
Ring-Opening of Cyclodextrins: An Efficient Route to Pure Maltohexa-, Hepta-, and Octaoses

 ,
,  , , , and
, , , and
Abstract
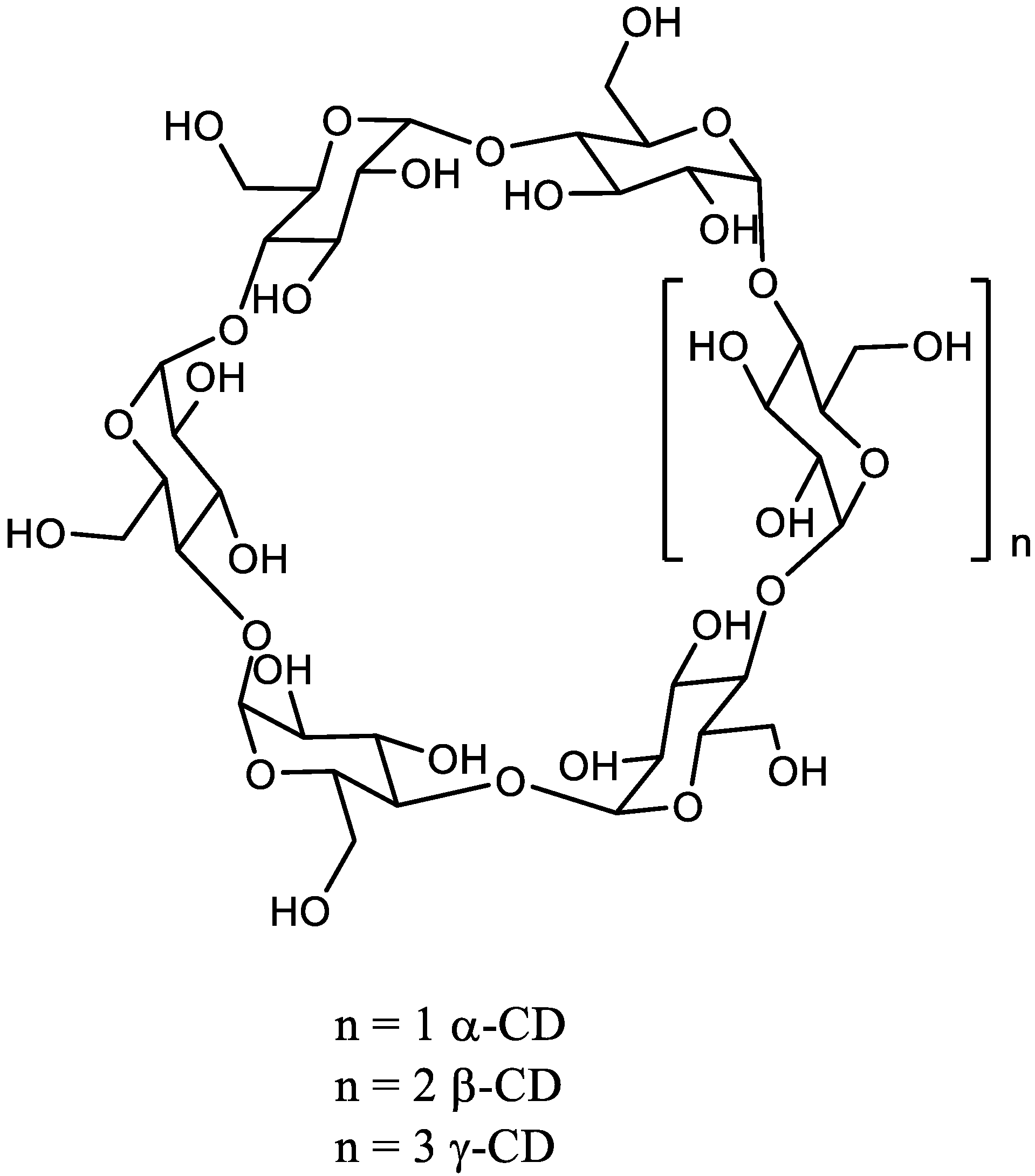
:1. Introduction
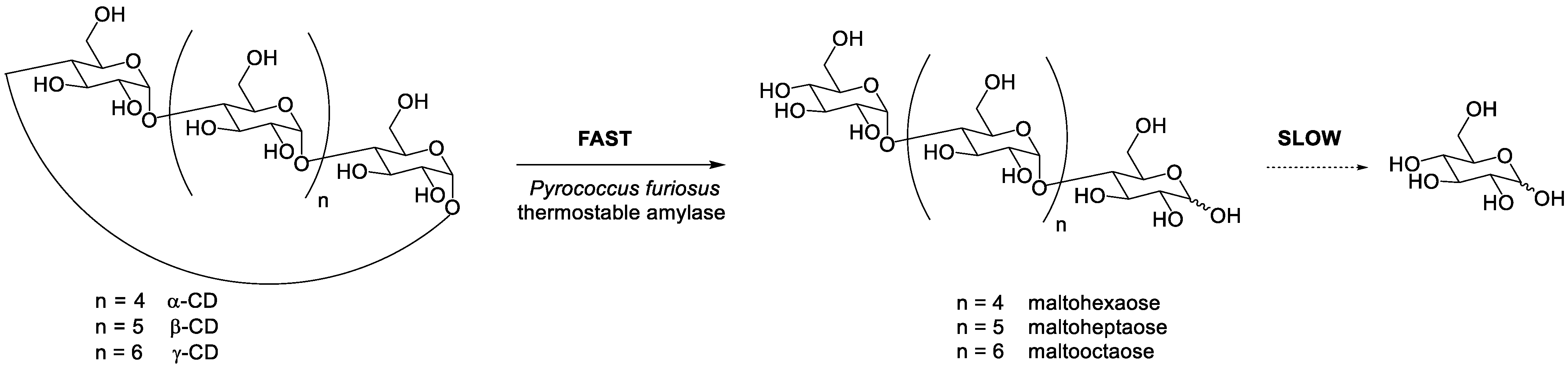
2. Enzymatic Ring Opening of Cyclodextrins
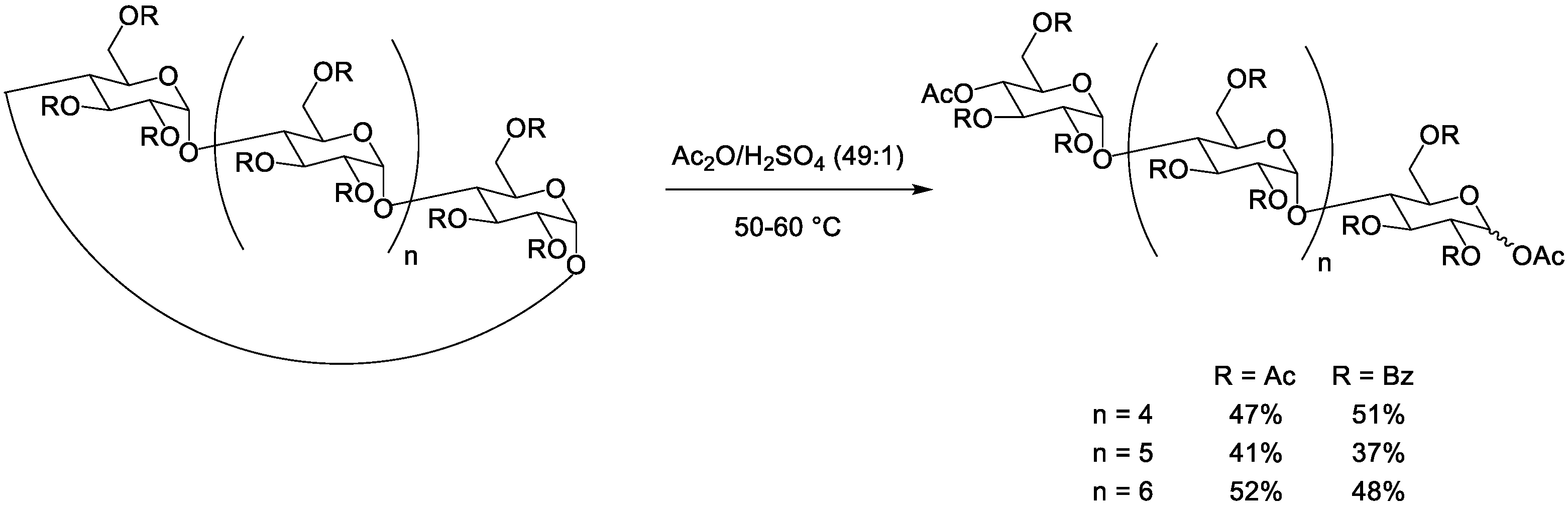
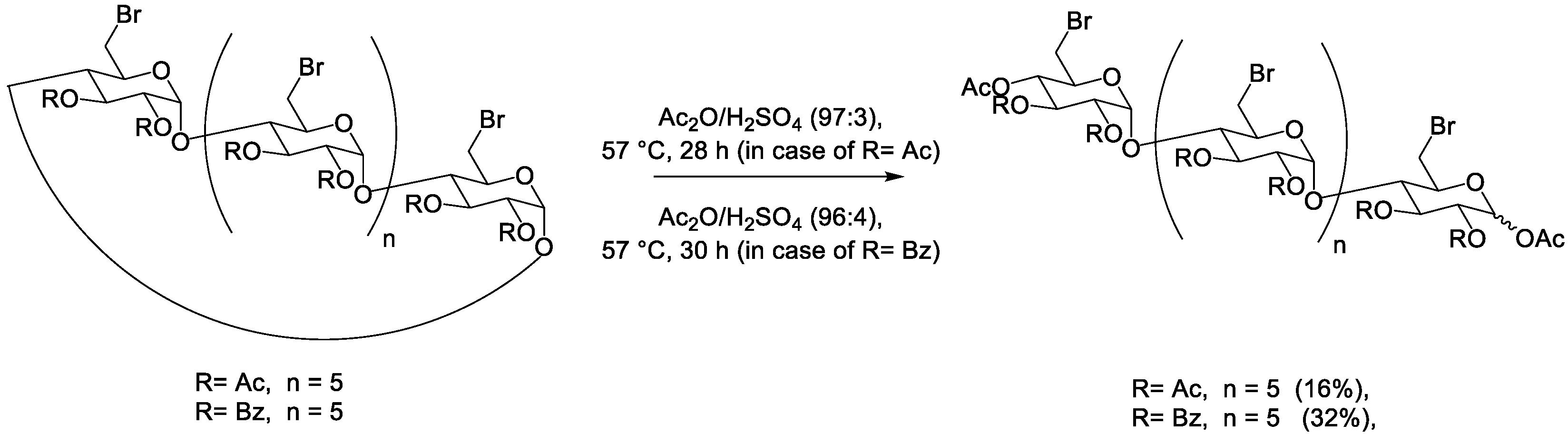
3. Acetolytic Cleavage of Cyclodextrins
4. Ring-Opening Cyclodextrins Using Other Acidic Conditions
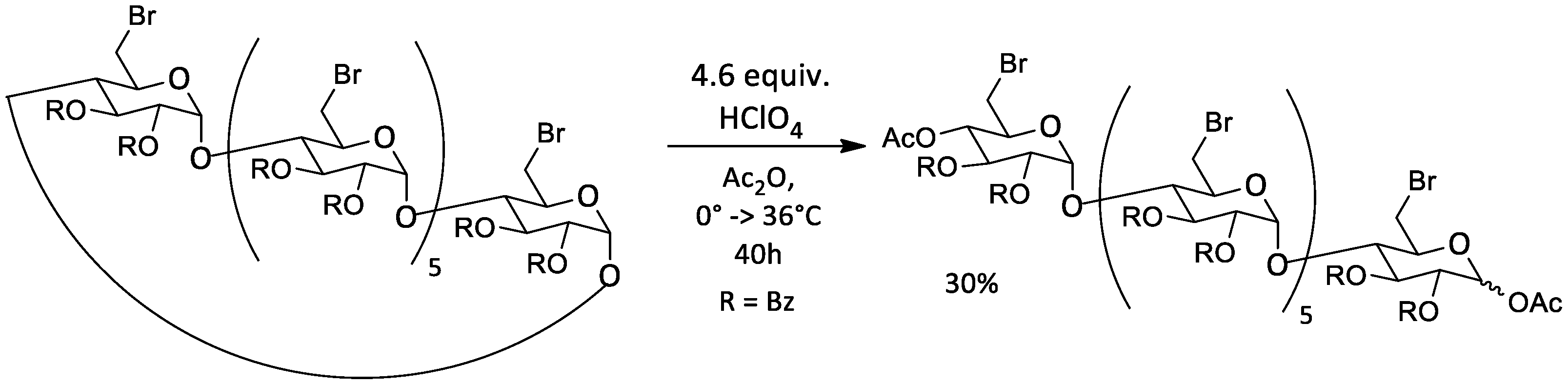
4.1. Ring-Opening Cyclodextrins Using Brønsted Acids (HClO4)
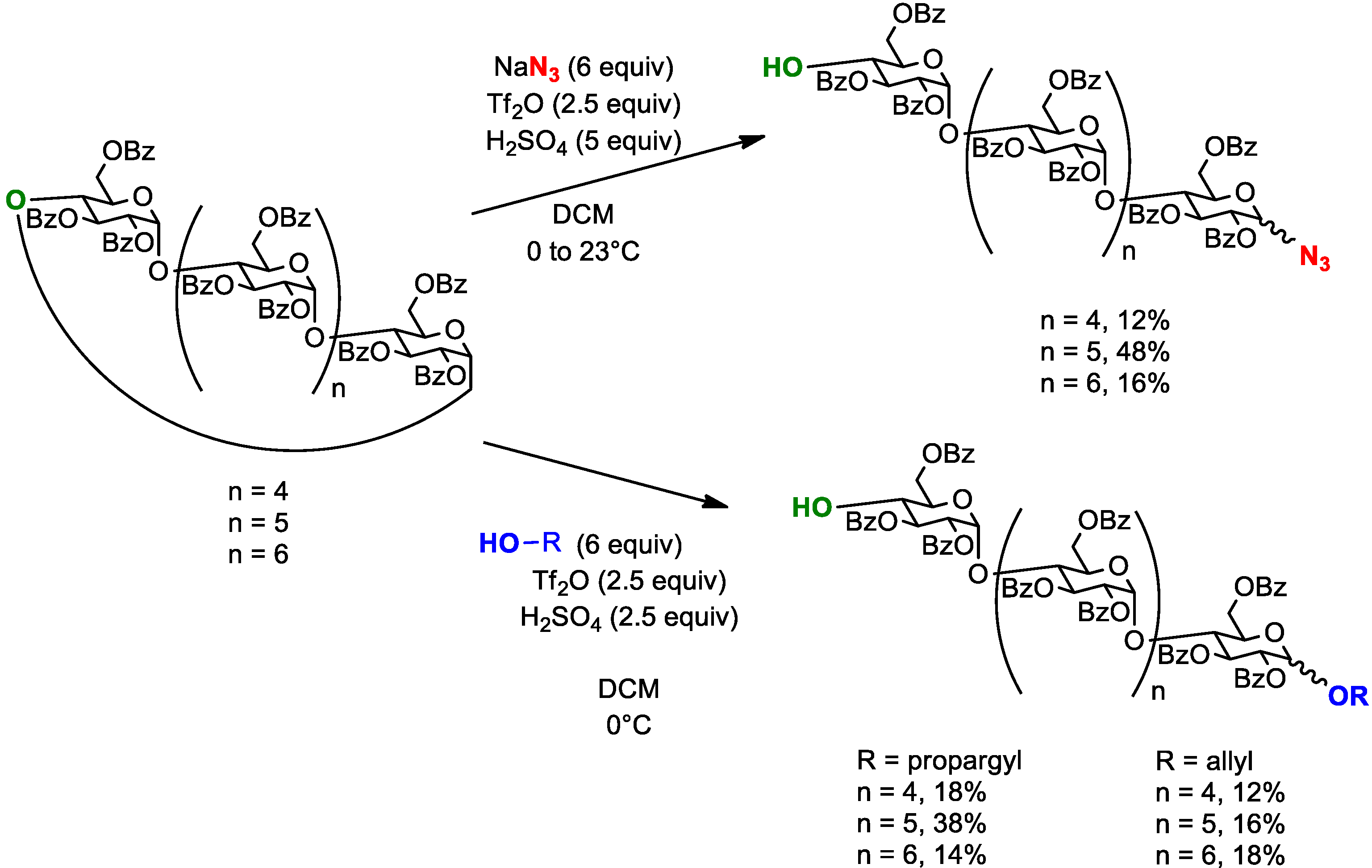
4.2. Ring-Opening of Cyclodextrins Using Sulfuric Acid and Triflic Anhydride
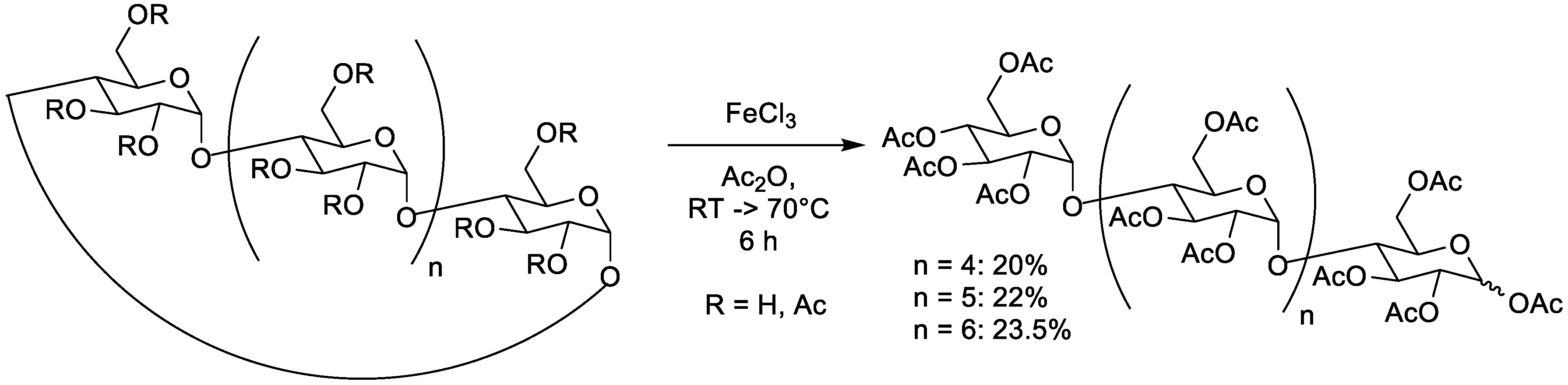
4.3. Lewis Acid FeCl3
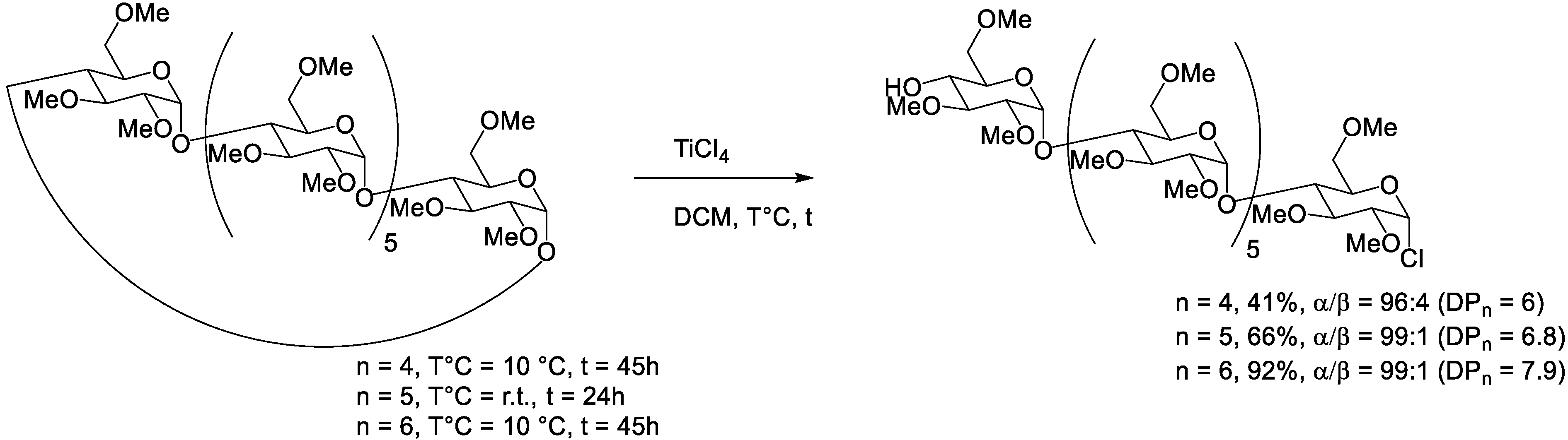
4.4. Lewis Acid TiCl4
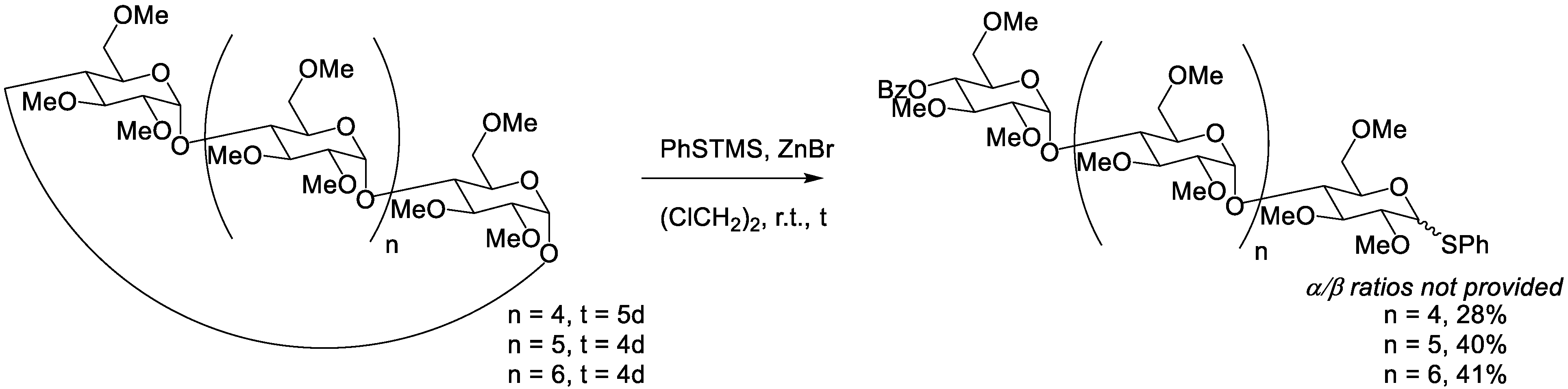
4.5. Lewis Acid ZnI/ZnBr and Thiolysis
5. Application Areas of Pure OM
5.1. Biological Target
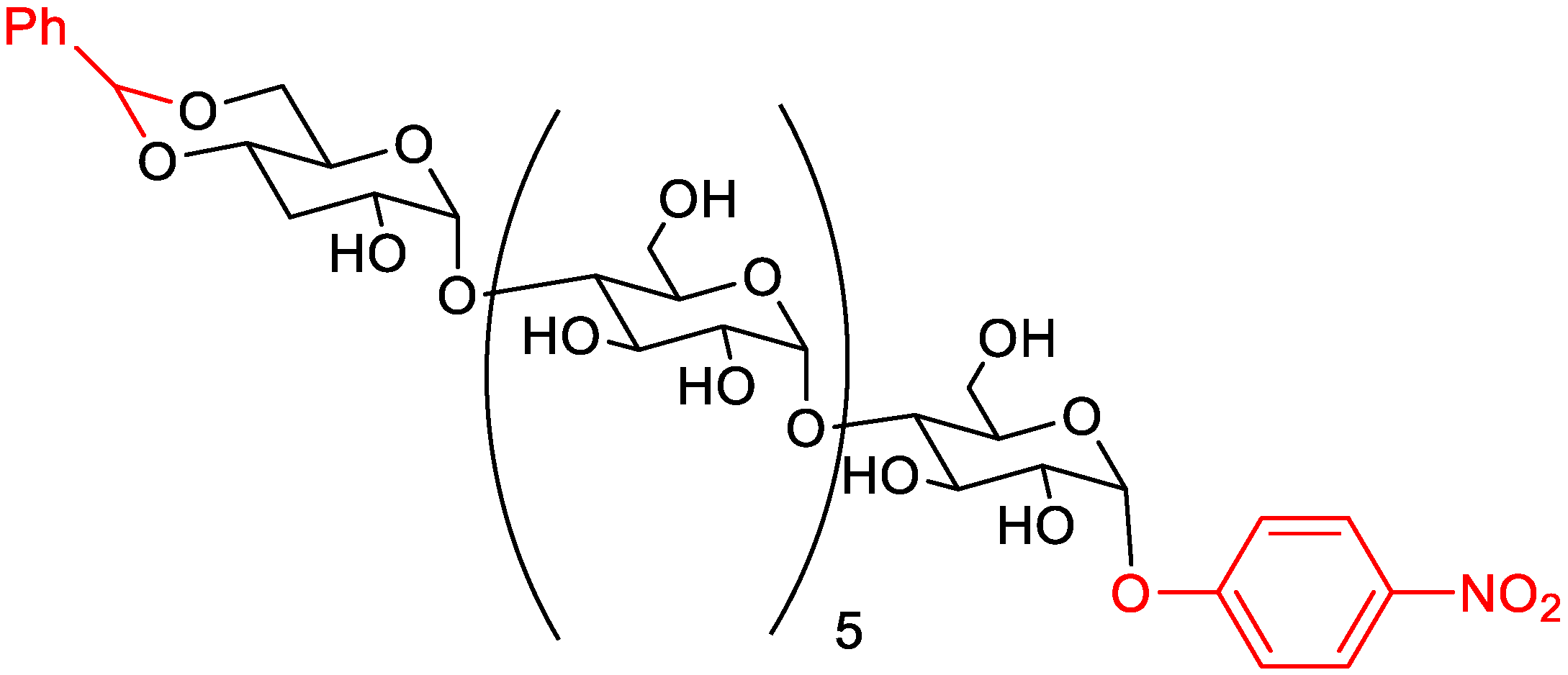
5.1.1. Chromogenic Substrates
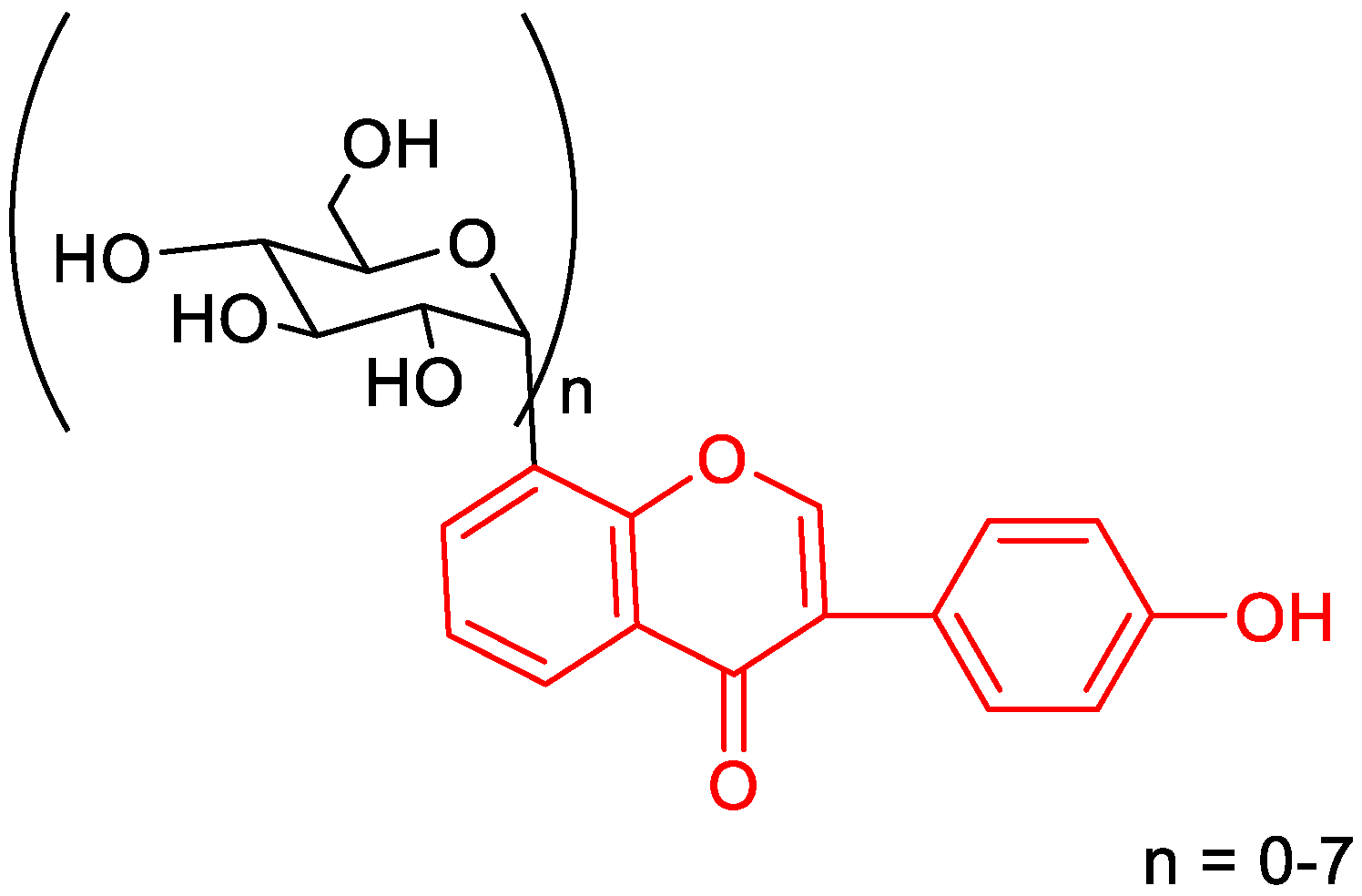
5.1.2. Antioxidant Property
5.2. Cyclodextrins Ring-Opening and Biopolymers
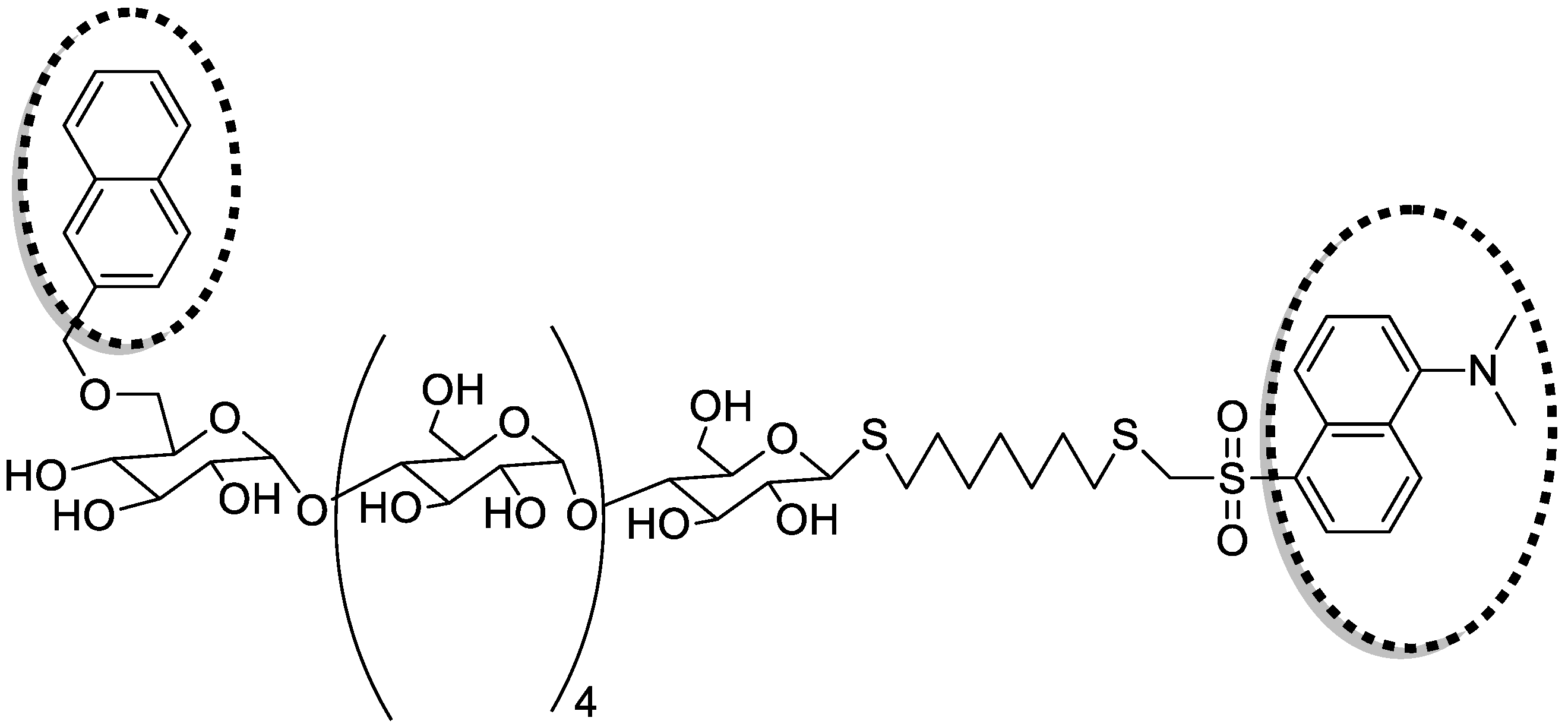
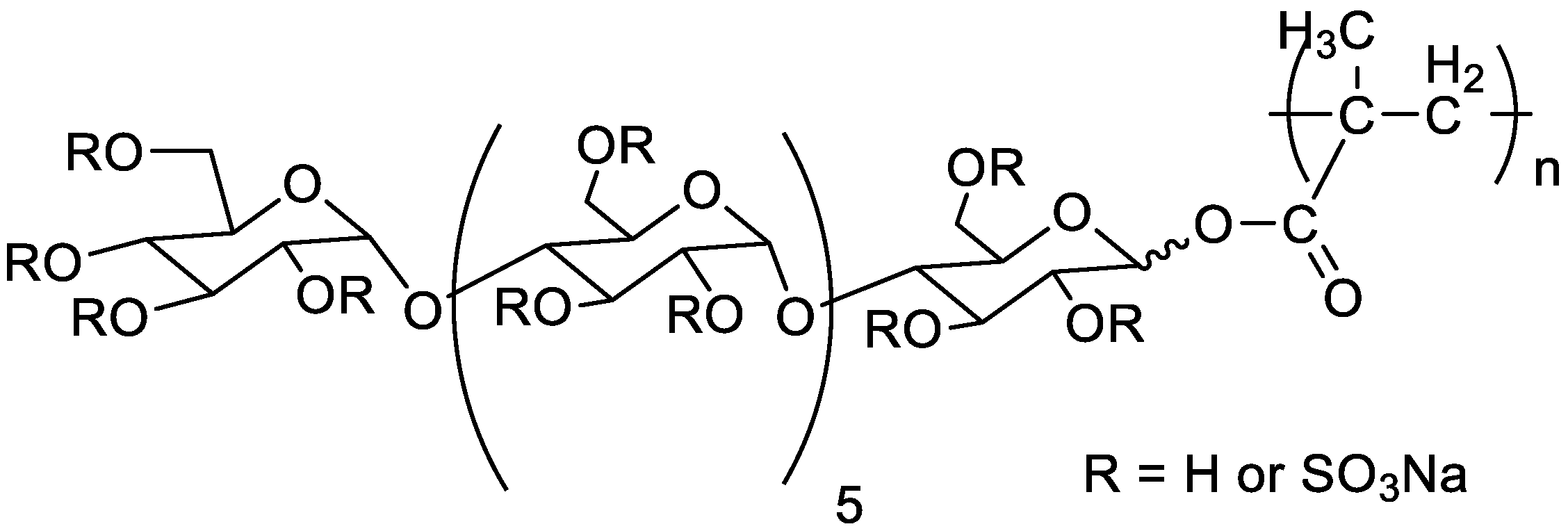
5.2.1. Anti HIV and Anticoagulant Activities
5.2.2. Lectin Binding Properties
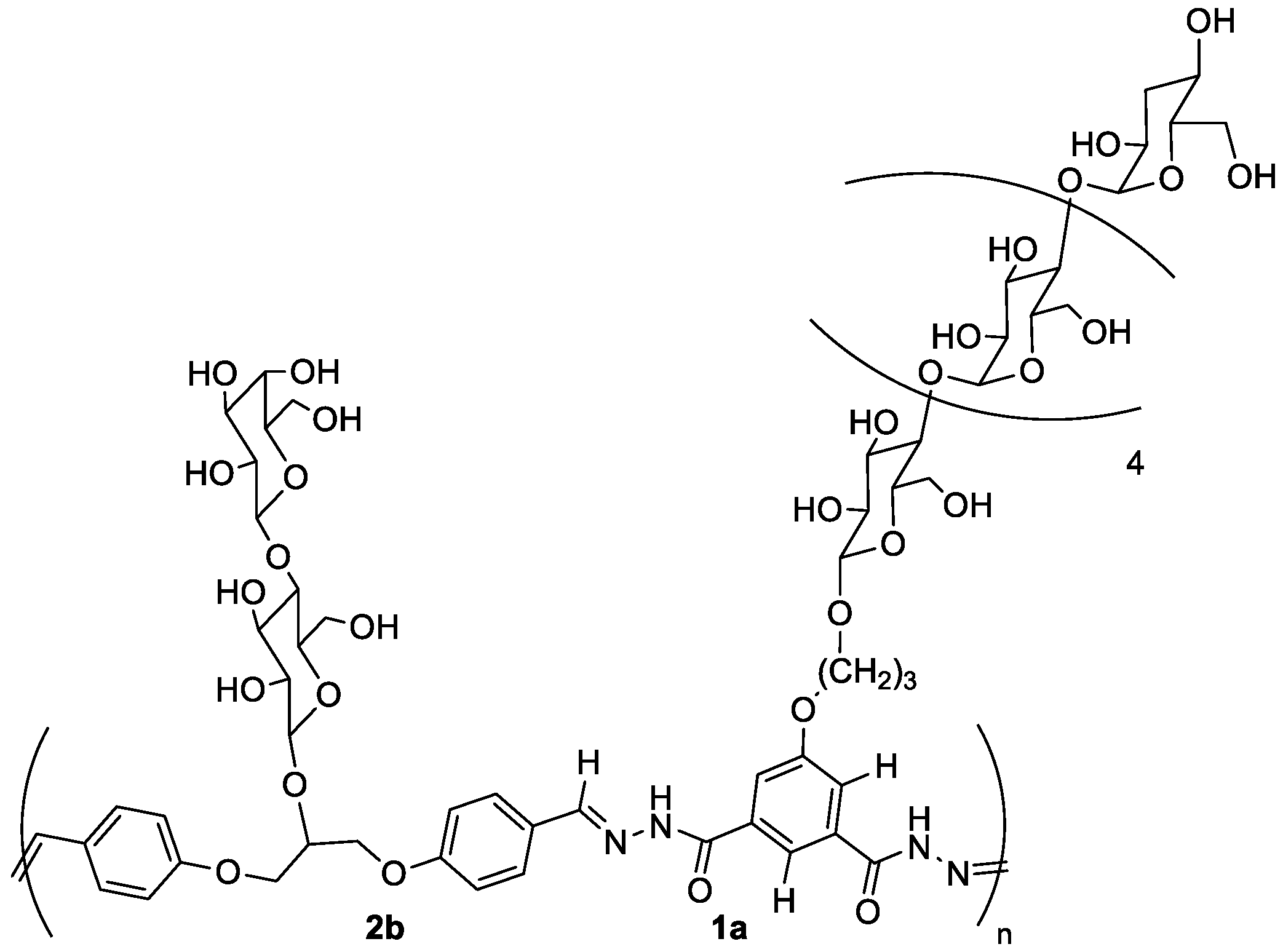
5.2.3. Block Copolymers
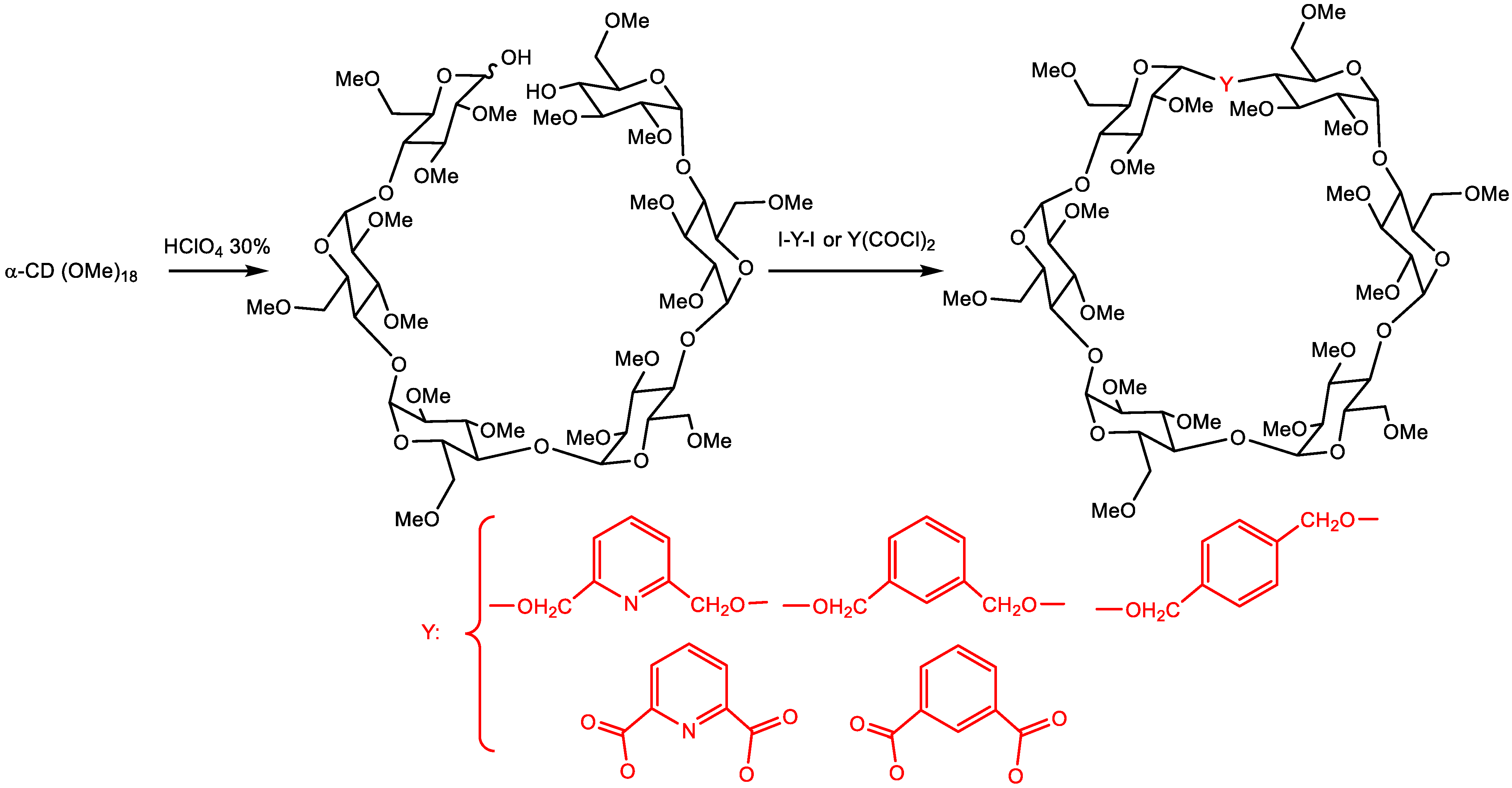
5.3. Modified Cyclodextrins
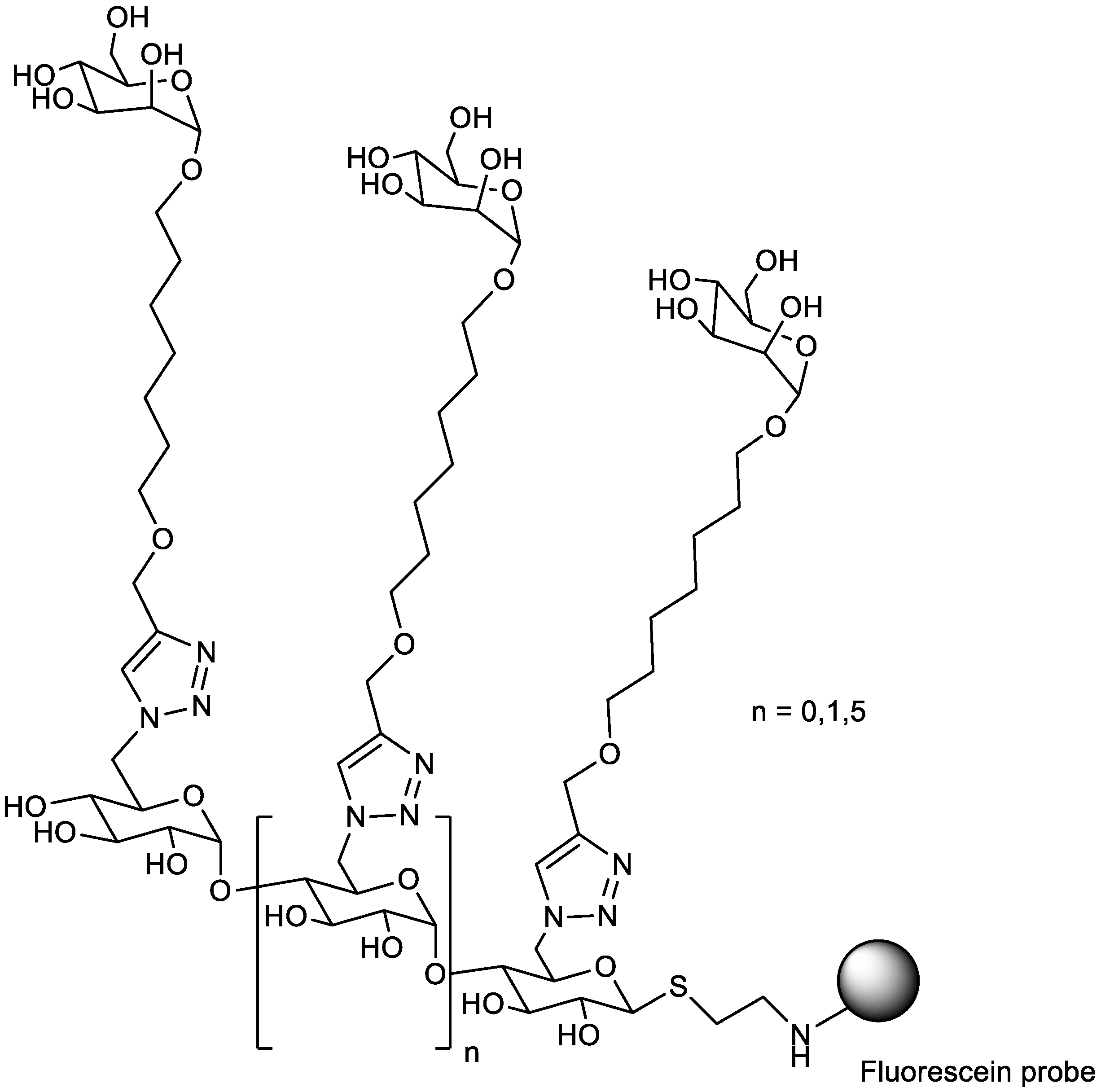
5.4. Multivalency Support
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Giri, K.V.; Nagabhushanam, A.; Nigam, V.N.; Belavadi, B. Synthesis of Oligosaccharides from Maltose by Rat Liver. Science 1955, 121, 898. [Google Scholar] [CrossRef]
- Nigam, V.N.; Giri, K.V. Enzymatic Synthesis of Oligosaccharides from Maltose by Germinated Green Gram (Phaseolus Radiatus). J. Biol. Chem. 1960, 235, 947–950. [Google Scholar] [CrossRef]
- Mangas-Sánchez, J.; Adlercreutz, P. Enzymatic Preparation of Oligosaccharides by Transglycosylation: A Comparative Study of Glucosidases. J. Mol. Catal. B Enzym. 2015, 122, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Abdul Manas, N.H.; Jonet, M.A.; Abdul Murad, A.M.; Mahadi, N.M.; Illias, R.M. Modulation of Transglycosylation and Improved Malto-Oligosaccharide Synthesis by Protein Engineering of Maltogenic Amylase from Bacillus lehensis G1. Process Biochem. 2015, 50, 1572–1580. [Google Scholar] [CrossRef]
