Amyloid Beta Peptides and Th1 Cytokines Modulate Human Brain Vascular Smooth Muscle Tonic Contractile Capacity In Vitro: Relevance to Alzheimer’s Disease?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Collagen Gel Contraction Assay
2.2.1. Preparation of Rat Tail Type 1 Collagen
2.2.2. Preparation of HBVSMC/Collagen Gel
2.3. Collagen Gel Treatment with Aβ and Cytokines
2.4. Gel Contraction Analysis
2.5. Data Analysis
3. Results
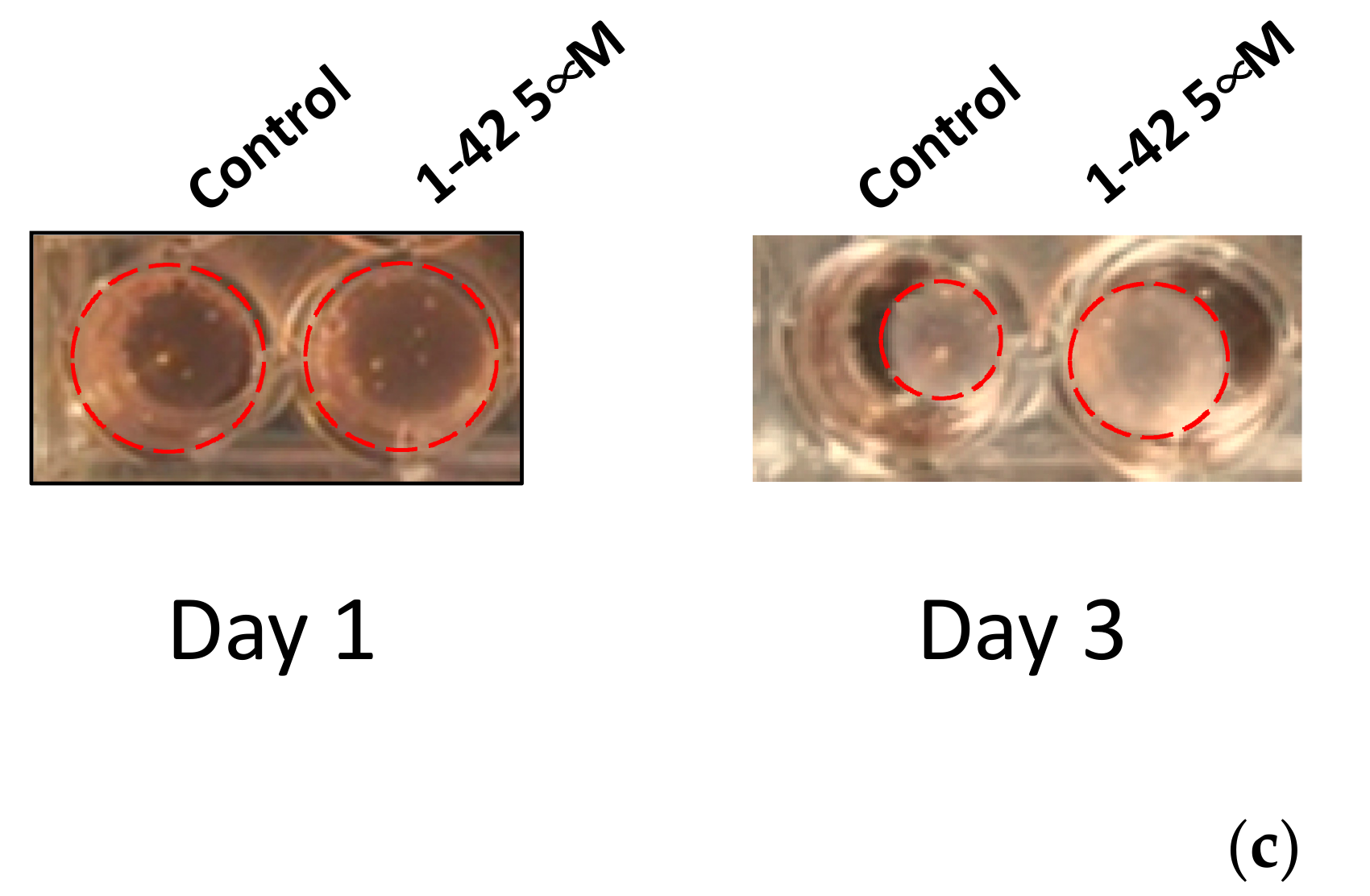
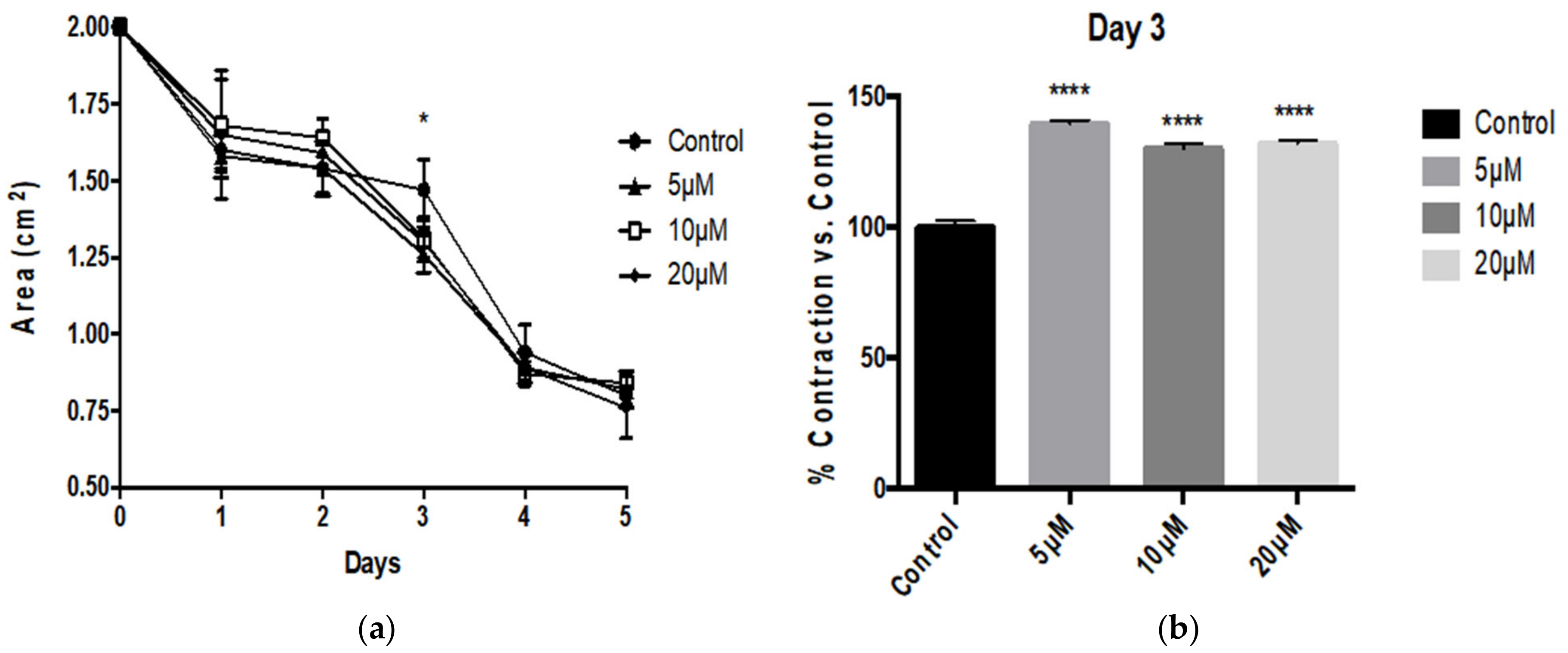
3.1. Aβ Peptides Disturb HBVSMC Contractility
3.2. Inflammatory Cytokines Suppress Human Vascular Smooth Muscle Tonic Contractility
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory Cytokines Induce Amyloid Beta Neurotoxicity through Modulating Amyloid Precursor Protein Levels/Metabolism. BioMed Res. Int. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Liao, Y.-F.; Wang, B.-J.; Cheng, H.-T.; Kuo, L.-H.; Wolfe, M.S. Tumor Necrosis Factor-α, Interleukin-1β, and Interferon-γ Stimulate γ-Secretase-mediated Cleavage of Amyloid Precursor Protein through a JNK-dependent MAPK Pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine—Searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Levine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- Wei, G.; Shea, J.-E. Effects of Solvent on the Structure of the Alzheimer Amyloid-β(25–35) Peptide. Biophys. J. 2006, 91, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Wakamiya, A.; Maeda, T.; Noguchi, K.; Takashima, A.; Imahori, K. Correlation among Secondary Structure, Amyloid Precursor Protein Accumulation, and Neurotoxicity of Amyloid β(25–35) Peptide as Analyzed by Single Alanine Substitution. J. Biochem. 1995, 118, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Nägga, K.; Wattmo, C.; Zhang, Y.; Wahlund, L.-O.; Palmqvist, S. Cerebral inflammation is an underlying mechanism of early death in Alzheimer’s disease: A 13-year cause-specific multivariate mortality study. Alzheimers Res. Ther. 2014, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhu, M.; Hjorth, E.; Cortés-Toro, V.; Eyjolfsdottir, H.; Graff, C.; Nennesmo, I.; Palmblad, J.; Eriksdotter, M.; Sambamurti, K.; et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement. 