Iron Regulation: Macrophages in Control


Medical University of Vienna, Center for Pathobiochemistry and Genetics, Vienna 1090, Austria
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2018, 11(4), 137; https://0-doi-org.brum.beds.ac.uk/10.3390/ph11040137
Submission received: 15 November 2018
/
Revised: 10 December 2018
/
Accepted: 12 December 2018
/
Published: 14 December 2018
(This article belongs to the Special Issue Iron as Therapeutic Targets in Human Diseases)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Macrophages are sentinel cells of the innate immune system and have important functions in development, tissue homeostasis, and immunity. These phylogenetically ancient cells also developed a variety of mechanisms to control erythropoiesis and the handling of iron. Red pulp macrophages in the spleen, Kupffer cells in the liver, and central nurse macrophages in the bone marrow ensure a coordinated metabolism of iron to support erythropoiesis. Phagocytosis of senescent red blood cells by macrophages in the spleen and the liver provide a continuous delivery of recycled iron under steady-state conditions and during anemic stress. Central nurse macrophages in the bone marrow utilize this iron and provide a cellular scaffold and niche to promote differentiation of erythroblasts. This review focuses on the role of the distinct macrophage populations that contribute to efficient iron metabolism and highlight important cellular and systemic mechanisms involved in iron-regulating processes.
1. Introduction
Iron is a multifaceted metal that plays a versatile role in many basic cellular processes in the body, including DNA synthesis, cellular metabolism and respiration, as well as cell growth and death mechanisms through iron-containing and iron-sequestering proteins and enzymes [1,2]. Moreover, iron is the main component of hemoglobin (Hgb) and is thus essential for oxygen transport within erythrocytes. As a redox-active metal, it can be found in two main oxidation states in the body that determine its features, functional roles, and toxicity. Whereas divalent ferrous iron (Fe2+) is the active compound in Hgb and in many enzymes, it can cause cellular toxicity by means of intracellular free radical production by Fenton reactions [3,4]. Therefore, iron is usually intracellularly stored or systemically transported as oxidized trivalent ferric iron (Fe3+). Iron metabolism is tightly regulated by a variety of redundant mechanisms that are often conserved throughout the mammalian kingdom to adjust iron concentrations on systemic and cellular levels. In response to iron and red blood cell (RBC) demand, an orchestrated interplay between iron-processing cells, including tissue macrophages, hepatocytes, erythrocytes, and duodenal epithelial cells controls and maintains iron homeostasis. In particular, macrophage populations play a key role as cellular components of iron metabolism to maintain the balance between the availability of sufficient iron levels and the prevention of toxic amounts of iron in the body. Tissue macrophages specialized for iron recycling include red pulp macrophages (RPMs) in the spleen, central nurse macrophages in bone marrow (BM), and Kupffer cells (KCs) in the liver. These macrophage populations have a tremendous ability to recycle between 90–95% of bodily iron and to maintain efficient erythropoiesis. Distortions in the equilibrium of iron demand and supply are sensed and corrected by a network of macrophages in their niches.
Erythrophagocytic macrophages that are in direct contact with labile heme and iron developed protective mechanisms to cope with their toxicity. On the contrary, erythropoiesis requires a continuous delivery of iron, supplied presumably through BM macrophages. In cases of blood loss, menstrual bleeding, or pregnancy, a higher iron requirement is adjusted by increasing the intestinal absorption, the only iron-regulatory process in which a role for macrophages is currently unknown. Recent observations, however, suggested a potential role of intestinal macrophages to transiently store iron [5]. Vertebrates do not possess active iron secretory mechanisms, but mainly lose iron due to shedding and renewing of the intestinal cell layer. Distinct types of macrophages control iron-processing mechanisms in interplay with other cellular components and environmental cues. In fact, in adipose tissues a macrophage population with a high iron recycling gene expression profile was identified [6]. In obesity, this macrophage population shows an impaired ability to handle iron and adipogenesis is linked to tissue iron overload [6,7,8]. These findings suggest the possible general importance of macrophages in tissue iron metabolism and the role of iron as a link to metabolic disease.
Here, we review the current knowledge of the cellular processes involved in iron metabolism and the regulatory mechanisms in iron-processing macrophages.
