Silibinin Restores NAD+ Levels and Induces the SIRT1/AMPK Pathway in Non-Alcoholic Fatty Liver

 ,
,  , , and
, , and 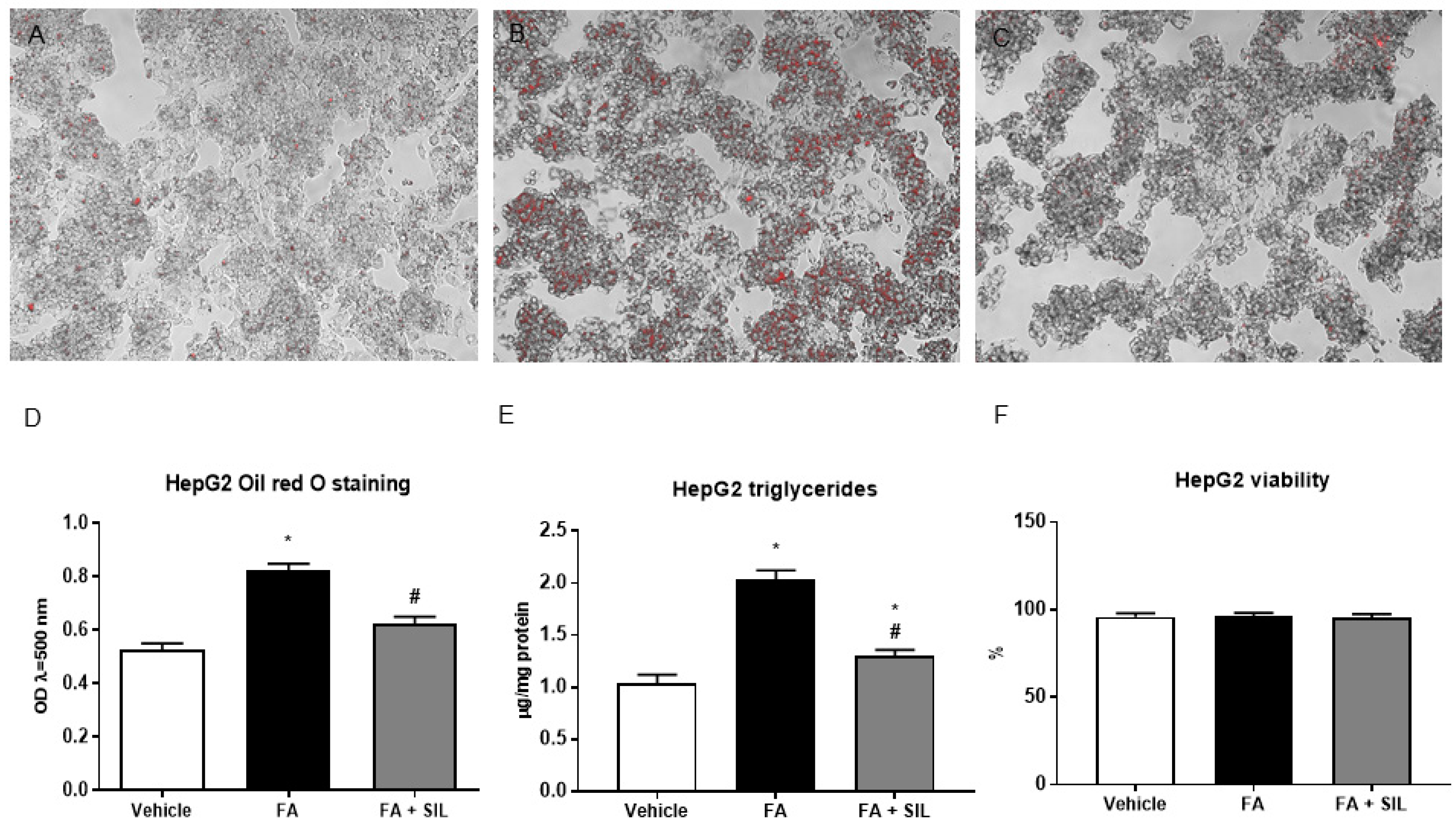{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Animals
2.3. Oil Red O and Hematoxylin/Eosin Staining
2.4. Biochemical Analyses
2.5. Real-Time PCR
2.6. ROS (Reactive Oxygen Species) Measurement
2.7. Statistical Analysis
3. Results
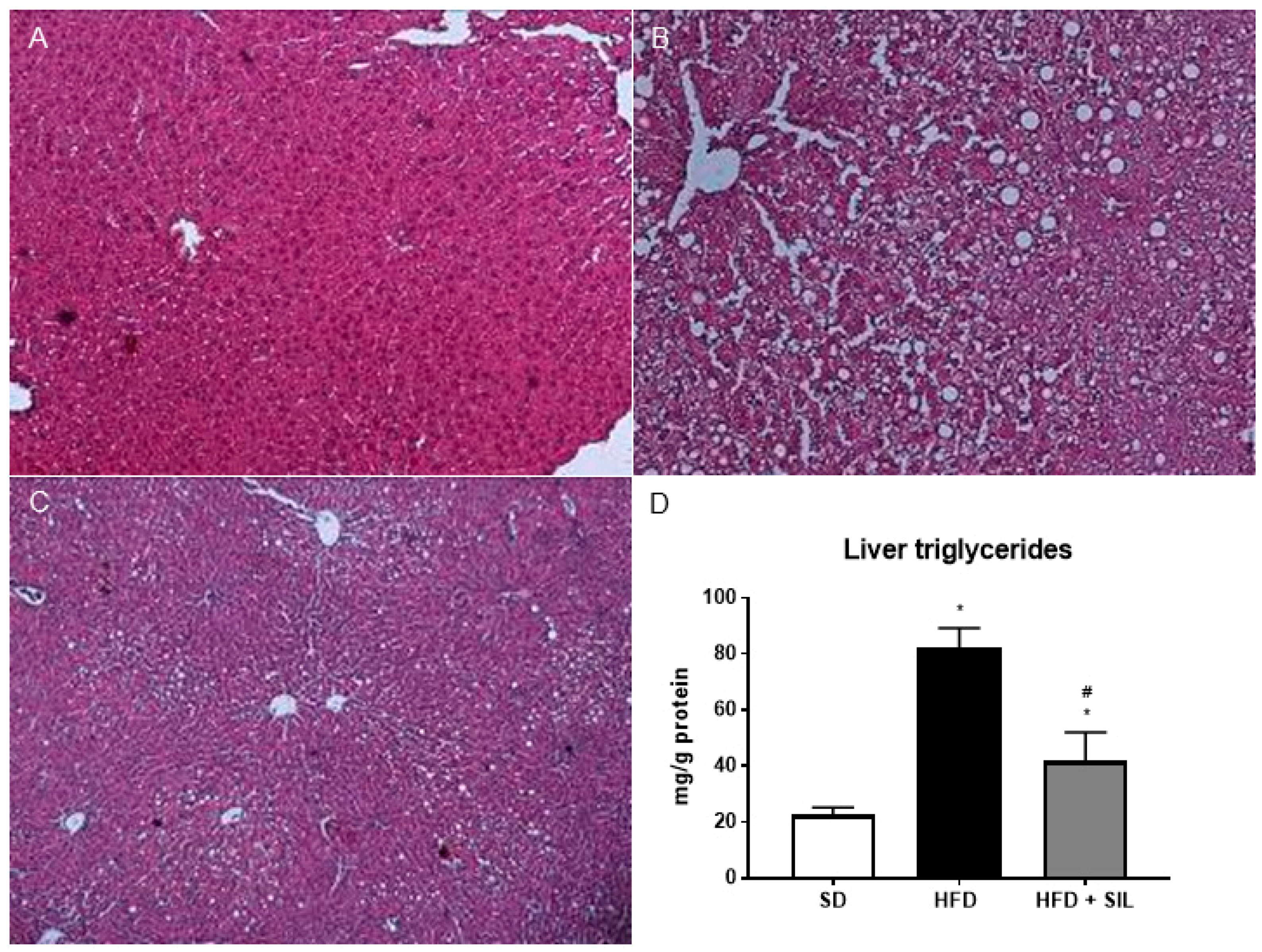
3.1. Silibinin Reduces Lipid Accumulation in HepG2 and Mice Liver
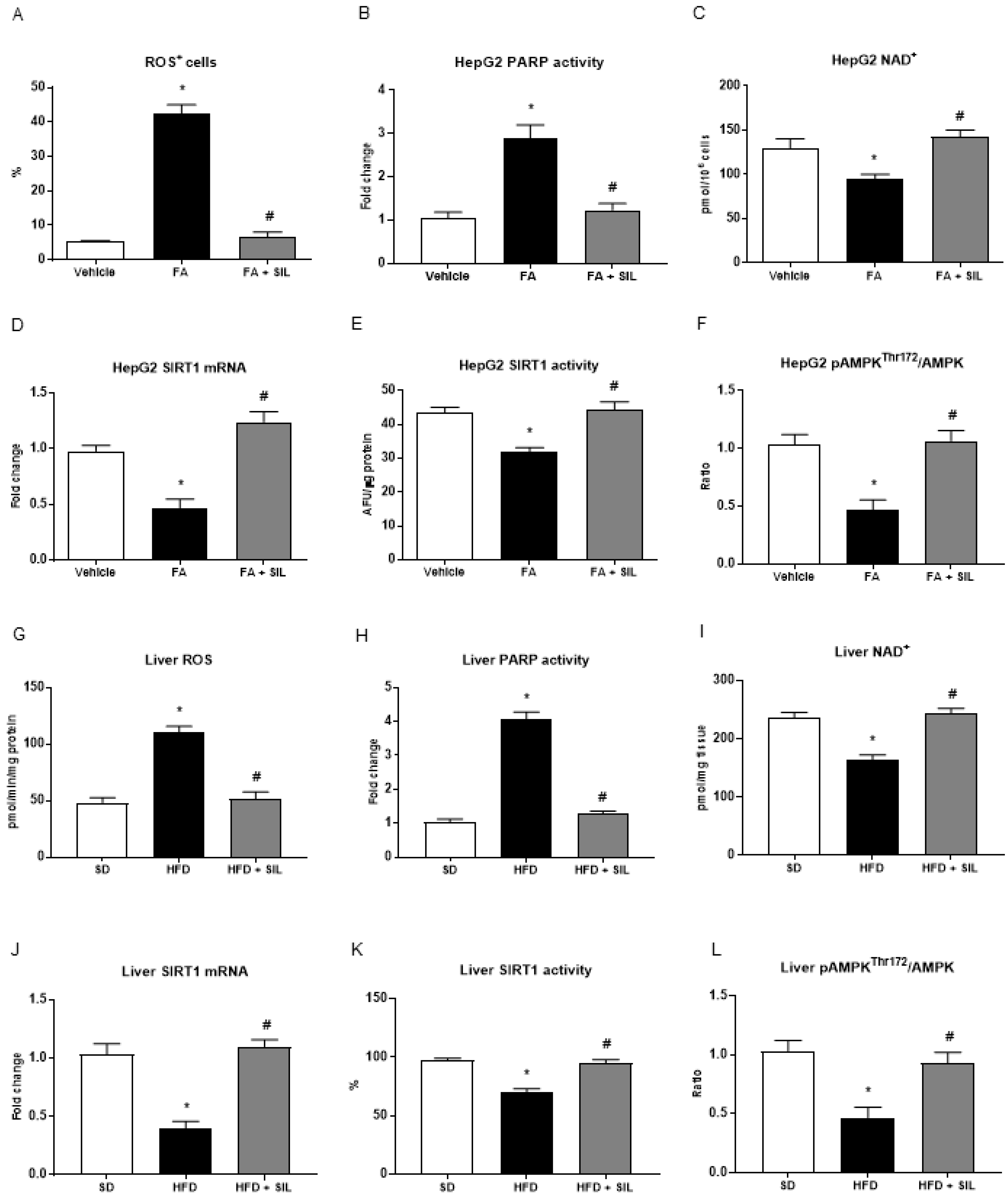
3.2. Silibinin Restores NAD+ Levels and Induces SIRT1/AMPK Signaling
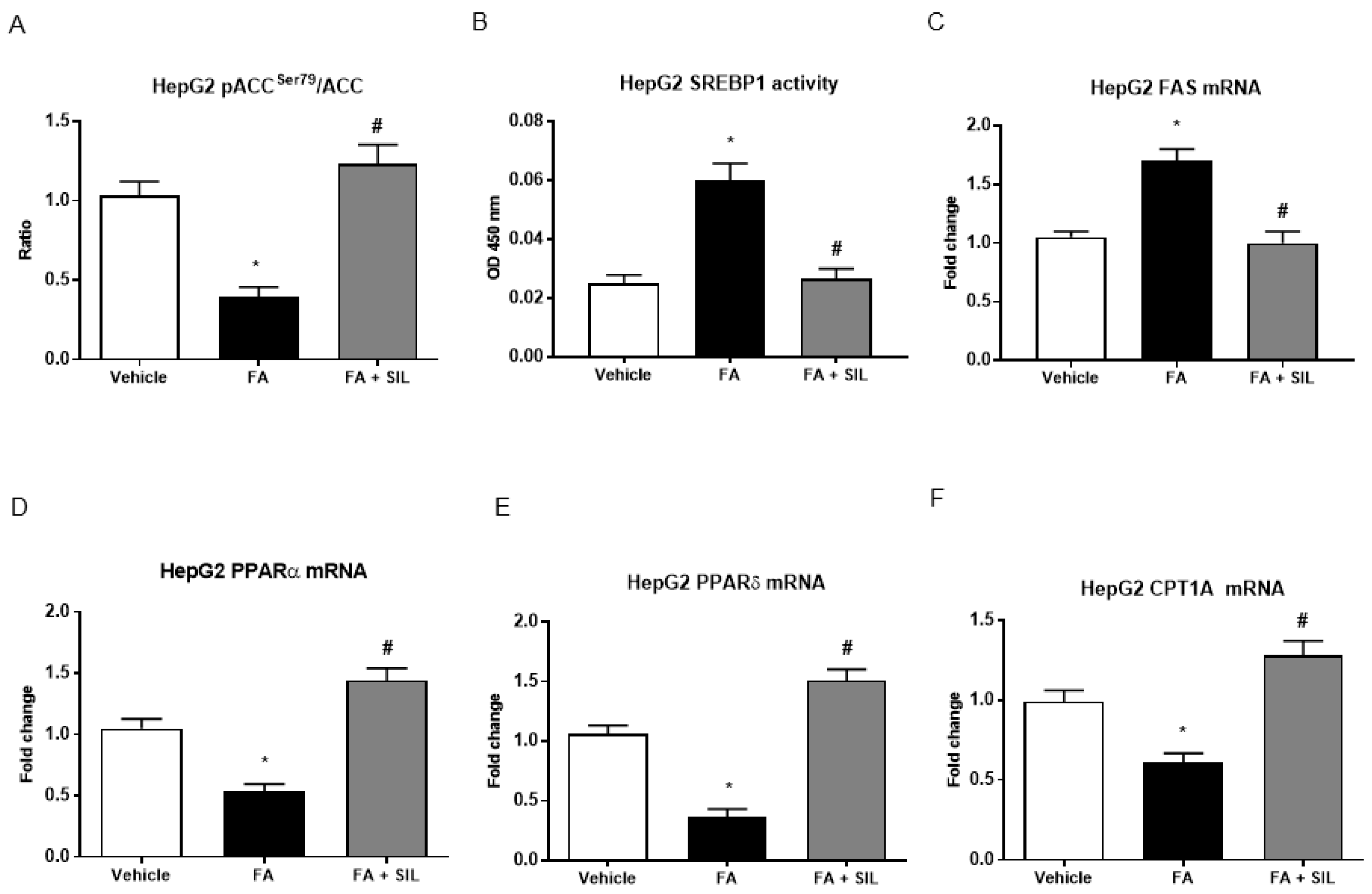
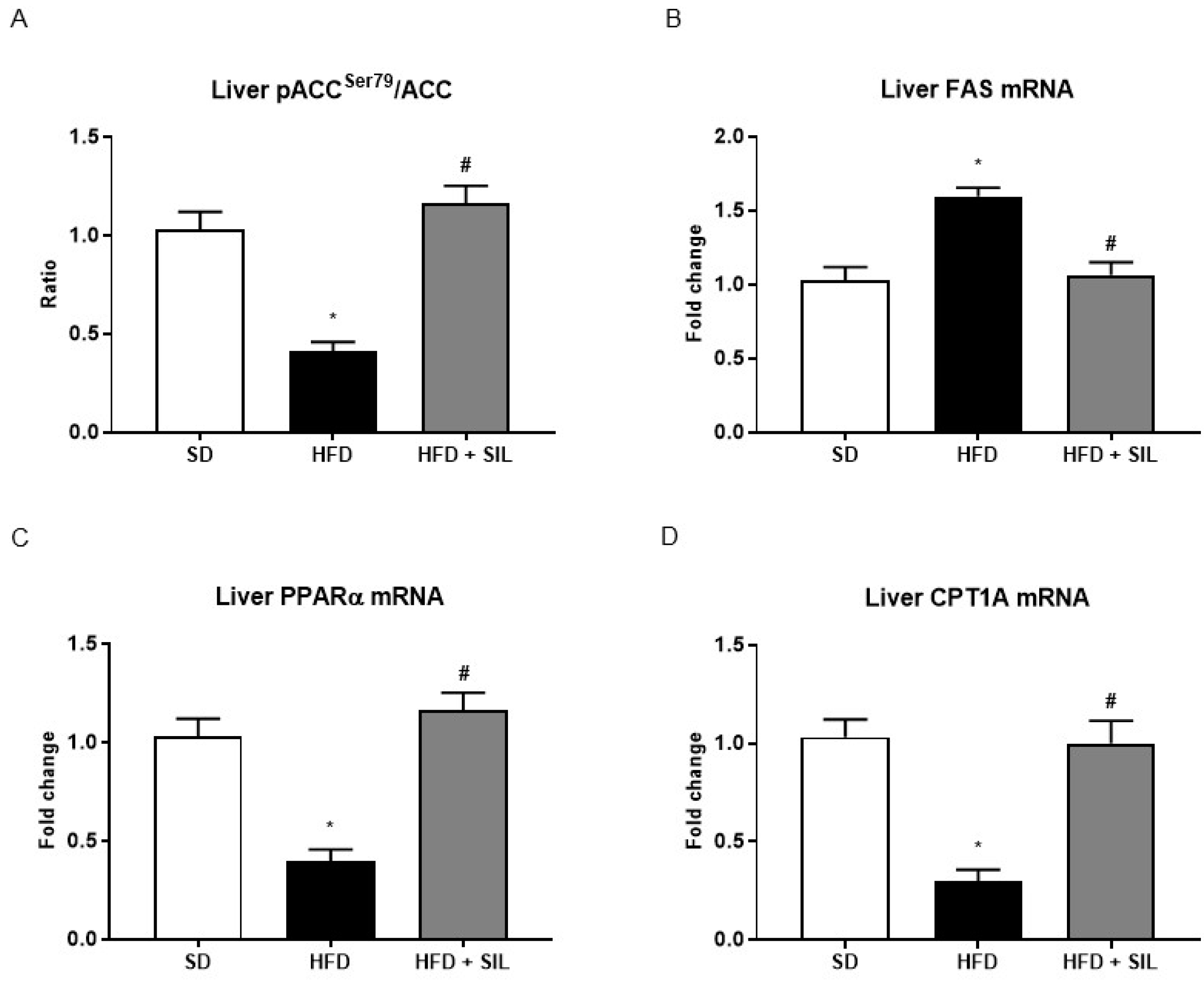
3.3. Silibinin Inhibits De Novo Lipogenesis and Promotes Mitochondrial β-Oxidation
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the united states. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Godos, J.; Salomone, F. Lifestyle changes for the treatment of nonalcoholic fatty liver disease: A review of observational studies and intervention trials. Therap. Adv. Gastroenterol. 2016, 9, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, M.; Loomba, R. State of the art: Treatment of nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2014, 30, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Godos, J.; Zelber-Sagi, S. Natural antioxidants for non-alcoholic fatty liver disease: Molecular targets and clinical perspectives. Liver Int. 2016, 36, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Auwerx, J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD+ metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 2012, 8, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Lagouge, M.; Canto, C.; Strehle, A.; Houten, S.M.; Milne, J.C.; Lambert, P.D.; Mataki, C.; Elliott, P.J.; Auwerx, J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008, 8, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tanuma, S.; Sato, A.; Oyama, T.; Yoshimori, A.; Abe, H.; Uchiumi, F. New insights into the roles of NAD+-poly(ADP-ribose) metabolism and poly(ADP-ribose) glycohydrolase. Curr. Protein Pept. Sci. 2016, 17, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Gual, P.; Postic, C. Therapeutic potential of nicotinamide adenine dinucleotide for nonalcoholic fatty liver disease. Hepatology 2016, 63, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; Festi, D. Silybin and the liver: From basic research to clinical practice. World J. Gastroenterol. WJG 2011, 17, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Eguchi, A.; Niesman, I.R.; Andronikou, N.; de Mollerat du Jeu, X.; Mulya, A.; Berk, M.; Lazic, M.; Thapaliya, S.; Parola, M.; et al. Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by endothelial cells. Sci. Signal. 2013, 6, ra88. [Google Scholar] [CrossRef] [PubMed]
- Trappoliere, M.; Caligiuri, A.; Schmid, M.; Bertolani, C.; Failli, P.; Vizzutti, F.; Novo, E.; di Manzano, C.; Marra, F.; Loguercio, C.; et al. Silybin, a component of sylimarin, exerts anti-inflammatory and anti-fibrogenic effects on human hepatic stellate cells. J. Hepatol. 2009, 50, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, I.; Vanella, L.; Cambria, M.T.; Tibullo, D.; Godos, J.; Guarnaccia, L.; Zappalà, A.; Galvano, F.; Li Volti, G. Silibinin regulates lipid metabolism and differentiation in functional human adipocytes. Front. Pharmacol. 2016, 6, 309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Fan, S.H.; Zheng, Y.L.; Lu, J.; Wu, D.M.; Shan, Q.; Hu, B. Troxerutin improves hepatic lipid homeostasis by restoring NAD+-depletion-mediated dysfunction of lipin 1 signaling in high-fat diet-treated mice. Biochem. Pharmacol. 2014, 91, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Assifi, M.M.; Suchankova, G.; Constant, S.; Prentki, M.; Saha, A.K.; Ruderman, N.B. AMP-activated protein kinase and coordination of hepatic fatty acid metabolism of starved/carbohydrate-refed rats. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E794–E800. [Google Scholar] [CrossRef] [PubMed]
- Saggerson, D.; Ghadiminejad, I.; Awan, M. Regulation of mitochondrial carnitine palmitoyl transferases from liver and extrahepatic tissues. Adv. Enzyme Regul. 1992, 32, 285–306. [Google Scholar] [CrossRef]
- Hsu, M.H.; Savas, U.; Griffin, K.J.; Johnson, E.F. Identification of peroxisome proliferator-responsive human genes by elevated expression of the peroxisome proliferator-activated receptor α in HepG2 cells. J. Biol. Chem. 2001, 276, 27950–27958. [Google Scholar] [CrossRef] [PubMed]