- Kobayashi, S.; Shoda, S.; Kashiwa, K.; Shimada, J. Method for the Preparation of Malto-Oligosaccharide. Eur. Patent 0,530,421 A1, 10 March 1993. [Google Scholar]
- Cottaz, S.; Driguez, H. First Regiospecific Synthesis of 6A,6C,6E-Tri-O-Methylyclomaltohexaose. J. Chem. Soc. Chem. Commun. 1989, 16, 1088–1089. [Google Scholar] [CrossRef]
- Park, J.Y.; Lee, S.O.; Lee, T.H. Syntheses of 1-O-Benzyl-α-Glucoside and 1-O-Benzyl-α-Maltoside by Transglycosylation of α-Amylase from Soluble Starch in Aqueous Solution. Biotechnol. Lett. 1999, 21, 81–86. [Google Scholar] [CrossRef]
- Niu, D.; Li, P.; Huang, Y.; Tian, K.; Liu, X.; Singh, S.; Lu, F. Preparation of Maltotriitol-Rich Malto-Oligosaccharide Alcohol from Starch. Process Biochem. 2017, 52, 159–164. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ogawa, T. Total Synthesis of Cyclomaltohexaose. Carbohydr. Res. 1987, 164, 277–296. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ogawa, T. Total Synthesis of Cyclomaltooctaose and an Isomer of Cyclomaltohexaose, Cyclo{→6)-[α-d-Glcp-(1→4)]5-α-d-Glcp-(1-}. Carbohydr. Res. 1987, 169, 127–149. [Google Scholar] [CrossRef]
- Crini, G. Review: A History of Cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef] [PubMed]
- Larsen, D.; Beeren, S.R. Building up Cyclodextrins from Scratch—Templated Enzymatic Synthesis of Cyclodextrins Directly from Maltose. Chem. Commun. 2021, 57, 2503–2506. [Google Scholar] [CrossRef]
- Saha, B.C.; Zeikus, J.G. Cyclodextrin Degrading Enzymes. Starch-Stärke 1992, 44, 312–315. [Google Scholar] [CrossRef]
- Bender, H. Purification and Characterization of a Cyclodextrin-Degrading Enzyme from Flavobacterium sp. Appl. Microbiol. Biotechnol. 1993, 39, 714–719. [Google Scholar] [CrossRef]
- Uchida, R.; Nasu, A.; Tobe, K.; Oguma, T.; Yamaji, N. A Convenient Preparation of Maltooctaose and Maltononaose by the Coupling Reaction of Cyclomaltodextrinase. Carbohydr. Res. 1996, 287, 271–274. [Google Scholar] [CrossRef]