2. Cellular Uptake and Metabolism of Iron in Macrophages
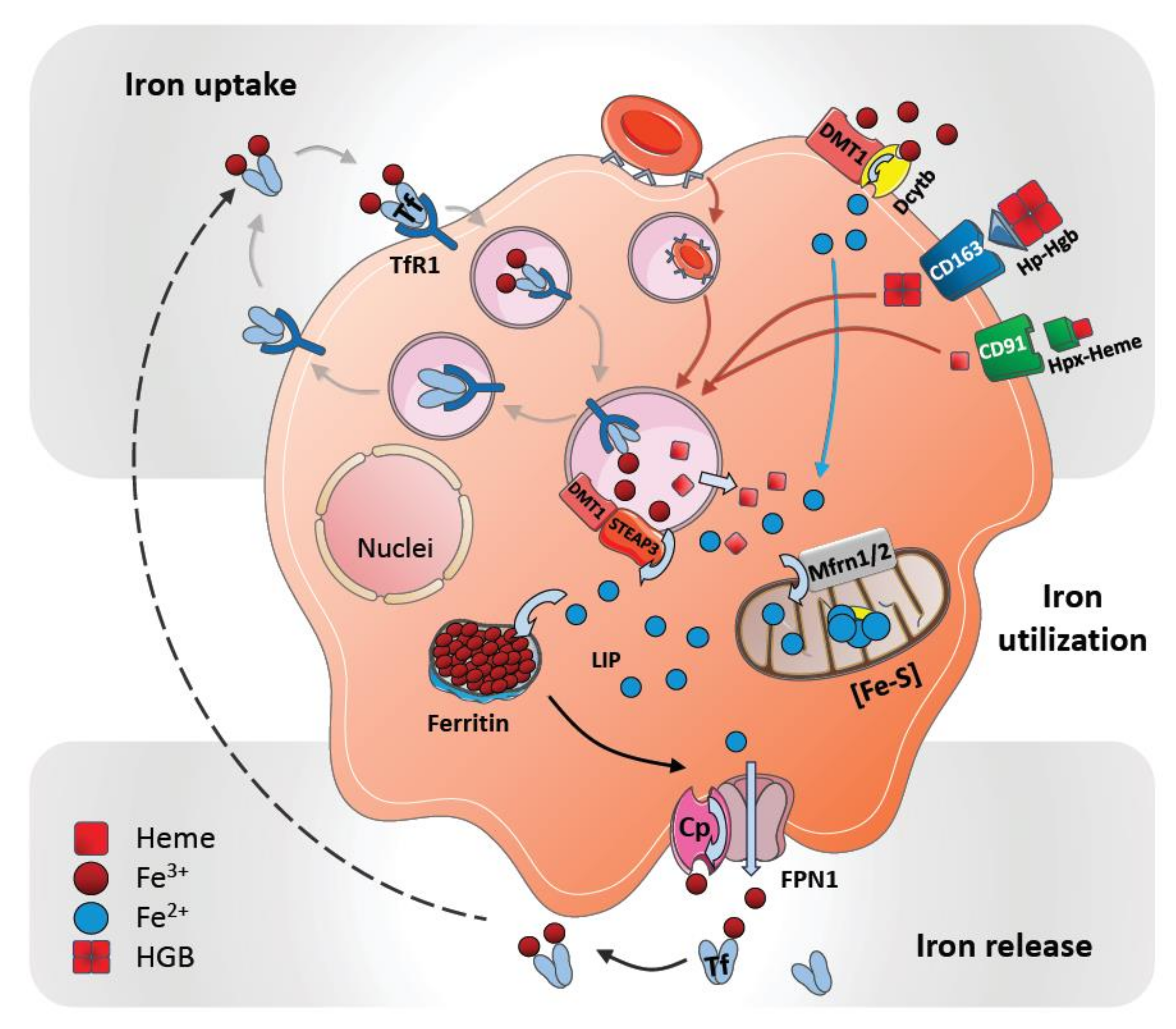
Macrophages are important regulators of iron metabolism by controlling cellular iron import and export. While iron is taken up by macrophages in different forms, such as transferrin-bound iron, heme iron, Hgb-bound iron, and as free iron, iron egress from macrophages occurs only through ferroportin-1 (FPN1); encoded by the gene SLC40A1 (Figure 1).