2015, 11, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular Dysfunction and Faulty Amyloid-Peptide Clearance in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef] [Green Version]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 2011, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingues, C.; Silva, O.A.D.C.E.; Henriques, A.G. Impact of Cytokines and Chemokines on Alzheimer’s Disease Neuropathological Hallmarks. Curr. Alzheimer Res. 2017, 14, 870–882. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kiyota, T.; Horiba, M.; Buescher, J.L.; Walsh, S.M.; Gendelman, H.E.; Ikezu, T. Interferon-γ and Tumor Necrosis Factor-α Regulate Amyloid-β Plaque Deposition and β-Secretase Expression in Swedish Mutant APP Transgenic Mice. Am. J. Pathol. 2007, 170, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [PubMed]
- Reinhard, M.; Lorenz, L.; Sommerlade, L.; Allignol, A.; Urbach, H.; Weiller, C.; Egger, K. Impaired dynamic cerebral autoregulation in patients with cerebral amyloid angiopathy. Brain Res. 2019, 1717, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Goos, J.D.; Kester, M.; Barkhof, F.; Klein, M.; Blankenstein, M.A.; Scheltens, P.; van der Flier, W.M. Patients with Alzheimer Disease With Multiple Microbleeds. Stroke 2009, 40, 3455–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.W.; Barzegar, M.; Boyer, C.J.; Minagar, A.; Couraud, P.O.; Alexander, J.S. Brain Endothelial Cells Release Apical and Basolateral Microparticles in Response to Inflammatory Cytokine Stimulation: Relevance to Neuroinflammatory Stress? Front. Immunol. 2019, 10, 1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanekiyo, T.; Liu, C.-C.; Shinohara, M.; Li, J.; Bu, G. LRP1 in Brain Vascular Smooth Muscle Cells Mediates Local Clearance of Alzheimer’s Amyloid-β. J. Neurosci. 2012, 32, 16458–16465. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, E.; Sánchez-Maldonado, C.; Mayoral, M.A.; Mendieta, L.; Alatriste, V.; Patricio-Martínez, A.; Limón, I.D. Neuroinflammation induced by the peptide amyloid-β (25–35) increase the presence of galectin-3 in astrocytes and microglia and impairs spatial memory. Neuropeptides 2019, 74, 11–23. [Google Scholar] [CrossRef]
