Mutational Frequencies of SARS-CoV-2 Genome during the Beginning Months of the Outbreak in USA

 and
and
Abstract
:1. Introduction
2. Results
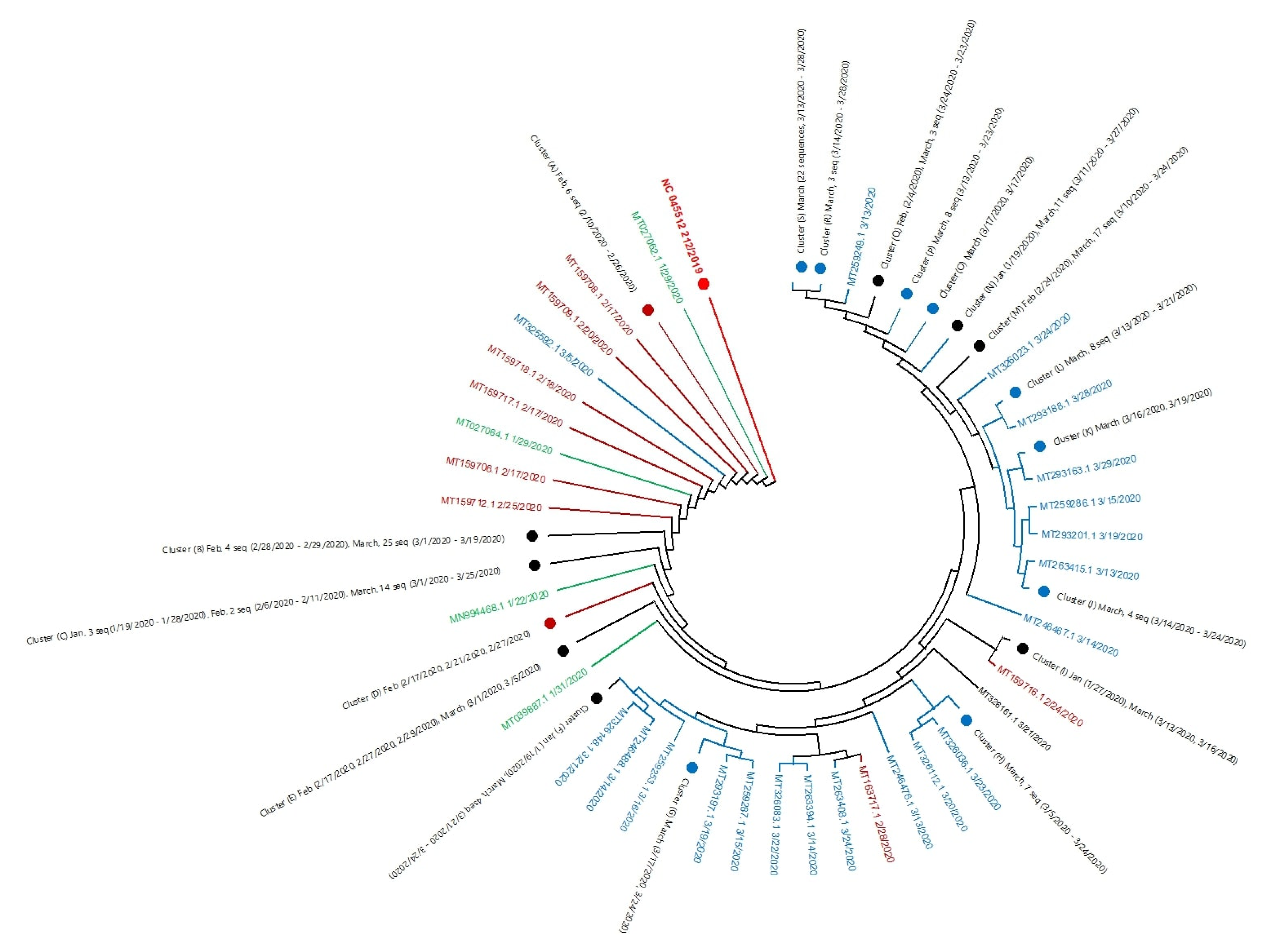
2.1. Genome Analysis
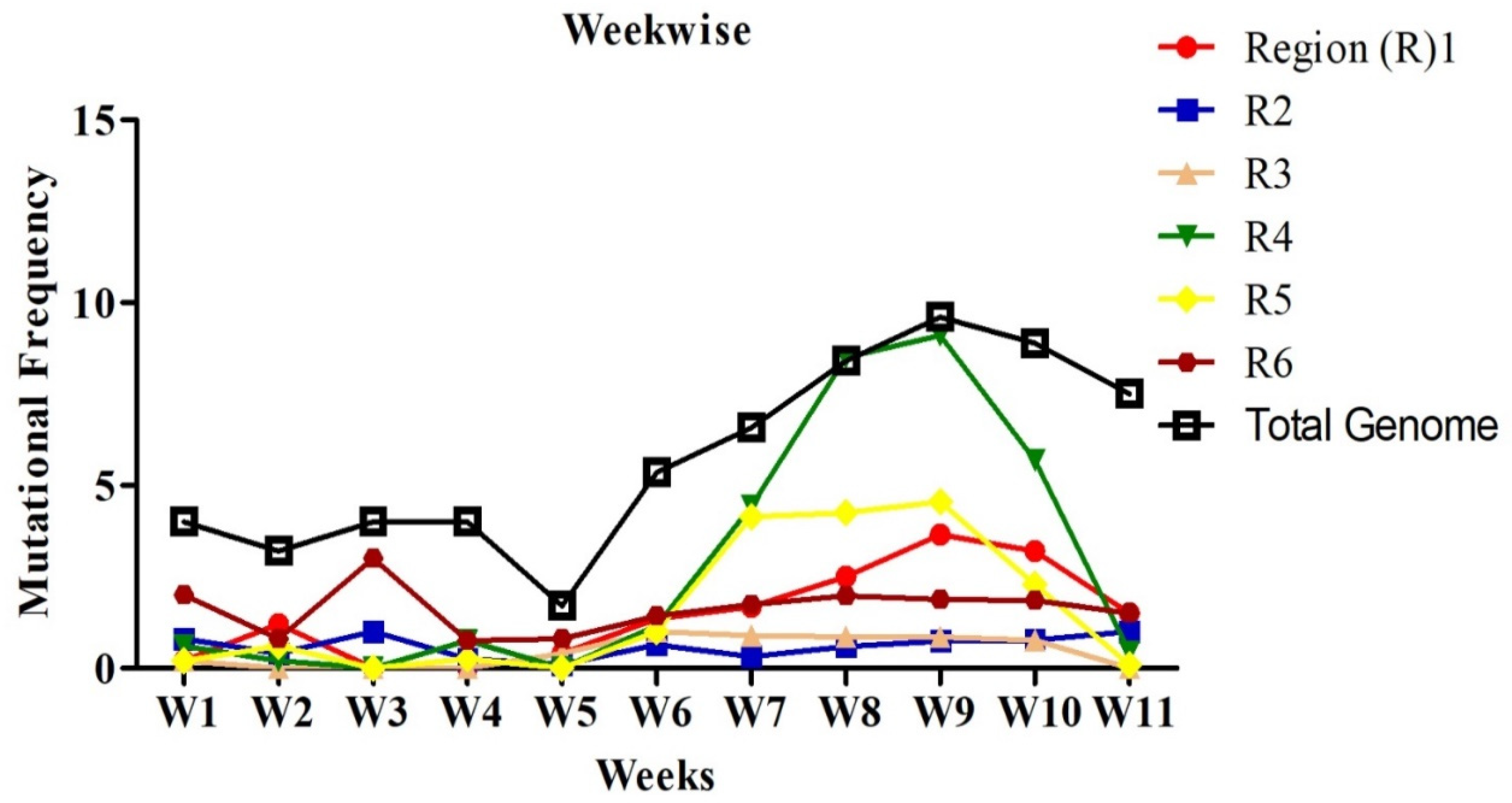
2.2. Mutational Analysis in Genome
2.3. Mutational Analysis of COVID19 Proteins
2.4. Mutational Analysis in Spike Protein
3. Discussion
4. Material and Methods
4.1. Genome Sequences Retrieval
4.2. Genome Analysis
4.3. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Novel Coronavirus—China. Available online: https://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/ (accessed on 17 May 2020).