Iron uptake mechanisms that are common to macrophages include the isolation of iron from senescent erythrocytes after phagocytosis; the import of transferrin-bound iron via transferrin receptor (TfR) or non-transferrin-bound iron (NTBI) via the divalent metal transporter 1 (DMT1), as well as zinc transporter ZIP14 (SLC39A14). Heme-bound iron can be taken up via different means. Heme-hemopexin (Hpx) complexes are internalized via its receptor CD91 (LDL receptor related protein 1, LRP1) and Hgb-bound iron in complex with its scavenger haptoglobin (Hp) is imported via its receptor CD163 (scavenger receptor cysteine-rich SRCR superfamily) (Figure 1). Additional pathways of iron transport into macrophages include natural resistance-associated macrophage protein 1 (NRAMP1/SLC11A1) and divalent cation transporter 1 (DCT1/NRAMP2) [9,10]. NRAMP1 is a divalent metal transporter that is expressed on the surface to mediate iron import, as well as within late endosomes and phagolysosomes to support intracellular iron recycling after erythrophagocytosis [11,12,13]. Other transporters that have been involved in cellular heme transportation include the heme-carrier protein 1 (HCP1) and the feline leukemia virus group C cellular receptor 1 (FLVCR1). HCP1 was first characterized as a cellular heme transporter, and later studies identified it as a proton-coupled folate transporter (PCFT/SLC46A1) [14,15]. Because HCP1 is abundantly expressed in macrophages and colocalizes with Hgb-Hp complexes, which are taken up via CD163, indeed suggest its dual role in cellular transport of heme and folate [16]. Besides implicated in heme uptake, other studies link FLVCR1 to cytoplasmic heme export that prevents intracellular heme overload [17,18]. Furthermore, heme responsive gene 1 (HRG1/SLC48A1) enables the transport of heme out of the phagolysosomes in erythrocyte-digesting macrophages. HRG1 is strongly expressed in macrophages and colocalizes with NRAMP1 to erythrophagolysosomes [19,20,21].
Ferritin is the main iron storage complex in cells (see below), but it can also be found in plasma. Two different ferritin receptors have been described that mediate uptake of ferritin-bound iron: Heavy chain H-ferritin (FtH) receptor T cell immunoglobulin and mucin domain 2 (TIM2) and light chain L-ferritin (FtL) receptor scavenger receptor, member 5 (Scara5) [22,23].
After uptake, iron is liberated from the various iron-containing proteins via different mechanisms (described below). Afterwards, ferrous iron (Fe2+) can be either translocated through the cytosol for further utilization in cellular processes, transported from the cell into the circulation or stored. In the cytosol iron becomes a part of a poorly described labile iron pool (LIP) [24,25]. It is assumed that the LIP directly triggers iron-utilizing cellular processes when cellular iron levels increase.
Excess iron is stored mainly in hepatocytes and macrophages as ferritin—a multimeric sphere-forming protein filled with up to 4500 iron atoms [26]. Ferritin consists of two distinct subunits: FtH and FtL [27]. Due to the ferroxidase activity of FtH, cytosolic ferrous iron (Fe2+) is oxidized into ferric iron (Fe3+) prior to storage in the ferritin cavity [28]. Furthermore, inside splenic macrophages an iron deposition complex known as hemosiderin can be found; these deposits form paracrystalline structures and are surrounded by intralysosomal formations. It has been interpreted as an aggregated form of partially digested ferritin, which may be only poorly available to supply iron when needed [29,30].
Iron within the cytosol of macrophages is utilized as a cofactor in many different cellular proteins that regulate energy production, hypoxic regulation, detoxification, and host defense and inflammation [31]. These proteins contain iron as part of two main cofactors: Iron-sulfur clusters (ISC) and heme groups. Both cofactors are synthesized in the mitochondria, where excess iron can also be stored as mitochondrial ferritin (FtMt). Iron is transported across the mitochondrial membrane in its ferrous form through mitoferrin 1 (Mfrn1) and Mfrn2 transporters [32]. Most complexes of the electron transport chain contain several ISC proteins that drive energy production. Important defense and inflammation-related proteins, such as myeloperoxidase, NADPH oxidase, indoleamine 2,3-dioxygenase, nitric oxide synthases or lipoxygenases that are produced by macrophages also contain iron.
Under steady-state conditions, iron is released from the main iron stores by means of FPN1, the only known iron exporter known until now. FPN1 is abundantly expressed on all iron-metabolizing macrophages [2,33]. In individuals with an FPN1 mutation—as well as when FPN1 is inactivated genetically in mice—a macrophage-specific iron overload in the spleen and liver has been observed [34,35,36]. These observations suggest a macrophage-specific release of iron through FPN1.