- Preston, J.E.; Hipkiss, A.R.; Himsworth, D.T.; Romero, I.A.; Abbott, J.N. Toxic effects of β-amyloid(25–35) on immortalised rat brain endothelial cell: Protection by carnosine, homocarnosine and β-alanine. Neurosci. Lett. 1998, 242, 105–108. [Google Scholar] [CrossRef]
- Mukhamediarov, M.A.; Volkov, E.M.; Leushina, A.V.; Kochunova, I.O.; Palotas, A.; Zefirov, A.L. Ionic and molecular mechanisms of beta-amyloid-induced depolarization of the mouse skeletal muscle fibres. Российский Физиологический Журнал И М Сеченова 2011, 97, 795–803. [Google Scholar]
- Haase, N.; Herse, F.; Spallek, B.; Haase, H.; Morano, I.; Qadri, F.; Szijártó, I.A.; Rohm, I.; Yilmaz, A.; Warrington, J.P.; et al. Amyloid-β Peptides Activate α1-Adrenergic Cardiovascular Receptors. Hypertension 2013, 62, 966–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchor, J.P.; van Nostrand, W.E. Fibrillar Amyloid β-Protein Mediates the Pathologic Accumulation of Its Secreted Precursor in Human Cerebrovascular Smooth Muscle Cells. J. Biol. Chem. 2000, 275, 9782–9791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coma, M.; Guix, F.X.; Ill-Raga, G.; Uribesalgo, I.; Alameda, F.; Valverde, M.A.; Muñoz, F.J. Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiol. Aging 2008, 29, 969–980. [Google Scholar] [CrossRef]
- Hald, E.S.; Timm, C.D.; Alford, P.W. Amyloid Beta Influences Vascular Smooth Muscle Contractility and Mechanoadaptation. J. Biomech. Eng. 2016, 138, 111007. [Google Scholar] [CrossRef] [PubMed]
- Rios, M.A.E.; Etoral-Rios, D.; Efranco-Bocanegra, D.; Evilleda-Hernández, J.; Ecampos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Blaise, R.; Mateo, V.; Rouxel, C.; Zaccarini, F.; Glorian, M.; Béréziat, G.; Golubkov, V.S.; Limon, I. Wild-type amyloid beta 1-40 peptide induces vascular smooth muscle cell death independently from matrix metalloprotease activity. Aging Cell 2012, 11, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Fang, C.; Crawford, F.; Mullan, M. Superoxide free radical and intracellular calcium mediate A β1–42 induced endothelial toxicity. Brain Res. 1997, 762, 144–152. [Google Scholar] [CrossRef]
