobiol. 2016, 53, 6882–6896. [Google Scholar] [CrossRef] [PubMed]
- Vignoli, B.; Battistini, G.; Melani, R.; Blum, R.; Santi, S.; Berardi, N.; Canossa, M. Peri-Synaptic Glia Recycles Brain-Derived Neurotrophic Factor for LTP Stabilization and Memory Retention. Neuron 2016, 92, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 2015. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Astroglial cradle in the life of the synapse. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.J.; Terzieva, S.; Olabarria, M.; Lanza, R.G.; Verkhratsky, A. Enriched environment and physical activity reverse astrogliodegeneration in the hippocampus of AD transgenic mice. Cell Death Dis. 2013, 4, e678. [Google Scholar] [CrossRef]
- Lalo, U.; Bogdanov, A.; Pankratov, Y. Diversity of Astroglial Effects on Aging- and Experience-Related Cortical Metaplasticity. Front. Mol. Neurosci. 2018, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Palygin, O.; Rasooli-Nejad, S.; Andrew, J.; Haydon, P.G.; Pankratov, Y. Exocytosis of ATP from astrocytes modulates phasic and tonic inhibition in the neocortex. PLoS Biol. 2014, 12, e1001747. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Palygin, O.; Verkhratsky, A.; Grant, S.G.; Pankratov, Y. ATP from synaptic terminals and astrocytes regulates NMDA receptors and synaptic plasticity through PSD-95 multi-protein complex. Sci. Rep. 2016, 6, 33609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankratov, Y.; Lalo, U. Role for astroglial alpha1-adrenoreceptors in gliotransmission and control of synaptic plasticity in the neocortex. Front. Cell. Neurosci. 2015, 9, 230. [Google Scholar] [CrossRef] [PubMed]
- Rasooli-Nejad, S.; Palygin, O.; Lalo, U.; Pankratov, Y. Cannabinoid receptors contribute to astroglial Ca2+-signalling and control of synaptic plasticity in the neocortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20140077. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Pankratov, Y. Exploring the Ca2+-dependent synaptic dynamics in vibro-dissociated cells. Cell Calcium 2017, 64, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Li, C.; Shevchuk, A.I.; Stepanyan, R.; Caldwell, M.; Hughes, S.; Smart, T.G.; Gorelik, J.; Ostanin, V.P.; Lab, M.J.; et al. Nanoscale live-cell imaging using hopping probe ion conductance microscopy. Nat. Methods 2009, 6, 279–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikora, A.; Rodak, A.; Unold, O.; Klapetek, P. The development of the spatially correlated adjustment wavelet filter for atomic force microscopy data. Ultramicroscopy 2016, 171, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, P.; Liu, Y.; Hu, Y.; Wang, T.; Zhao, Y.P.; Liu, Q.S. BDNF interacts with endocannabinoids to regulate cocaine-induced synaptic plasticity in mouse midbrain dopamine neurons. J. Neurosci. 2015, 35, 4469–4481. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, E.; Kanhema, T.; Soule, J.; Tiron, A.; Dagyte, G.; da Silva, B.; Bramham, C.R. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J. Neurosci. 2007, 27, 10445–10455. [Google Scholar] [CrossRef] [PubMed]
- Waung, M.W.; Pfeiffer, B.E.; Nosyreva, E.D.; Ronesi, J.A.; Huber, K.M. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 2008, 59, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Korchev, Y.E.; Negulyaev, Y.A.; Edwards, C.R.; Vodyanoy, I.; Lab, M.J. Functional localization of single active ion channels on the surface of a living cell. Nat. Cell Biol. 2000, 2, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Galluzzi, L.; Freije, J.M.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef] [PubMed]
- Paukert, M.; Agarwal, A.; Cha, J.; Doze, V.A.; Kang, J.U.; Bergles, D.E. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 2014, 82, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, R.; Jahn, H.M.; Courjaret, R.; Messemer, N.; Kirchhoff, F.; Deitmer, J.W. The inhibitory input to mouse cerebellar Purkinje cells is reciprocally modulated by Bergmann glial P2Y1 and AMPA receptor signaling. Glia 2016, 64, 1265–1280. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Palygin, O.; North, R.A.; Verkhratsky, A.; Pankratov, Y. Age-dependent remodelling of ionotropic signalling in cortical astroglia. Aging Cell 2011, 10, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalo, U.; Rasooli-Nejad, S.; Pankratov, Y. Exocytosis of gliotransmitters from cortical astrocytes: Implications for synaptic plasticity and aging. Biochem. Soc. Trans. 