- WHO. Naming the Coronavirus Disease (COVID-19) and the Virus that Causes It. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it (accessed on 17 May 2020).
- Zhu, N.; Zhang, D.; Wang, W. China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, Q.; Zhang, T. Pangolin homology associated with 2019-nCoV. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.-W.; Wong, B.H.; Wong, S.S.; Leung, S.-Y.; Chan, K.-H.; Yuen, K.-Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagaili, A.N.; Briese, T.; Mishra, N.; Kapoor, V.; Sameroff, S.C.; de Wit, E.; Munster, V.J.; Hensley, L.E.; Zalmout, I.S.; Kapoor, A. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. MBio 2014, 5, e00884-00814. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Zheng, B.; He, Y.; Liu, X.; Zhuang, Z.; Cheung, C.; Luo, S.; Li, P.; Zhang, L.; Guan, Y. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Ruan, S. Likelihood of survival of coronavirus disease 2019. Lancet Infec. Dis. 2020, 20, 630–631. [Google Scholar] [CrossRef]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2020. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [Green Version]
- Bjedov, I.; Tenaillon, O.; Gerard, B.; Souza, V.; Denamur, E.; Radman, M.; Taddei, F.; Matic, I. Stress-induced mutagenesis in bacteria. Science 2003, 300, 1404–1409. [Google Scholar] [CrossRef]
- Lai, M.M.; Cavanagh, D. The molecular biology of coronaviruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 1997; Volume 48, pp. 1–100. [Google Scholar]
- Hon, C.-C.; Lam, T.-Y.; Shi, Z.-L.; Drummond, A.J.; Yip, C.-W.; Zeng, F.; Lam, P.-Y.; Leung, F.C.-C. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 2008, 82, 1819–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; Van der Hoek, L. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Niu, P.; Wang, L.; Zhou, H.; Zhao, X.; Wang, W.; Wang, J.; Ji, C.; Ding, X.; Wang, X. Mutations, Recombination and Insertion in the Evolution of 2019-nCoV. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Shen, G.; Zhang, Y.; Huang, K.-S.; Ho, H.-Y.; Hor, W.-S.; Yang, C.-H.; Li, C.; Wang, W.-L. Analysis of the mutation dynamics of SARS-CoV-2 reveals the spread history and emergence of RBD mutant with lower ACE2 binding affinity. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef]
- Maitra, A.; Sarkar, M.C.; Raheja, H.; Biswas, N.K.; Chakraborti, S.; Singh, A.K.; Ghosh, S.; Sarkar, S.; Patra, S.; Mondal, R.K. Mutations in SARS-CoV-2 viral RNA identified in Eastern India: Possible implications for the ongoing outbreak in India and impact on viral structure and host susceptibility. J. Biosci. 2020, 45, 76. [Google Scholar] [CrossRef]
- Lokman, S.M.; Rasheduzzaman, M.; Salauddin, A.; Barua, R.; Tanzina, A.Y.; Rumi, M.H.; Hossain, M.I.; Siddiki, A.Z.; Mannan, A.; Hasan, M.M. Exploring the genomic and proteomic variations of SARS-CoV-2 spike glycoprotein: A computational biology approach. Infect. Genet. Evol. 2020, 104389. [Google Scholar] [CrossRef]
- Coppee, F.; Lechien, J.R.; Decleves, A.E.; Tafforeau, L.; Saussez, S. Severe acute respiratory syndrome coronavirus 2: Virus mutations in specific European populations. New Microbes New Infect. 2020, 36, 100696. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Ou, J.; Zhou, Z.; Dai, R.; Zhang, J.; Lan, W.; Zhao, S.; Wu, J.; Seto, D.; Cui, L.; Zhang, G. Emergence of RBD mutations in circulating SARS-CoV-2 strains enhancing the structural stability and human ACE2 receptor affinity of the spike protein. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Banerjee, A.K.; Tripathi, P.P.; Srivastava, A.K.; Ray, U. A virus that has gone viral: Amino acid mutation in S protein of Indian isolate of Coronavirus COVID-19 might impact receptor binding, and thus, infectivity. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed]
- Johns Hopkins University. COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). 2020. Available online: https://0-coronavirus-jhu-edu.brum.beds.ac.uk/map.html (accessed on 17 May 2020).