- Miners, J.S.; Kehoe, P.; Love, S. Neprilysin Protects against Cerebral Amyloid Angiopathy and Aβ-Induced Degeneration of Cerebrovascular Smooth Muscle Cells. Brain Pathol. 2011, 21, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.M.; de Waal, R.M.W.; Schipper, J.J.; van Nostrand, W.E. Rapid Degeneration of Cultured Human Brain Pericytes by Amyloid β Protein. J. Neurochem. 2002, 68, 1135–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carare, R.O.; Aldea, R.; Agarwal, N.; Bacskai, B.J.; Bechman, I.; Boche, D.; Bu, G.; Bulters, D.; Clemens, A.; Counts, S.E.; et al. Clearance of interstitial fluid (ISF) and CSF (CLIC) group—Part of Vascular Professional Interest Area (PIA). Alzheimers Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12053. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, J.W.; Washington, C.; McCormick, J.; Stevenson, E.; Alexander, J.S. Amyloid Beta Peptides and Th1 Cytokines Modulate Human Brain Vascular Smooth Muscle Tonic Contractile Capacity In Vitro: Relevance to Alzheimer’s Disease? Pathophysiology 2021, 28, 64-75. https://0-doi-org.brum.beds.ac.uk/10.3390/pathophysiology28010006
Yun JW, Washington C, McCormick J, Stevenson E, Alexander JS. Amyloid Beta Peptides and Th1 Cytokines Modulate Human Brain Vascular Smooth Muscle Tonic Contractile Capacity In Vitro: Relevance to Alzheimer’s Disease? Pathophysiology. 2021; 28(1):64-75. https://0-doi-org.brum.beds.ac.uk/10.3390/pathophysiology28010006
Chicago/Turabian StyleYun, J. Winny, Caretia Washington, Joi McCormick, Emily Stevenson, and J. Steven Alexander. 2021. "Amyloid Beta Peptides and Th1 Cytokines Modulate Human Brain Vascular Smooth Muscle Tonic Contractile Capacity In Vitro: Relevance to Alzheimer’s Disease?" Pathophysiology 28, no. 1: 64-75. https://0-doi-org.brum.beds.ac.uk/10.3390/pathophysiology28010006