2014, 42, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mitogen- and stress-activated protein kinase 1 and brain-derived neurotrophic factor mediate astroglia-induced synaptic scaling in hippocampal neurons. (A) Synaptic scaling was induced in cultured hippocampal pyramidal neurons via incubation with the selective astroglia activator TFLLR (PAR-1 receptor agonist). Upper and middle panels show representative mEPSCs from wild-type neurons in control media (upper left) and after 24 h incubation with either 3 µM TFLLR alone (upper right) or the TrkB inhibitor Cyclotraxin B (CTX B) alone (middle left) or TFLLR and Cyclotraxin B together (middle right). Bottom panels show mEPSCs recorded in neurons of MSK1 KD mice. The insets on the right show corresponding average mEPSC waveforms. Note that incubation with TFLLR upregulated only mEPSCs recorded in the wild-type mice under control conditions. Synaptic currents were recorded at a membrane potential of −80 mV in the presence of picrotoxin (100 µM), TTX (1 µM), and PPADS (10 µM). (B) Corresponding amplitude distributions recorded in the same neurons as in (A). Note that the unitary quantal size, indicated by the position of the main peak in the amplitude distribution, undergoes a considerable increase, suggesting a postsynaptic effect of exposure to TFLLR in the wild-type, but not MSK1 KD neurons or in the WT neurons in the presence of BDNF TrkB receptor blocker. (C) Bar graphs show the quantal size and average frequency of mEPSCs recorded as described above. Data show mean ± SD for 9–12 neurons from three to four primary neuronal cultures. Note the significant increase (p < 0.005, unpaired t-test) in the quantal mEPSC amplitude in wild-type neurones after incubation with TFLLR. The lack of significant changes in the mEPSC frequency supports the postsynaptic origin of the effect. In contrast, activation of astrocytes with TFLLR did not cause significant alterations in mEPSCs in MSK KD1 neurons or WT neurons in the presence of BDNF TrkB receptor blocker. * p < 0.005.
Figure 1.
Mitogen- and stress-activated protein kinase 1 and brain-derived neurotrophic factor mediate astroglia-induced synaptic scaling in hippocampal neurons. (A) Synaptic scaling was induced in cultured hippocampal pyramidal neurons via incubation with the selective astroglia activator TFLLR (PAR-1 receptor agonist). Upper and middle panels show representative mEPSCs from wild-type neurons in control media (upper left) and after 24 h incubation with either 3 µM TFLLR alone (upper right) or the TrkB inhibitor Cyclotraxin B (CTX B) alone (middle left) or TFLLR and Cyclotraxin B together (middle right). Bottom panels show mEPSCs recorded in neurons of MSK1 KD mice. The insets on the right show corresponding average mEPSC waveforms. Note that incubation with TFLLR upregulated only mEPSCs recorded in the wild-type mice under control conditions. Synaptic currents were recorded at a membrane potential of −80 mV in the presence of picrotoxin (100 µM), TTX (1 µM), and PPADS (10 µM). (B) Corresponding amplitude distributions recorded in the same neurons as in (A). Note that the unitary quantal size, indicated by the position of the main peak in the amplitude distribution, undergoes a considerable increase, suggesting a postsynaptic effect of exposure to TFLLR in the wild-type, but not MSK1 KD neurons or in the WT neurons in the presence of BDNF TrkB receptor blocker. (C) Bar graphs show the quantal size and average frequency of mEPSCs recorded as described above. Data show mean ± SD for 9–12 neurons from three to four primary neuronal cultures. Note the significant increase (p < 0.005, unpaired t-test) in the quantal mEPSC amplitude in wild-type neurones after incubation with TFLLR. The lack of significant changes in the mEPSC frequency supports the postsynaptic origin of the effect. In contrast, activation of astrocytes with TFLLR did not cause significant alterations in mEPSCs in MSK KD1 neurons or WT neurons in the presence of BDNF TrkB receptor blocker. * p < 0.005.