- Fraschini, C.; Greffe, L.; Driguez, H.; Vignon, M.R. Chemoenzymatic Synthesis of 6ω-Modified Maltooligosaccharides from Cyclodextrin Derivatives. Carbohydr. Res. 2005, 340, 1893–1899. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-J.; Lee, H.-S.; Park, C.-S.; Kim, Y.-R.; Moon, T.-W.; Park, K.-H. Enzymatic Analysis of an Amylolytic Enzyme from the Hyperthermophilic Archaeon Pyrococcus Furiosus Reveals Its Novel Catalytic Properties as Both an Alpha-Amylase and a Cyclodextrin-Hydrolyzing Enzyme. Appl. Environ. Microbiol. 2004, 70, 5988–5995. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-J.; Lee, H.-S.; Kim, J.-W.; Lee, M.-H.; Auh, J.-H.; Lee, B.-H.; Park, K.-H. Enzymatic Preparation of Maltohexaose, Maltoheptaose, and Maltooctaose by the Preferential Cyclomaltooligosaccharide (Cyclodextrin) Ring-Opening Reaction of Pyrococcus Furiosus Thermostable Amylase. Carbohydr. Res. 2006, 341, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Koo, Y.-S.; Lee, H.-W.; Jeon, H.-Y.; Choi, H.-J.; Choung, W.-J.; Shim, J.-H. Development and Characterization of Cyclodextrin Glucanotransferase as a Maltoheptaose-Producing Enzyme Using Site-Directed Mutagenesis. Protein Eng. Des. Sel. 2015, 28, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, D.; Levine, M.L.; Pazur, J.H. Studies on the Schardinger Dextrins. II. Preparation and Properties of Amyloheptaose. J. Am. Chem. Soc. 1949, 71, 356–358. [Google Scholar] [CrossRef] [PubMed]
- French, D.; Knapp, D.W.; Pazur, J.H. Studies on the Schardinger Dextrins. VI. The Molecular Size and Structure of the γ-Dextrin1. J. Am. Chem. Soc. 1950, 72, 5150–5152. [Google Scholar] [CrossRef]
- Freudenberg, K. Hydrolysis and Optical Rotation of Cellulose, Starch, and Cycloglucans. J. Polym. Sci. 1957, 23, 791–799. [Google Scholar] [CrossRef]
- Freudenberg, K.; Blomqvist, G.; Ewald, L.; Soff, K. Hydrolyse Und Acetolyse Der Stärke Und Der Schardinger-Dextrine. Chem. Ber. 1936, 69, 1258–1266. [Google Scholar] [CrossRef]
- Liptak, A.; Nanasi, P.; Szejtli, J.; Kari, Z.M. Process for Producing Linear Maltooligosaccharides Comprising 6, 7 or 8 Glucose Units and Their Peracetylated Derivatives. Hungarian Patent 204857 B, 28 May 1990. [Google Scholar]
- Sakairi, N.; Wang, L.-X.; Kuzuhara, H. Insertion of a D-Glucosamine Residue into the α-Cyclodextrin Skeleton; A Model Synthesis of ‘Chimera Cyclodextrins’. J. Chem. Soc. Chem. Commun. 1991, 5, 289–290. [Google Scholar] [CrossRef]
- Sakairi, N.; Matsui, K.; Kuzuhara, H. Acetolytic Fission of a Single Glycosidic Bond of Fully Benzoylated α-, β-, and γ-Cyclodextrins. A Novel Approach to the Preparation of Maltooligosaccharide Derivatives Regioselectively Modified at Their Nonreducing Ends. Carbohydr. Res. 1995, 266, 263–268. [Google Scholar] [CrossRef]