- Handa, P.; Maliken, B.D.; Nelson, J.E.; Morgan-Stevenson, V.; Messner, D.J.; Dhillon, B.K.; Klintworth, H.M.; Beauchamp, M.; Yeh, M.M.; Elfers, C.T.; et al. Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology 2014, 60, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Xue, Y.; Xue, L.; Jiang, X.; Wang, X.; Zhang, Z.; Yang, J.; Lu, J.; Zhang, C.; Wang, W.; et al. Hepatic menin recruits SIRT1 to control liver steatosis through histone deacetylation. J. Hepatol. 2013, 59, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Gao, Z.; Zhang, J.; Rivera, C.A.; Yin, J.; Weng, J.; Ye, J. Lack of SIRT1 (Mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/- mice: A role of lipid mobilization and inflammation. Endocrinology 2010, 151, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Ruan, J.; Zhang, W.; Qian, F.; Yu, Z. Targeting NAD+ degradation: The therapeutic potential of flavonoids for Alzheimer’s disease and cognitive frailty. Pharmacol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Fry, J.L.; Han, J.; Hou, X.; Pimentel, D.R.; Matsui, R.; Cohen, R.A.; Bachschmid, M.M. A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 2014, 289, 7293–7306. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor α/δ agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Giudetti, A.M.; Gnoni, G.V.; Petrella, A.; Tamborra, R.; Romano, A.D.; Rollo, T.; Vendemiale, G.; Altomare, E. A silybin-phospholipid complex prevents mitochondrial dysfunction in a rodent model of nonalcoholic steatohepatitis. J. Pharmacol. Exp. Ther. 2010, 332, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Haddad, Y.; Vallerand, D.; Brault, A.; Haddad, P.S. Antioxidant and hepatoprotective effects of silibinin in a rat model of nonalcoholic steatohepatitis. Evid. Based Complement. Altern. Med. 2011, 2011, nep164. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhi, M.; Minhu, C. Effect of silybin on high-fat-induced fatty liver in rats. Braz. J. Med. Biol. Res. 2011, 44, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Salamone, F.; Galvano, F.; Cappello, F.; Mangiameli, A.; Barbagallo, I.; Li Volti, G. Silibinin modulates lipid homeostasis and inhibits nuclear factor kappa B activation in experimental nonalcoholic steatohepatitis. Transl. Res. 2012, 159, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Salamone, F.; Galvano, F.; Marino Gammazza, A.; Paternostro, C.; Tibullo, D.; Bucchieri, F.; Mangiameli, A.; Parola, M.; Bugianesi, E.; Li Volti, G. Silibinin improves hepatic and myocardial injury in mice with nonalcoholic steatohepatitis. Dig. Liver Dis. 2012, 44, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hai, J.; Cao, M.; Zhang, Y.; Pei, S.; Wang, J.; Zhang, Q. Silibinin ameliorates steatosis and insulin resistance during non-alcoholic fatty liver disease development partly through targeting IRS-1/PI3K/AKt pathway. Int. Immunopharmacol. 