Figure 2.
Astroglia and brain-derived neurotrophic factor (BDNF) regulate synaptic size. (A) The principle of tomographic imaging of synaptic boutons using high-resolution hopping probe scanning ion conductance microscopy (SICM). Synapses of live hippocampal neurons were prestained with synaptic vesicle marker FM1-43. (B) High-resolution SICM 3D images of hippocampal synapses of wild-type neurons in control and after 24 h incubation with 3 µM TFLLR. The X- and Y-scales are indicated on the images, the Z-scale is indicated as pseudo-colour (same scale for both graphs). (C) The pooled data on the volume (V; µm3 ) and size (ΔL, µm) of synaptic boutons of wild-type neurons in control and after 24 h-incubation with the PAR1 agonist TFLLR alone, TFLLR and the TrkB antagonist Cyclotraxin B (CTX B), and BDNF. Open symbols indicate individual boutons, the closed symbols show mean ± standard deviation for the whole group (n = 30–35 boutons for three preparations). Note the significant increase in the size and volume of synapses after incubation with either TFLLR or BDNF.
Figure 2.
Astroglia and brain-derived neurotrophic factor (BDNF) regulate synaptic size. (A) The principle of tomographic imaging of synaptic boutons using high-resolution hopping probe scanning ion conductance microscopy (SICM). Synapses of live hippocampal neurons were prestained with synaptic vesicle marker FM1-43. (B) High-resolution SICM 3D images of hippocampal synapses of wild-type neurons in control and after 24 h incubation with 3 µM TFLLR. The X- and Y-scales are indicated on the images, the Z-scale is indicated as pseudo-colour (same scale for both graphs). (C) The pooled data on the volume (V; µm3 ) and size (ΔL, µm) of synaptic boutons of wild-type neurons in control and after 24 h-incubation with the PAR1 agonist TFLLR alone, TFLLR and the TrkB antagonist Cyclotraxin B (CTX B), and BDNF. Open symbols indicate individual boutons, the closed symbols show mean ± standard deviation for the whole group (n = 30–35 boutons for three preparations). Note the significant increase in the size and volume of synapses after incubation with either TFLLR or BDNF.

Figure 3.
Age- and experience-dependent changes in astrocytic Ca2+ signalling. Astroglial Ca2+-signalling was evaluated in the neocortex of 6–12 week-old (young) and 9–15 month-old mice (old) as described previously [26,27,28]. The MSK1 KD and their wild-type littermates were kept either in standard housing (SH) or exposed to environmental enrichment (EE) or caloric restriction (CR) as described in the Methods. (A) Representative multiphoton images of astrocytes of old MSK1 KD mouse preloaded with Oregon Green BAPTA-2 AM (OGB-2) and stained with fluorescent astroglial marker SR101 and pseudo-colour images of OGB-2 fluorescence recorded before and after application of noradrenaline (NA, 1 μM). Graphs below show the time course of OGB-2 fluorescence averaged over regions indicated in the fluorescent images. Note the increase in the amplitude and frequency of spontaneous Ca2+-elevations and in response to NA. (B) The pooled data on peak amplitude and frequency of the baseline spontaneous Ca2+-transients recorded in astrocytes of WT, MSK1, and dn-SNARE mice of different ages and treatments. Number and size of spontaneous events were pooled for the whole cell image. (C) The pooled data on the net responses to application of 1 μM noradrenaline and 10 μM ATP. Net response was evaluated as an integral Ca2+-signal measured within 3 min after stimulation, averaged over the whole cell image and normalized to the baseline integral Ca2+ signal. Data in the panels (B, C) are shown as mean ± SD for the 6 to 12 astrocytes from 3 to 4 animals. Asterisks (*, **) correspondingly indicate statistical significance (p < 0.05) of the effect of EE or CR treatment (as compared to SH) and difference between the old and young mice of the same treatment group and genotype. Note the significant increase in spontaneous and evoked Ca2+-signalling in astrocytes of mice exposed to EE and CR, and the lack of difference in Ca2+-signalling in the WT, MSK1 KD, and dnSNARE mice.