- Sakairi, N.; Wang, L.-X.; Kuzuhara, H. Modification of Cyclodextrins by Insertion of a Heterogeneous Sugar Unit into Their Skeletons. Synthesis of 2-Amino-2-Deoxy-β-Cyclodextrin from α-Cyclodextrin. J. Chem. Soc. Perkin Trans. 1 1995, 4, 437–443. [Google Scholar] [CrossRef]
- Yoshida, T.; Akasaka, T.; Choi, Y.; Hattori, K.; Yu, B.; Mimura, T.; Kaneko, Y.; Nakashima, H.; Aragaki, E.; Premanathan, M.; et al. Synthesis of Polymethacrylate Derivatives Having Sulfated Maltoheptaose Side Chains with Anti-HIV Activities. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 789–800. [Google Scholar] [CrossRef]
- Haddleton, D.M.; Ohno, K. Well-Defined Oligosaccharide-Terminated Polymers from Living Radical Polymerization. Biomacromolecules 2000, 1, 152–156. [Google Scholar] [CrossRef]
- Lesur, D.; Gassama, A.; Moreau, V.; Pilard, S.; Djedaïni-Pilard, F. Synthesis of Regioselectively and Uniformly Modified Maltoheptaose Derivatives from Cyclomaltoheptaose Precursors. Carbohydr. Res. 2005, 340, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Lesur, D.; Gassama, A.; Brique, A.; Thiebault, N.; Djedaïni-Pilard, F.; Pilard, S.; Moreau, V. Synthesis and Characterization of Regioselectively Monoderivatized Maltooligosaccharides through a Combination of Tandem Mass Spectrometry and Enzymatic Hydrolysis Studies. Arkivoc 2013, 2, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, B.; Zanini, D.; Ripoche, I.; Bürli, R.; Vasella, A. Oligosaccharide Analogues of Polysaccharides, Part 22, Synthesis of Cyclodextrin Analogues Containing a Buta-1,3-Diyne-1,4-Diyl or a Butane-1,4-Diyl Unit. Helv. Chim. Acta 2001, 84, 1862–1888. [Google Scholar] [CrossRef]
- Ruff, Y.; Buhler, E.; Candau, S.-J.; Kesselman, E.; Talmon, Y.; Lehn, J.-M. Glycodynamers: Dynamic Polymers Bearing Oligosaccharides Residues—Generation, Structure, Physicochemical, Component Exchange, and Lectin Binding Properties. J. Am. Chem. Soc. 2010, 132, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Bernet, B.; Vasella, A. Oligosaccharide Analogues of Polysaccharides. Helv. Chim. Acta 2002, 85, 265–287. [Google Scholar] [CrossRef]