- Johns Hopkins University. Mortality Analyses. Available online: https://0-coronavirus-jhu-edu.brum.beds.ac.uk/data/mortality (accessed on 17 May 2020).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Walls, A.C.; Xiong, X.; Park, Y.-J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell 2019, 176, 1026–1039.e1015. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef]
- WHO Organization. WHO Director-General’s opening remarks at the media briefing on COVID-19-11 March 2020. 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 17 May 2020).
- Guo, Y.-R.; Cao, Q.-D.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.-S.; Wang, D.-Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak–an update on the status. Mil. Med. Res. 2020, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawood, A.A. Mutated COVID-19, may foretells mankind in a great risk in the future. New Microbes New Infect. 2020, 35, 100673. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Peng, G.; Wilken, M.; Geraghty, R.J.; Li, F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2012, 287, 8904–8911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef]
- Fung, S.-Y.; Yuen, K.-S.; Ye, Z.-W.; Chan, C.-P.; Jin, D.-Y. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: Lessons from other pathogenic viruses. Emerg. Microb. Infect. 2020, 9, 558–570. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M.V. Positive selection of ORF3a and ORF8 genes drives the evolution of SARS-CoV-2 during the 2020 COVID-19 pandemic. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Benvenuto, D.; Giovanetti, M.; Ciccozzi, A.; Spoto, S.; Angeletti, S.; Ciccozzi, M. The 2019-new coronavirus epidemic: Evidence for virus evolution. J. Med. Virol. 2020, 92, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Consortium, C.S.M.E. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 2004, 303, 1666–1669. [Google Scholar]
- Tang, J.W.; Cheung, J.L.; Chu, I.M.; Sung, J.J.; Peiris, M.; Chan, P.K. The large 386-nt deletion in SARS-associated coronavirus: Evidence for quasispecies? J. Infect. Dis. 2006, 194, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Nabar, N.R.; Huang, N.-N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.H.; Fung, T.S.; Fang, S.; Huang, M.; Le, M.T.; Liu, D.X. Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin-dependent rapid degradation of interferon regulatory factor 3. Virology 2018, 515, 165–175. [Google Scholar] [CrossRef]
- Zhang, Q.; Shi, K.; Yoo, D. Suppression of type I interferon production by porcine epidemic diarrhea virus and degradation of CREB-binding protein by nsp1. Virology 2016, 489, 252–268. [Google Scholar] [CrossRef] [Green Version]