Figure 3.
Age- and experience-dependent changes in astrocytic Ca2+ signalling. Astroglial Ca2+-signalling was evaluated in the neocortex of 6–12 week-old (young) and 9–15 month-old mice (old) as described previously [26,27,28]. The MSK1 KD and their wild-type littermates were kept either in standard housing (SH) or exposed to environmental enrichment (EE) or caloric restriction (CR) as described in the Methods. (A) Representative multiphoton images of astrocytes of old MSK1 KD mouse preloaded with Oregon Green BAPTA-2 AM (OGB-2) and stained with fluorescent astroglial marker SR101 and pseudo-colour images of OGB-2 fluorescence recorded before and after application of noradrenaline (NA, 1 μM). Graphs below show the time course of OGB-2 fluorescence averaged over regions indicated in the fluorescent images. Note the increase in the amplitude and frequency of spontaneous Ca2+-elevations and in response to NA. (B) The pooled data on peak amplitude and frequency of the baseline spontaneous Ca2+-transients recorded in astrocytes of WT, MSK1, and dn-SNARE mice of different ages and treatments. Number and size of spontaneous events were pooled for the whole cell image. (C) The pooled data on the net responses to application of 1 μM noradrenaline and 10 μM ATP. Net response was evaluated as an integral Ca2+-signal measured within 3 min after stimulation, averaged over the whole cell image and normalized to the baseline integral Ca2+ signal. Data in the panels (B, C) are shown as mean ± SD for the 6 to 12 astrocytes from 3 to 4 animals. Asterisks (*, **) correspondingly indicate statistical significance (p < 0.05) of