The function of FPN1 in iron-recycling macrophages is aided by a multicopper oxidase, ceruloplasmin (Cp, IREG1), which is synthesized and secreted by the liver and macrophages [37]. The transport of iron across all biological membranes is requires the oxidation and reduction of iron. Accordingly, different ferroxidases and ferroreductases are inevitable for iron transportation. Cp is commonly present on macrophage membranes and is thought to mediate iron oxidation to facilitate its export out of the cell by FPN1 [38]. Cp-deficient mice and humans with Cp mutations show hepatocyte- and macrophage-specific iron accumulation and develop anemia indicating a critical role of Cp in the process of iron release from macrophages [39,40,41]. Cp can be expressed both as a circulating plasma protein and as a membrane-bound GPI-linked protein [42]. Interestingly, FPN1 internalization and degradation are specifically performed in the absence of membrane-bound Cp; this may suggest that Cp is involved in hepcidin-mediated iron regulation [39].
After oxidation to ferric iron (Fe3+) and cellular egress, it is presumed that iron is loaded directly onto the glycoprotein transferrin, the main systemic iron transporter of ferric iron (Figure 1). Transferrin-bound iron is mostly utilized by developing erythrocytes and nurse macrophages in the BM to support erythropoiesis.
3. Systemic Iron Metabolism
Iron enters the body through epithelial cells of the duodenum and upper ilium in the small intestine. Iron absorption is adjusted to iron demands and can be increased to maintain iron requirements. The transfer of non-heme Fe3+ iron from the lumen of the duodenum into the epithelium is carried out by DMT1 (Figure 1).
Dietary ferric iron (Fe3+) is first reduced at the apical brush border of the epithelium to enter the enterocytes; this relies on the activity of iron-reducing ferric reductase duodenal cytochrome B (Dcytb or CYBRD1) [43]. DMT1 is colocalized with Dcytb at the apical side of enterocytes when absorption is elevated, and their expression is induced under iron-deficient conditions. This suggests that at the apical surface of the epithelium, DMT1 and Dcytb serve as main transport coordinators [44,45]. As an important source of dietary iron, heme containing ferrous iron (Fe2+) can also enter enterocytes through the heme transporter HCP1.
Once taken up, iron in the cytosol of the enterocytes might be transferred from the apical to the basolateral site by means of vesicular trafficking [46]. An alternative mechanism is that iron transfer might be mediated by “chaperones”, such as the calreticulin-like protein mobilferrin [47].
The release of iron at the basolateral site of epithelium is facilitated by FPN1. The selective inactivation of FPN1 in the intestine results in a marked iron accumulation in duodenal enterocytes and systemic iron deficiency [48]. This confirms the essential function of FPN1 in the dietary iron release into the circulation [34,49]. Of particular relevance is the contribution of iron-oxidizing ferroxidases in FPN1-mediated egress of iron from the cellular environment. The membrane-bound ferroxidase hephaestin (HEPH), a closely related homolog to Cp, is presumed to be responsible for iron oxidation of FPN1-exported iron specifically in the enterocytes [50,51]. In addition, several studies have revealed a significant role of Cp for iron absorption in enterocytes under iron-stress conditions, such as bleeding and iron deficiency [52].
Interestingly, HEPH colocalizes with apo-transferrin in enterocytes [53]. This suggests a direct transfer of iron to transferrin after its release from the intestinal epithelium. Upon binding of two ferric iron (Fe3+) atoms, a holo-transferrin is formed, which enables a secure transport of ferric iron (Fe3+) through plasma. In healthy human individuals, approximately 30% of the total transferrin is saturated [33]. Holo-transferrin can bind to its receptor TfR (CD71) on macrophages, erythrocytes, and other cell types, which is then internalized through receptor-mediated endocytosis.