2013, 17, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Dehmlow, C.; Erhard, J.; de Groot, H. Inhibition of kupffer cell functions as an explanation for the hepatoprotective properties of silibinin. Hepatology 1996, 23, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Ezhilarasan, D.; Evraerts, J.; Brice, S.; Buc-Calderon, P.; Karthikeyan, S.; Sokal, E.; Najimi, M. Silibinin inhibits proliferation and migration of human hepatic stellate LX-2 cells. J. Clin. Exp. Hepatol. 2016, 6, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [PubMed]
- Colak, Y.; Yesil, A.; Mutlu, H.H.; Caklili, O.T.; Ulasoglu, C.; Senates, E.; Takir, M.; Kostek, O.; Yilmaz, Y.; Yilmaz Enc, F.; et al. A potential treatment of non-alcoholic fatty liver disease with SIRT1 activators. J. Gastrointest. Liver Dis. JGLD 2014, 23, 311–319. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; Andreone, P.; Brisc, C.; Brisc, M.C.; Bugianesi, E.; Chiaramonte, M.; Cursaro, C.; Danila, M.; de Sio, I.; Floreani, A.; et al. Silybin combined with phosphatidylcholine and vitamin E in patients with nonalcoholic fatty liver disease: A randomized controlled trial. Free Radic. Biol. Med. 2012, 52, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; Izaola, O.; Gomez, S.; Tafur, C.; Gonzalez, G.; Berroa, E.; Mora, N.; Gonzalez, J.M.; de Luis, D.A. Effect of silymarin plus vitamin e in patients with non-alcoholic fatty liver disease. A randomized clinical pilot study. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3118–3124. [Google Scholar] [PubMed]