the effect of EE or CR treatment (as compared to SH) and difference between the old and young mice of the same treatment group and genotype. Note the significant increase in spontaneous and evoked Ca2+-signalling in astrocytes of mice exposed to EE and CR, and the lack of difference in Ca2+-signalling in the WT, MSK1 KD, and dnSNARE mice.

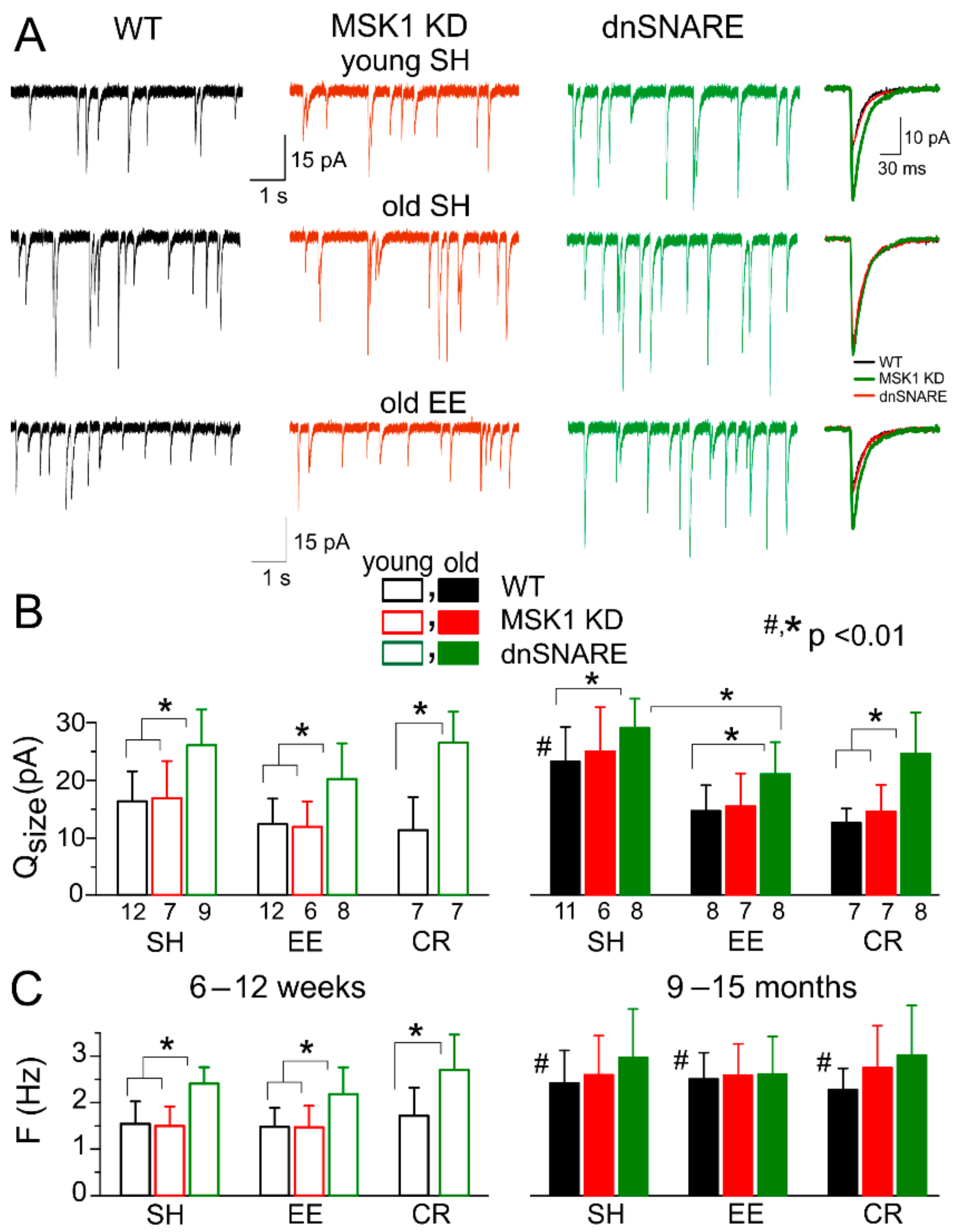
Figure 4.
Experience-dependent alterations in excitatory synaptic transmission. AMPA receptor-mediated spontaneous miniature excitatory postsynaptic currents (mEPSCs) were recorded in neocortical layer 2/3 pyramidal neurons at −80 mV in the presence of 100 µM picrotoxin, 1 µM TTX and 10 µM PPADS. (A) Representative whole-cell currents recorded in young (top row) and old SH (middle row) and EE (bottom row) WT, MSK1 KD and dnSNARE mice (left, middle, and right columns, respectively). The inserts on the right shows average mEPSC waveforms. Note the significant decrease in mEPSC amplitude in the old WT mice of standard housing, and upregulation of mEPSCs in the EE mice of the same age. The effect of EE is impaired in the MSK1 KD and dnSNARE mice. (B, C) Pooled data on the quantal size (B) and frequency (C) of mEPSCs in mice of different age and experience groups. Data are shown as mean ± SD for the number of neurons indicated in (B). The statistical significance (2-population unpaired t-test) for the difference between young and old mice of same genotype and experience is indicated by (#) symbols; asterisks (*) indicate statistical significance of the difference between different genotype and experience groups as indicated.
Figure 4.