- Kida, T.; Michinobu, T.; Zhang, W.; Nakatsuji, Y.; Ikeda, I. A Facile Synthesis of Novel Types of Cyclodextrin Derivatives by Insertion of an Aromatic Dicarbonyl Spacer into a Permethylated α-Cyclodextrin Skeleton. Chem. Commun. 2002, 8, 1596–1597. [Google Scholar] [CrossRef]
- Kida, T.; Kikuzawa, A.; Higashimoto, H.; Nakatsuji, Y.; Akashi, M. Synthesis of Novel Cyclodextrin Derivatives by Aromatic Spacer Insertion and Their Inclusion Ability. Tetrahedron 2005, 61, 5763–5768. [Google Scholar] [CrossRef]
- Almant, M.; Moreau, V.; Kovensky, J.; Bouckaert, J.; Gouin, S.G. Clustering of Escherichia Coli Type-1 Fimbrial Adhesins by Using Multimeric Heptyl α-D-Mannoside Probes with a Carbohydrate Core. Chem. Eur. J. 2011, 17, 10029–10038. [Google Scholar] [CrossRef]
- Ishida, Y.; Fukuhara, G. Efficient Cleavage of Permethylated Cyclodextrins. ACS Omega 2018, 3, 6279–6282. [Google Scholar] [CrossRef] [PubMed]
- Pélingre, M.; Smadhi, M.; Bil, A.; Bonnet, V.; Kovensky, J. One-Pot Synthesis of Asymmetrically Difunctionalized Oligomaltosides by Cyclodextrin Ring Opening. ChemistryOpen 2021, 10, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Jánossy, L.; Harangi, J.; Kandra, L.; Lipták, A. Synthesis of Chromogenic Substrates of α-Amylases on a Cyclodextrin Basis. Carbohydr. Res. 1997, 303, 407–415. [Google Scholar] [CrossRef]
- de Medeiros Modolon, S.; Otsuka, I.; Fort, S.; Minatti, E.; Borsali, R.; Halila, S. Sweet Block Copolymer NanoPicles: Preparation and Self-Assembly of Fully Oligosaccharide-Based Amphiphile. Biomacromolecules 2012, 13, 1129–1135. [Google Scholar] [CrossRef]
- Bösch, A.; Mischnick, P. Bifunctional Building Blocks for Glyco-Architectures by TiCl4-Promoted Ring Opening of Cyclodextrin Derivatives. Biomacromolecules 2007, 8, 2311–2320. [Google Scholar] [CrossRef] [PubMed]
- Sakairi, N.; Kuzuhara, H. Regioselective Mono-2-C-Iodination of Fully Methylated Cyclodextrins through Interconversions between Cyclic and Acyclic Structures. Chem. Lett. 1993, 22, 1093–1096. [Google Scholar] [CrossRef]
- Sakairi, N.; Kuzuhara, H. Facile Preparation of Phenyl 1-Thioglycosides of Partially Methylated Maltooligosaccharides by Restricted Thiolysis of Fully Methylated Cyclodextrins. Carbohydr. Res. 1996, 280, 139–143. [Google Scholar] [CrossRef]