4. Systemic Iron Regulation by Hepcidin
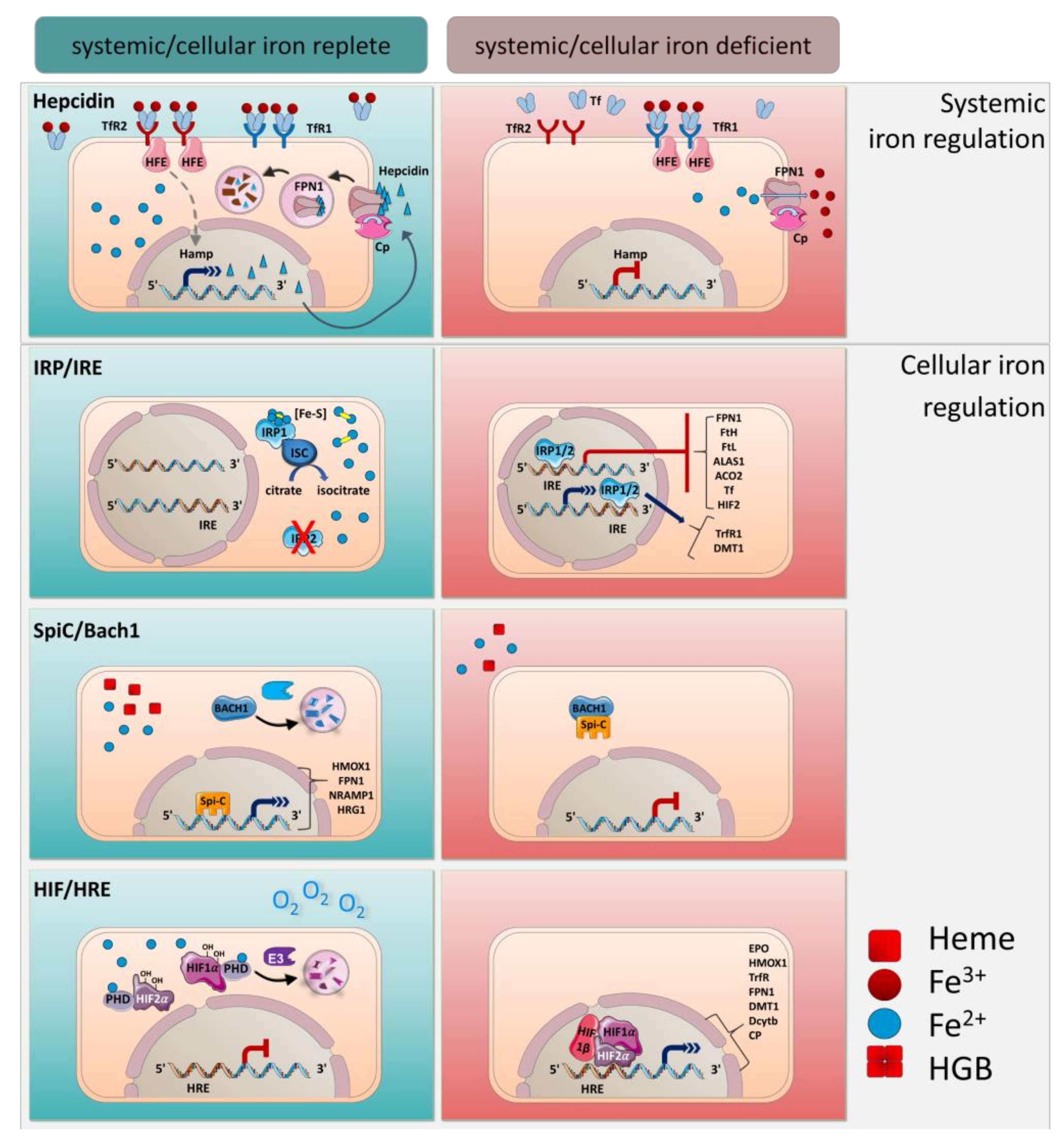
Iron homeostasis is regulated at the systemic and the cellular level by several well-coordinated mechanisms (Figure 2). Hepcidin is the main regulator of the systemic transportation of iron. It is a peptide hormone expressed mainly in the liver by hepatocytes and undergoes proteolytic processing to yield a bioactive molecule that is secreted into the bloodstream [31,54,55]. Hepcidin binds to FPN1 and mediates its phosphorylation, internalization and degradation. Thus, in FPN1-expressing cells, such as macrophages, hepatocytes, and duodenal epithelial cells, iron export is blocked [34,56,57].
Hepcidin expression is regulated by iron and iron-sensing mechanisms. Accordingly, the synthesis of hepcidin is increased by iron loading and decreased by anemia and hypoxia. In this manner, an increase of systemic transferrin iron leads to hepcidin expression that causes iron retention in the macrophages and blocks iron uptake in the enterocytes to avoid further systemic iron accumulation [34,56,58]. Inappropriate low hepcidin expression due to mutations in the hepcidin-encoding gene Hamp or mutations that impair hepcidin function are linked to hereditary hemochromatosis and total body iron overload [56,59,60,61,62]. On the contrary, pathological hepcidin excess caused by inflammatory disorders induces hypoferremia, anemia of inflammation, or anemia of chronic disease [31,63,64]. Therefore, hepcidin-mediated regulation is essential in maintaining iron homeostasis.
Hepcidin expression is controlled transcriptionally by various signals, such as transferrin saturation, erythropoietic activity, hypoxia and inflammation, rendering it an important player in the process of iron homeostasis. Organisms have evolved multitude mechanisms to sense iron and to activate hepcidin expression (Figure 2).