Experience-dependent alterations in excitatory synaptic transmission. AMPA receptor-mediated spontaneous miniature excitatory postsynaptic currents (mEPSCs) were recorded in neocortical layer 2/3 pyramidal neurons at −80 mV in the presence of 100 µM picrotoxin, 1 µM TTX and 10 µM PPADS. (A) Representative whole-cell currents recorded in young (top row) and old SH (middle row) and EE (bottom row) WT, MSK1 KD and dnSNARE mice (left, middle, and right columns, respectively). The inserts on the right shows average mEPSC waveforms. Note the significant decrease in mEPSC amplitude in the old WT mice of standard housing, and upregulation of mEPSCs in the EE mice of the same age. The effect of EE is impaired in the MSK1 KD and dnSNARE mice. (B, C) Pooled data on the quantal size (B) and frequency (C) of mEPSCs in mice of different age and experience groups. Data are shown as mean ± SD for the number of neurons indicated in (B). The statistical significance (2-population unpaired t-test) for the difference between young and old mice of same genotype and experience is indicated by (#) symbols; asterisks (*) indicate statistical significance of the difference between different genotype and experience groups as indicated.

Figure 5.
Experience-dependent alterations in inhibitory synaptic transmission. GABAA receptor-mediated miniature inhibitory postsynaptic currents (mIPSCs) were recorded in neocortical layer 2/3 pyramidal neurons at −80 mV in the presence of 30 µM DNQX, 1 µM TTX, and 10 µM PPADS. (A) Representative whole-cell currents recorded in young (top row) and old SH (middle row) and EE (bottom row) WT, MSK1 KD, and dnSNARE mice (left, middle, and right columns, respectively). The inserts on the right shows average mIPSCs waveforms. (B, C) Pooled data on the quantal size (B) and frequency (C) of mIPSCs in mice of different age and experience groups. Data are shown as mean ± SD for the number of neurons indicated in (B). The statistical significance (2-population unpaired t-test) for the difference between young and old mice of same genotype and experience is indicated by (#) symbols; asterisks (*) indicate statistically significance of the difference between different genotype and experience groups as indicated. Note the significant increase in mIPSC amplitude in the old standard housed WT mice and down-regulation of mIPSCs in the EE mice of the same age. The effect of EE is impaired in the MSK1 KD and dnSNARE mice.
Figure 5.
Experience-dependent alterations in inhibitory synaptic transmission. GABAA receptor-mediated miniature inhibitory postsynaptic currents (mIPSCs) were recorded in neocortical layer 2/3 pyramidal neurons at −80 mV in the presence of 30 µM DNQX, 1 µM TTX, and 10 µM PPADS. (A) Representative whole-cell currents recorded in young (top row) and old SH (middle row) and EE (bottom row) WT, MSK1 KD, and dnSNARE mice (left, middle, and right columns, respectively). The inserts on the right shows average mIPSCs waveforms. (B, C) Pooled data on the quantal size (B) and frequency (C) of mIPSCs in mice of different age and experience groups. Data are shown as mean ± SD for the number of neurons indicated in (B). The statistical significance (2-population unpaired t-test) for the difference between young and old mice of same genotype and experience is indicated by (#) symbols; asterisks (*) indicate statistically significance of the difference between different genotype and experience groups as indicated. Note the significant increase in mIPSC amplitude in the old standard housed WT mice and down-regulation of mIPSCs in the EE mice of the same age. The effect of EE is impaired in the MSK1 KD and dnSNARE mice.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lalo, U.; Bogdanov, A.; Moss, G.W.J.; Frenguelli, B.G.; Pankratov, Y. Role for Astroglia-Derived BDNF and MSK1 in Homeostatic Synaptic Plasticity. Neuroglia 2018, 1, 381-394. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020026
AMA Style
Lalo U, Bogdanov A, Moss GWJ, Frenguelli BG, Pankratov Y. Role for Astroglia-Derived BDNF and MSK1 in Homeostatic Synaptic Plasticity. Neuroglia. 2018; 1(2):381-394. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020026
Chicago/Turabian StyleLalo, Ulyana, Alexander Bogdanov, Guy W. J. Moss, Bruno G. Frenguelli, and Yuriy Pankratov. 2018. "Role for Astroglia-Derived BDNF and MSK1 in Homeostatic Synaptic Plasticity" Neuroglia 1, no. 2: 381-394. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020026