- Lorentz, K. α-Amylase Assay: Current State and Future Development. J. Clin. Chem. Clin. Biochem. 1979, 17, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Hanson, N.; Yasmineh, W. Alpha-Amylase Activity by the Beckman Reaction System and Suppression by Pyruvate. Clin. Chem. 1979, 25, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, G.; Hilaire, G.; Aubry, C. Rapid Determination of Alpha-Amylase Activity by Use of a New Chromogenic Substrate. Clin. Chem. 1987, 33, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Lorentz, K. Evaluation of α-Amylase Assays with 4-Nitrophenyl-α-Oligosaccharides as Substrates. J. Clin. Chem. Clin. Biochem. 1983, 21, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kandra, L.; Gyémánt, G.; Farkas, E.; Lipták, A. Action Pattern of Porcine Pancreatic Alpha-Amylase on Three Different Series of β-Maltooligosaccharide Glycosides. Carbohydr. Res. 1997, 298, 237–242. [Google Scholar] [CrossRef]
- Oka, H.; Koyama, T.; Hatano, K.; Terunuma, D.; Matsuoka, K. Simple and Conveniently Accessible Bi-Fluorescence-Labeled Substrates for Amylases. Bioorg. Med. Chem. Lett. 2010, 20, 1969–1971. [Google Scholar] [CrossRef]
- Li, X.; Li, D.; Park, S.-H.; Gao, C.; Park, K.-H.; Gu, L. Identification and Antioxidative Properties of Transglycosylated Puerarins Synthesised by an Archaeal Maltogenic Amylase. Food Chem. 2011, 124, 603–608. [Google Scholar] [CrossRef]
- Lin, B.J. Immersion Lithography and Its Impact on Semiconductor Manufacturing. J. Micro/Nanolith. MEMS MOEMS 2004, 3, 377. [Google Scholar] [CrossRef]
- Cushen, J.D.; Otsuka, I.; Bates, C.M.; Halila, S.; Fort, S.; Rochas, C.; Easley, J.A.; Rausch, E.L.; Thio, A.; Borsali, R.; et al. Oligosaccharide/Silicon-Containing Block Copolymers with 5 Nm Features for Lithographic Applications. ACS Nano 2012, 6, 3424–3433. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, I.; Fuchise, K.; Halila, S.; Fort, S.; Aissou, K.; Pignot-Paintrand, I.; Chen, Y.; Narumi, A.; Kakuchi, T.; Borsali, R. Thermoresponsive Vesicular Morphologies Obtained by Self-Assemblies of Hybrid Oligosaccharide-Block-Poly(N-Isopropylacrylamide) Copolymer Systems. Langmuir 2010, 26, 2325–2332. [Google Scholar] [CrossRef]
- Sakai-Otsuka, Y.; Zaioncz, S.; Otsuka, I.; Halila, S.; Rannou, P.; Borsali, R. Self-Assembly of Carbohydrate-Block-Poly(3-Hexylthiophene) Diblock Copolymers into Sub-10 Nm Scale Lamellar Structures. Macromolecules 2017, 50, 3365–3376. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Komiyama, M. β-Cyclodextrin Bearing Diethylenetriamine As Highly Active Phosphodiester Hydrolyzing Agent. Chem. Lett. 1990, 19, 469–472. [Google Scholar] [CrossRef]