- Rosso, N.; Marin, V.; Giordani, A.; Persiani, S.; Sala, F.; Cavicchioli, L.; Rovati, L.C.; Tiribelli, C. The pros and the cons for the use of silybin-rich oral formulations in treatment of liver damage (NAFLD in particular). Curr. Med. Chem. 2015, 22, 2954–2971. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salomone, F.; Barbagallo, I.; Godos, J.; Lembo, V.; Currenti, W.; Cinà, D.; Avola, R.; D’Orazio, N.; Morisco, F.; Galvano, F.; et al. Silibinin Restores NAD+ Levels and Induces the SIRT1/AMPK Pathway in Non-Alcoholic Fatty Liver. Nutrients 2017, 9, 1086. https://0-doi-org.brum.beds.ac.uk/10.3390/nu9101086
Salomone F, Barbagallo I, Godos J, Lembo V, Currenti W, Cinà D, Avola R, D’Orazio N, Morisco F, Galvano F, et al. Silibinin Restores NAD+ Levels and Induces the SIRT1/AMPK Pathway in Non-Alcoholic Fatty Liver. Nutrients. 2017; 9(10):1086. https://0-doi-org.brum.beds.ac.uk/10.3390/nu9101086
Chicago/Turabian StyleSalomone, Federico, Ignazio Barbagallo, Justyna Godos, Vincenzo Lembo, Walter Currenti, Diana Cinà, Roberto Avola, Nicolantonio D’Orazio, Filomena Morisco, Fabio Galvano, and et al. 2017. "Silibinin Restores NAD+ Levels and Induces the SIRT1/AMPK Pathway in Non-Alcoholic Fatty Liver" Nutrients 9, no. 10: 1086. https://0-doi-org.brum.beds.ac.uk/10.3390/nu9101086