The molecular mechanism by which transferrin iron serves as a sensor for hepcidin expression is based on a coordinated interplay between TfR1 and TfR2 with the homeostatic iron regulator HFE, a major histocompatibility complex class I integral membrane protein [65,66]. Circulating transferrin-bound iron interacts with high affinity to TfR1, whereas TfR2 has a lower iron-binding affinity. Accordingly, under low transferrin saturation only TfR1 is engaged to transferrin iron. In this case only, HFE is able to associate with TfR1 and hepcidin expression is not induced, while TfR2 is directed to a lysosomal degradation pathway [1,66,67]. When transferrin iron levels increase in plasma, transferrin iron interacts also with TfR2, which leads to detachment of HFE from TfR1 and binding to TfR2 to induce hepcidin transcription [68,69,70]. Consequently, the competitive interaction between HFE and TfR1 or TfR2 determines iron regulation through hepcidin. In hemochromatosis patients with HFE gene mutations or in HFE-deficient mice, hepcidin synthesis is inappropriately low that induces systemic iron overload [65,71].
Furthermore, liver iron stimulates the bone morphogenetic protein (BMP) BMP6 signaling pathway to trigger hepcidin production [72,73]. Hepcidin induction by classical BMP signaling is dependent on the BMP co-receptor hemojuvelin (HJV or HFE2), a GPI-linked membrane protein. HJV binds to BMP6 and activates the Smad family of transcription factors to stimulate Hamp transcription [58].
Inflammatory mediators are major hepcidin-inducing factors. Lipopolysaccharide, interleukin-6 (IL-6), IL-1, or IL-22 induce hepcidin partially via signal transducer and activator of transcription 3 (STAT3) [64,74,75,76]. Hepcidin expression in response to infections rapidly lowers plasma iron to prevent iron sequestration of microbes that need iron for growth [77]. Additionally, macrophages are able to produce hepcidin locally in response to bacterial signals at the side of the infection to limit iron bioavailability for pathogens [78,79]. Thus, iron regulation by macrophages is an important defense mechanism during an infection [80].
On the contrary, hepcidin expression is blocked by hypoxia to stimulate iron mobilization and erythropoiesis [81,82]. Other hepcidin inhibitory factors were identified, including members of the growth factor beta (TGF-β) superfamily comprising growth differentiation factor 15 (GDF15), transforming growth factor beta (TGF-β), twisted gastrulation BMP signaling modulator 1 (TWSG1) and erythroid factor erythropoietin (Epo) [83,84].
5. Cellular Regulation of Iron by IRE/IRPs
Compared to the transcriptional regulation of hepcidin, many proteins involved in controlling intracellular iron metabolism are governed by posttranscriptional mechanisms. The main cellular iron uptake, transport, storage, utilization, and release processes are governed by the interplay of distinct mRNA-binding iron-regulatory proteins (IRPs), which have been extensively studied in macrophages and hepatocytes [1,85]. Two different IRPs are known: IRP1 and IRP2. IRPs bind to their target transcripts that contain cis-regulatory iron-regulatory elements (IREs), conserved RNA stem loop structures [86,87,88]. Besides iron status, the localization of IREs on transcripts determines whether a given IRP-IRE interaction stimulates or suppresses protein translation (Figure 2).
In cellular iron deficiency, IRPs bind to IREs at the 5’ untranslated region (UTR) of FPN1, FtH, FtL, delta-aminolevulinate synthase 1 (ALAS1), aconitase 2 (ACO2), hypoxia-inducible factor 2 (HIF2) mRNAs to mediate the degradation of the transcripts of the IREs to decrease iron storage and export [89]. In addition, the binding of IRPs to IREs at 3’ UTR of TfR1 and DMT1 mRNA stabilizes these transcripts and supports their translation to increase iron import [37,85].
Conversely, under cellular iron-replete conditions, IRP2 gets ubiquitinated and degraded by the proteasome to inhibit binding to IREs [90,91,92,93]. IRP1 similarly loses its IRE-binding activity but acquires an ISC [4Fe-4S] to obtain cytosolic aconitase activity that converts citrate to isocitrate in the citric acid cycle [94]. Because the bi-functional IRP1 shifts between iron-regulation and metabolism-regulation, this mechanism suggests an interesting link between iron and cellular energy metabolism [95,96,97,98]. IRP1−/− and IRP2−/− mice show massive impaired iron homeostasis suggesting that the IRP/IRE system is critical in cellular iron control [95,98,99].