- Singh, S. Nanomaterials Exhibiting Enzyme-Like Properties (Nanozymes): Current Advances and Future Perspectives. Front. Chem. 2019, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, S.B.; Costa Duarte, F.Í.; Heimfarth, L.; Siqueira Quintans, J.D.S.; Quintans-Júnior, L.J.; Veiga Júnior, V.F.D.; Neves de Lima, Á.A. Cyclodextrin-Drug Inclusion Complexes: In Vivo and In Vitro Approaches. Int. J. Mol. Sci. 2019, 20, 642. [Google Scholar] [CrossRef] [Green Version]
- Nepogodiev, S.A.; Dedola, S.; Marmuse, L.; de Oliveira, M.T.; Field, R.A. Synthesis of Triazole-Linked Pseudo-Starch Fragments. Carbohydr. Res. 2007, 342, 529–540. [Google Scholar] [CrossRef] [PubMed]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Temperature (°C) | Time (h) | Sulfuric Acid Conc. (vol%) | Yield % |
|---|---|---|---|---|
| 1 | 0 | 72 | 6 | 35 |
| 2 | 30 | 30 | 6 | 35 |
| 3 | 40 | 11 | 6 | 38 |
| 4 | 50 | 10 | 2 | 35 |
| 5 | 50 | 4 | 6 | 49 |
| 6 | 50 | 1.5 | 10 | 35 |
| 7 | 60 | 2 | 6 | 43 |
| 8 | 78 | 0.5 | 6 | 40 |
| 9 | 100 | 0.1 | 6 | 40 |
| Acid. | [Acid] | Cd | [cd] | Functional Groups | T °C | Time | α/β | Yield | Starting Material Recovered | Ref | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C-2 et C-3 | C-6 | ||||||||||
| FeCl3 | 0.074 M | α | 0.3 M | none | RT then 70 °C | 2.5 + 3.5h | / | 20% | / | [40] | |
| 0.074 M | β | 0.3 M | none | RT then 70 °C | 2.5 + 3.5h | 9:1 | 22% | / | [40] | ||
| 0.074 M | γ | 0.3 M | none | RT then 70 °C | 2.5 + 3.5h | / | 23.5% | / | [40] | ||
| H2SO4 | 0.373 M | α | / | Ac | 50–60 °C | 20 h | 5.3:1 | 47% | 46% | [25] | |
| 1.2 M | α | / | Ac | 50 °C | 4 h | / | 48% | / | [24] | ||
| 0.373 M | β | / | Ac | 50–60 °C | 20 h | / | 41% | 49% | [25] | ||
| 0.373 M | γ | / | Ac | 50–60 °C | 20 h | / | 52% | 37% | [25] | ||
| 0.560 M | β | 0.021 M | Ac | Br | 57 °C | 28 h | / | 16% | 78% | [30] | |
| 0.373 M | β | 0.043 M | Bz | 1 I and 6 Bz | 55 °C | 24 h | / | 70% * | 9% | [31] | |
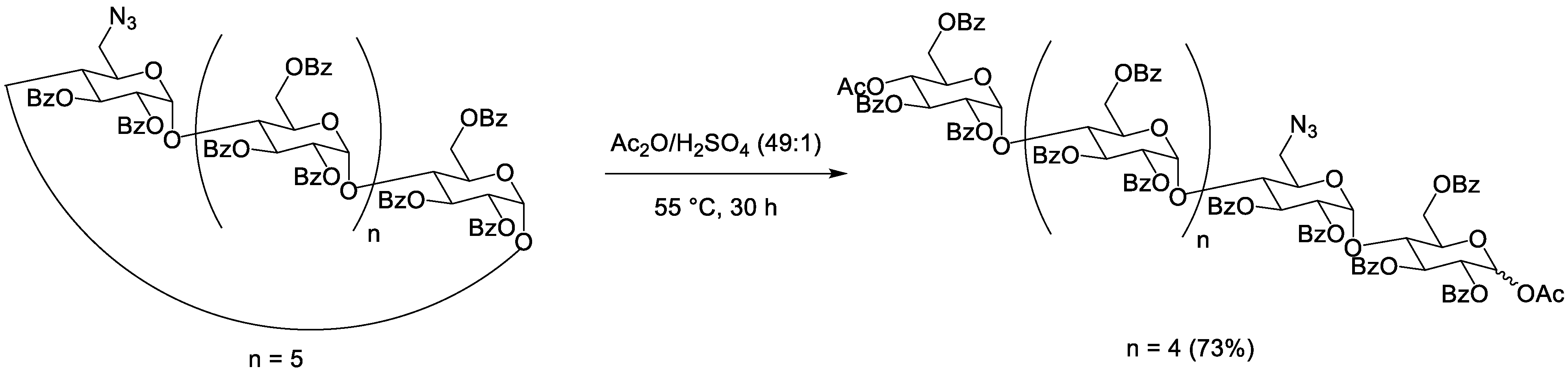
| 0.373 M | β | 0.044 M | Bz | 1 N3 and 6 Bz | 55 °C | 24 h | / | 73% | 10% | [31] | |
| 0.666 M | β | 0.099 M | Bz | 57 °C | 30 h | / | 32% | 58% | [30] | ||
| 0.373 M | α | 0.035 M | Bz | 60 °C | 30 h | / | 82% | 15% | [30] | ||