- Van Hemert, M.J.; Van Den Worm, S.H.; Knoops, K.; Mommaas, A.M.; Gorbalenya, A.E.; Snijder, E.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008, 4, e1000054. [Google Scholar] [CrossRef] [Green Version]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weißbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Marchand, B.; Te Velthuis, A.J.; Snijder, E.J.; Weiss, S.; Eoff, R.L.; Singh, K.; Sarafianos, S.G. Mechanism of nucleic acid unwinding by SARS-CoV helicase. PloS ONE 2012, 7, e36521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, R.W.; Chim, S.S.; Tong, Y.-K.; Fung, K.S.; Chan, P.K.; Zhao, G.-P.; Lo, Y.D. Tracing SARS-coronavirus variant with large genomic deletion. Emerg. Infect. Dis. 2005, 11, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. In Coronaviruses; Humana Press: New York, NY, USA, 2015; pp. 1–23. [Google Scholar]
- Pfefferle, S.; Krähling, V.; Ditt, V.; Grywna, K.; Mühlberger, E.; Drosten, C. Reverse genetic characterization of the natural genomic deletion in SARS-Coronavirus strain Frankfurt-1 open reading frame 7b reveals an attenuating function of the 7b protein in-vitro and in-vivo. Virol. J. 2009, 6, 131. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Li, J.; Guo, M.; Tian, X.; Liu, C.; Wang, X.; Yang, X.; Wu, P.; Xiao, Z.; Qu, Y. Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wright, H.L.; Cross, A.L.; Edwards, S.W.; Moots, R.J. Effects of IL-6 and IL-6 blockade on neutrophil function in vitro and in vivo. Rheumatology 2014, 53, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microb. Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.L.; Millet, J.K.; Daniel, S.; Freed, J.H.; Whittaker, G.R. The SARS-CoV fusion peptide forms an extended bipartite fusion platform that perturbs membrane order in a calcium-dependent manner. J. Mol. Biol. 2017, 429, 3875–3892. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [Green Version]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Unique Mutations |
|---|---|
| Leader | G82_V86del (509_523delGGTCATGTTATGGTT), K141_F143del (686_694delAAGTCATTT), D75*E (490**T > A), A117T (614G > A), M1L (266A > T), L21S (327T > C) [6 ***] |
| NSP2 | F10L (833T > C), D43N (932G > A), T85I (1059C > T), N98S (1098A > G), P129L (1191C > T), H194Y (1385C > T), G199E (1401G > A), G212D (1440G > A), T223I (1473C > T), S248N (1548G > A), V311M (1736G > A), K337R (1815A > G), G339S (1820G > A), A361V (1887C > T), T429I (2091C > T), V480A (2244T > C), M609I (2632G > T) [17] |
| NSP3 | A58T (2891G > A) **, A655V (4683C > T), T127I (3099C > T), P153L (3177C > T), Q180H (3259G > T), T217I (3369C > T), D218E (3373C > A), A231V (3411C > T), P340L (3738C > T), D410Y (3947G > T), K412N (3955G > T), N506S (4236A > G), S697F (4809C > T), S721del (4880_4882delAGT), T763M (5007C > T), P778L (5052C > T), I789V (5084A > G), M951I (5572G > T), T1004I (5730C > T), T1022I (5784C > T), K1042N (5845A > T), S1106G (6035A > G), A1179V (6255C > T), V1209A (6345T > C), T1306I (6636C > T), K1325R (6693A > G), T1482I (7164C > T), N1587S (7479A > G), A1600V (7518C > T), R1614K (7560G > A), K1771R (8031A > G) [31] |
| NSP4 | A307V (9474C > T), T327N (9534C > A), A128V (8937C > T), I43V (8681A > G) [4] |
| NSP5 | L89F (10319C > T), A173V (10572C > T, T190I (10623C > T), A255V (10818C > T) [4] |
| NSP6 | L37F (11083G > T), L260F (11750C > T), V149F (11417G > T), F191del (11543_11545delTTT)I189T, (11538T > C) [5] |
| NSP7 | No mutations |
| NSP8 | T187I (12651C > T), S41F (12213C > T) [2] |
| NSP9 | No mutations |