6. Transcriptional Regulation by Spi-C and HIF
Iron homeostasis on the cellular level is also regulated transcriptionally by the induction of the transcription factors Spi-C and HIFs. Endogenous labile heme derived from e.g., hemolysis binds to and liberates Spi-C from its repressor BTB domain and CNC homolog 1 (BACH1), which is subsequently degraded by the ubiquitin−protein ligase HOIL-1 [100,101,102,103]. Unbound Spi-C can enter the nucleus and directly stimulates important heme- and iron-regulating proteins, such as heme oxygenase 1 (HMOX1), FPN1, NRAMP1, and HRG1. Spi-C-mediated control of iron genes in macrophages has been reported to be particularly important when rapid iron metabolism is needed; for example, through increased erythrophagocytosis or hemolysis [102,104]. Iron-driven Spi-C activation is a critical factor for the development of iron-specific macrophages. Moreover, the number of BM macrophages and RPMs is drastically reduced in Spic−/− mice suggesting that Spi-C-mediated regulation is indispensable for iron metabolism in these cells and for protection against iron toxicity [100,105].
Additionally, iron metabolism is regulated in response to oxygen by means of HIF1α and HIF2α. If sufficient levels of ferrous iron (Fe2+) and oxygen are present, HIF1α and HIF2α are hydroxylated by oxygen prolyl hydroxylases (PHDs) and are then degraded by Ligase E3 [106,107]. In contrast, low iron levels in cells, as well as hypoxia, induce an accumulation of HIF1α and HIF2α in the nucleus and subsequently triggers the recruitment of HIF1β. Accordingly, the HIFα/HIFβ complex binds to hypoxia-responsive elements (HRE) and promotes transcription of genes that are involved in iron metabolism such transferrin, TfR, DMT1, FPN1, Dcytb, Cp, and HMOX1 [108,109]. In line, the HIF1/2-HRE interaction in response to hypoxia and to low iron induces iron release from macrophages [110,111]. This process is not only important in macrophages, but also induces RBC generation and maturation through Epo [112]. HIF2α contains IREs in its 5’ UTR of mRNA and is thus under control of IRP1. These findings show that iron homeostasis depends on complex, global and cellular regulations at various levels of gene expression.
7. Steady-State Erythrophagocytosis by Macrophages in the Spleen
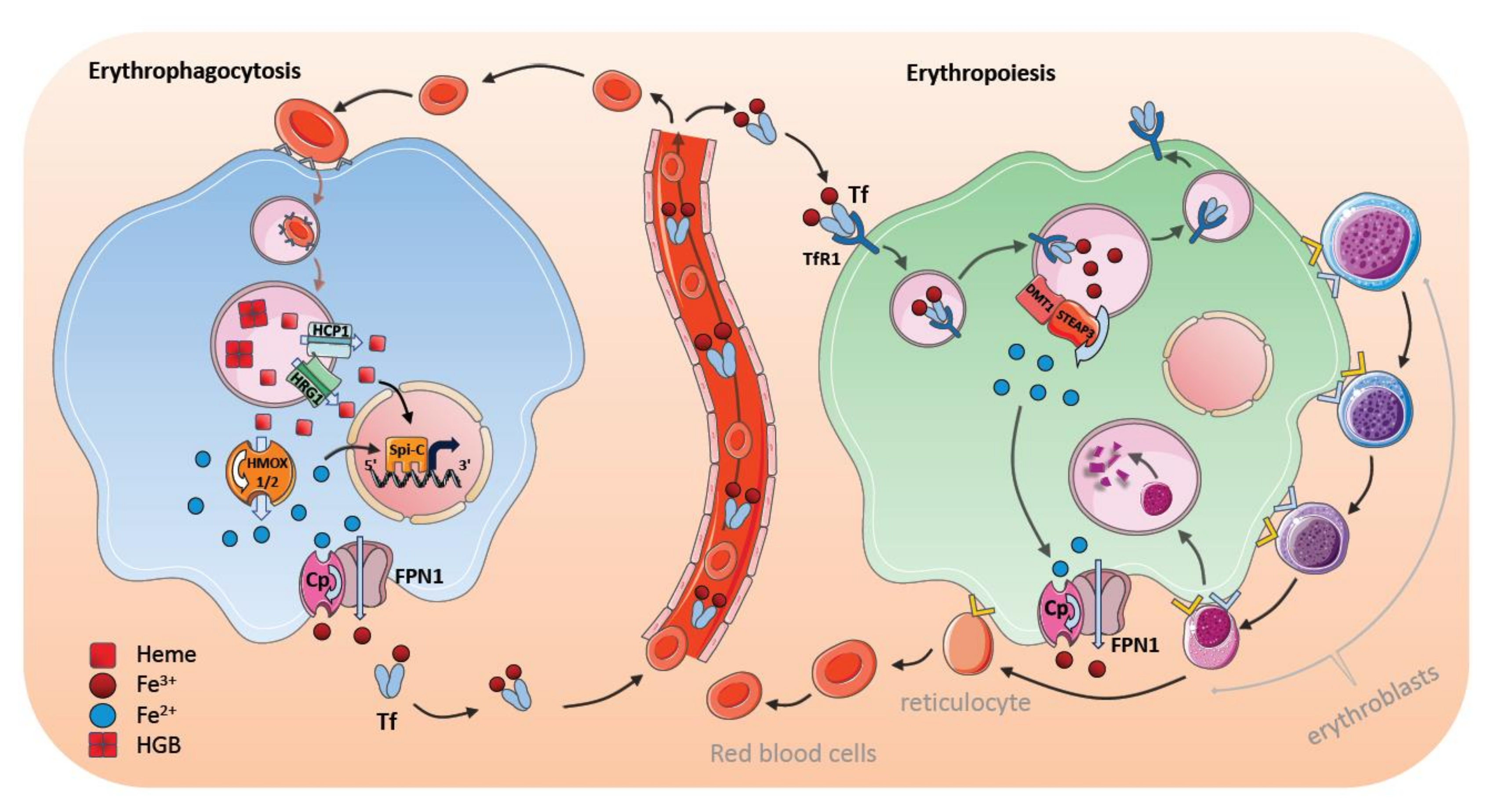
The majority of iron (20–21 mg per day) is recycled from heme of erythrocytes in the spleen, whereas only 1–2 mg of iron is absorbed by the diet. Macrophages extract iron from senescent erythrocytes by disassembling Hgb. Approximately 1 billion iron atoms are extracted from 250–280 million Hgb molecules per erythrocyte [113]. Thus, almost all iron atoms pass through macrophages for further cellular and systemic utilization.