| 1.373 M | α | 0.036 M | Bz | 50 °C | 32 h | / | 51% | 36% | [26] | ||
| 0.373 M | β | 0.080 M | Bz | 55 °C | 42 h | / | 76% | 12% | [30] | ||
| 1.373 M | β | 0.086 M | Bz | 50 °C | 29 h | / | 37% | 54% | [26] | ||
| 0.373 M | γ | 0.015 M | Bz | 50 °C | 35 h | / | 70% | 17% | [30] | ||
| 1.373 M | γ | 0.015 M | Bz | 50 °C | 27 h | / | 48% | 39% | [26] | ||
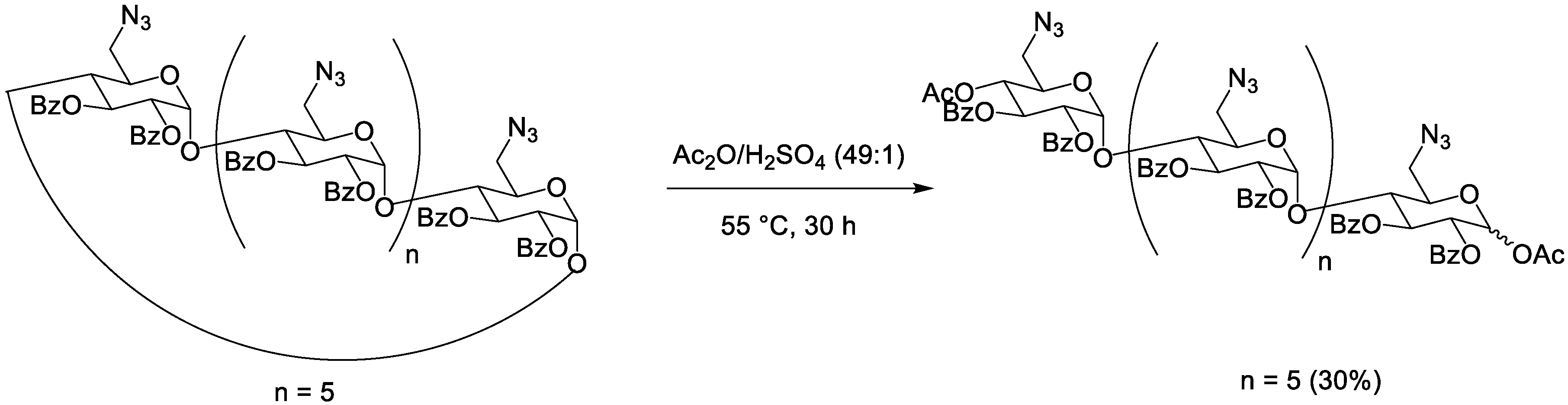
| 0.373 M | β | 0.102 M | Bz | N3 | 55 °C | 30 h | / | 30% | 66% | [30] | |
| HClO4 | 0.086 M | α | 0.019 M | Ac | 0 °C | 22 h | / | 60% | 30% | [30] | |
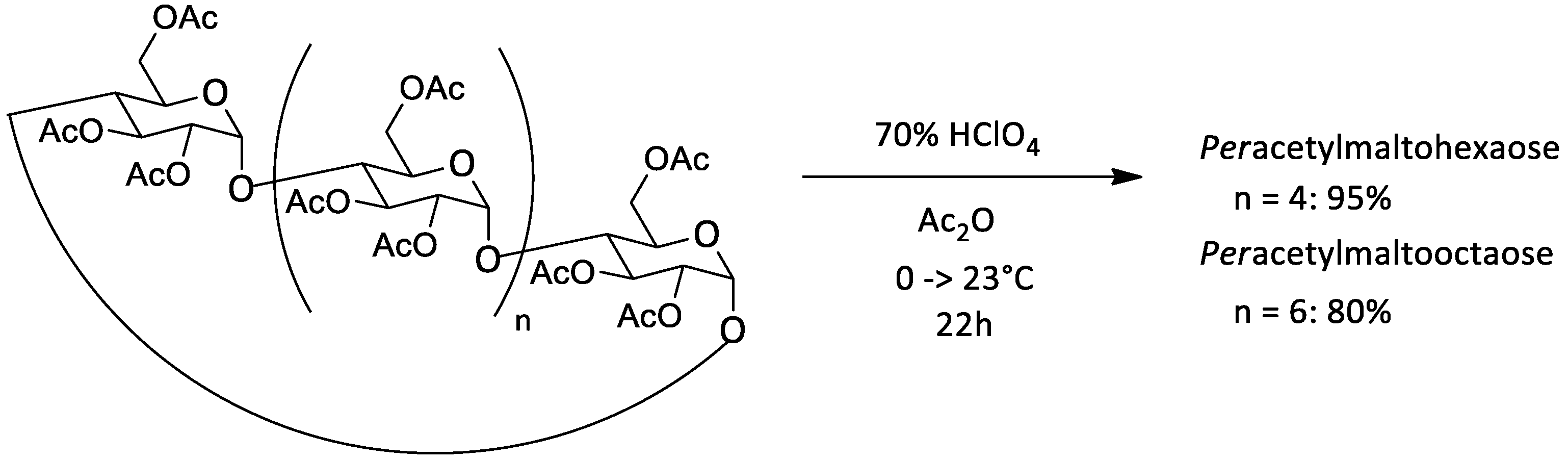
| 0.087 M | α | 0.019 M | Ac | 0 °C | 45 h | >9:1 | 95% | / | [32] | ||
| 0.087 M | α | 0.019 M | Ac | 0 °C | 45 h | >9:1 | 95% | / | [32] | ||
| 0.084 M | β | 0.018 M | Ac | 0 °C | 20 h | / | 35% | 55% | [30] | ||
| 0.086 M | β | / | Bz | Br | 0 °C then 36 °C | 2 × 20 h | / | 30% | / | [30] | |
| 0.036 M | β | 0.0072 M | Bz | N3 | −20 °C | 16 h | / | 85% | / | [37] | |
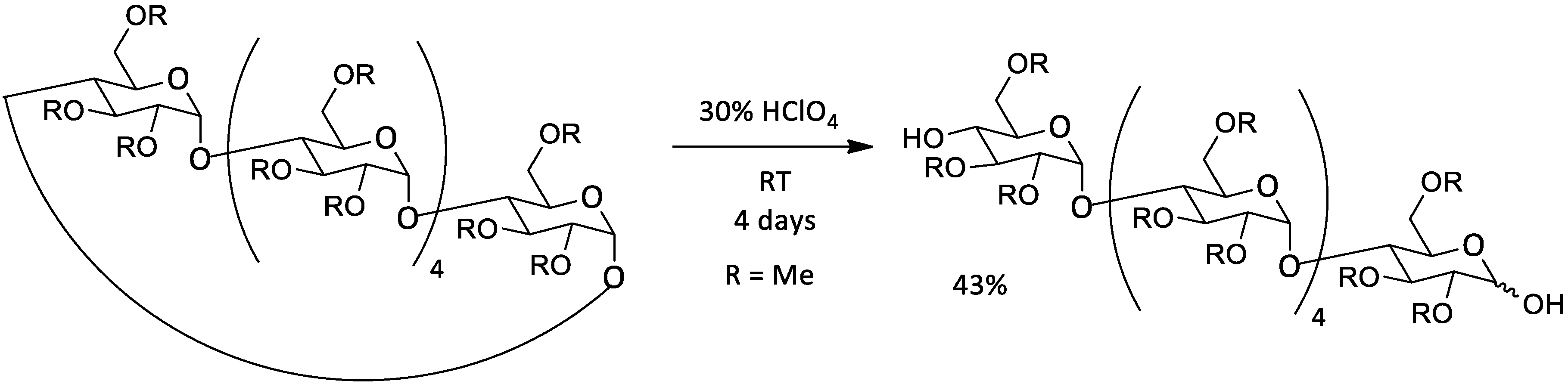
| ZnBr(+PhSTMS) | 0.266 M | α | 0.066 M | Me | RT | 5 days | 1:1 | 28% | 68% | [44] | |
| 0.2 M | β | 0.2 M | Me | RT | 4 days | 1:1 | 40% | / | [44] | ||
| 0.2 M | γ | 0.05 M | Me | RT | 4 days | 2:8 | 41% | / | [44] | ||
| TiCl4 | 0.4 M | α | 0.1 M | Me, Et or All | 10 °C | 45h | 96:4 | 41% * | / | [42] | |
| 0.4 M | β | 0.1 M | Me, Et or All | RT | 24h | 99:1 | 66% * | / | [42] | ||
| 0.4 M | γ | 0.1 M | Me, Et or All | 10 °C | 45h | 99:1 | 92%* | / | [42] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pélingre, M.; Koffi Teki, D.S.-E.; El-Abid, J.; Chagnault, V.; Kovensky, J.; Bonnet, V. Ring-Opening of Cyclodextrins: An Efficient Route to Pure Maltohexa-, Hepta-, and Octaoses. Organics 2021, 2, 287-305. https://0-doi-org.brum.beds.ac.uk/10.3390/org2030015
Pélingre M, Koffi Teki DS-E, El-Abid J, Chagnault V, Kovensky J, Bonnet V. Ring-Opening of Cyclodextrins: An Efficient Route to Pure Maltohexa-, Hepta-, and Octaoses. Organics. 2021; 2(3):287-305. https://0-doi-org.brum.beds.ac.uk/10.3390/org2030015
Chicago/Turabian StylePélingre, Matthieu, Dindet Steve-Evanes Koffi Teki, Jamal El-Abid, Vincent Chagnault, José Kovensky, and Véronique Bonnet. 2021. "Ring-Opening of Cyclodextrins: An Efficient Route to Pure Maltohexa-, Hepta-, and Octaoses" Organics 2, no. 3: 287-305. https://0-doi-org.brum.beds.ac.uk/10.3390/org2030015