| NSP10 | No mutations |
| NSP11 | No mutations |
| RNA dependent RNA polymerase | G44V (13571G > T), K103R (13748A > G), M110V (13768A > G), Q191L (14012A > T), P323L (14408C > T), L372F (14554C > T), K426N (14718G > T), N491S (14912A > G), E744D (15672G > T), G774S (15760G > A), K780T (15779A > C), H810L (15869A > T) [12] |
| Helicase | A18V (16289C > T), G54C (16396G > T), P77L (16466C > T), P78S (16468C > T), K131R (16628A > G), V226L (16912G > T), T255I (17000C > T), P364S (17326C > T), R392C (17410C > T), T413I (17474C > T), K460R (17615A > G), S468L (17639C > T), P504L (17747C > T), Y541C (17858A > G), T550A (17884A > G), A553V (17894C > T), M576I (17964G > A) [17] |
| 3′to 5′ exonuclease | F233L (18736T > C), V287F (18898 G > T), D379A (19175A > C), M501I (19542G > T) [4] |
| EndoRnase | N4K (19632T > A), V9F (19645G > T), V22L (19684G > T), G76D (19847G > A), V127F (19999G > T), F221L (20281T > C), L227del (20299_20301delTTA) [7] |
| 2′O Ribose methyltransferase | Y242_S243insF (21384_21385insTTC), S243F (21386C > T), G265V (21452G > T) [3] |
| Spike | L5F (21575C > T), P9L (21588C > T), H49Y (21707C > T), L54F (21724G > C), F157L (22033C > A), W258L (22335G > T), A348T (22604G > A), G476S (22988G > A), V483A (23010T > C), H519Q (23119T > A), D614G (23403A > G), V615F (23405G > T), V622I (23426G > A), D936Y (24368G > T), S939F (24378C > T), A1078S (24794G > T) [16] |
| ORF3a | V13L (25429G > T), L53F (25549C > T), F56C (25559T > G), Q57H (25563G > T), V88A (25655T > C), A99V (25688C > T), T151I (25844C > T), E239D (26109G > T), G251V (26144G > T) [9] |
| Envelope | No mutations |
| Matrix | D3G (26530A > G) [1] |
| ORF6 | V9F (27226G > T) [1] |
| ORF 7a | S81L (27635C > T), I110T (27722T > C) [2] |
| ORF7b | No mutations |
| ORF8 | M1T (27895T > C), T11A (27924A > G), T11I (27925C > T), S24L (27964C > T), P36S (27999C > T), V62L (28077G > C), S69L (28099C > T), L84S (28144T > C) [8] |
| Nucleocapsid | P6T (28289C > A), S23T (28340T > A), P46S (28409C > T), A152S (28727G > T), S183Y (28821C > A), R185C (28826C > T), S194L (28854C > T), S202N (28878G > A),R203K (28881G > A 28882G > A), G204R (28883G > C), T205I (28887C > T), S232T (28968G > C 28969C > T) [12] |
| ORF10 | No mutations |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaushal, N.; Gupta, Y.; Goyal, M.; Khaiboullina, S.F.; Baranwal, M.; Verma, S.C. Mutational Frequencies of SARS-CoV-2 Genome during the Beginning Months of the Outbreak in USA. Pathogens 2020, 9, 565. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9070565
Kaushal N, Gupta Y, Goyal M, Khaiboullina SF, Baranwal M, Verma SC. Mutational Frequencies of SARS-CoV-2 Genome during the Beginning Months of the Outbreak in USA. Pathogens. 2020; 9(7):565. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9070565
Chicago/Turabian StyleKaushal, Neha, Yogita Gupta, Mehendi Goyal, Svetlana F. Khaiboullina, Manoj Baranwal, and Subhash C. Verma. 2020. "Mutational Frequencies of SARS-CoV-2 Genome during the Beginning Months of the Outbreak in USA" Pathogens 9, no. 7: 565. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9070565