The first signal that stimulates iron digestion in macrophages is the recognition of senescent RBCs and the uptake of these cells by means of receptor-mediated erythrophagocytosis (Figure 3). The macrophage surface is equipped with various scavenging receptor proteins to recognize the “eat me” signals from aged RBCs [114,115]; these include, among others, signal-regulatory protein alpha (SIRPα), glucose-6-phosphate dehydrogenase, phosphatidylserine (PS) receptors (such as TIM4 and TIM1), scavenger receptor type A member I (SR-AI), and CD36. The most popular presenters of the “eat me” signals are CD47 and PS, which are exposed to aging RBCs [10,116,117]. After internalization, RBCs-containing phagosomes merge with lysosomal vesicles to form phagolysosomes, where the RBCs are digested. Hgb breaks down into heme and heme is then transferred from the erythrophagolysosomes into the cytosol [21,118]. The transport of heme out of the erythrophagolysosome is facilitated by a heme transporter, known as HRG1, which is regulated posttranscriptionally by heme and iron [19]. HRG1 expression is high in the macrophages of the spleen, liver, as well as BM, and it localizes in erythrophagolysosomes one hour after erythrophagocytosis. In addition, the putative heme transporter HCP1 has been suggested to transport heme into the cytosol [16]. In the cytosol of macrophages, iron is immediately processed from the protoporphyrin ring of heme by means of HMOX1 [119]. Degradation of heme by HMOX1 generates equivalent amounts of CO and biliverdin in addition to iron [120]. HMOX1 is not present on the erythrophagocyte membrane, but has been located in the endoplasmic reticulum with its active site directed to cytosol; this process supports the idea that iron is extracted in the cytosol [19,121]. Moreover, HMOX1 expression can be detected immediately after erythrophagocytosis or after hemolysis corroborating its crucial role in iron and heme detoxication [122,123]. The role of HMOX1 is underscored by the finding that HMOX1 deficiency causes a depletion of RPM and BM macrophages; suggesting that the survival and function of iron-metabolizing macrophages is dependent on HMOX1. In addition, there is a second heme oxygenase HMOX2, which is involved in removing intracellular heme. In contrast to HMOX1, which is inducible, HMOX2 is continuously expressed in most tissues and is involved in extracting iron in different cell types [122,123].
Recently, NRAMP1 (SLC11A1) has been identified as an important iron transporter in the macrophages, which is localized to erythrophagolysosomes after erythrophagocytosis [21,124]. NRAMP1-knockout mice retain iron in the macrophages, but release storage iron from hepatocytes to compensate for the impaired iron efflux from macrophages [124].
Depending on erythropoietic activity and other iron-consuming processes, iron can be either stored or released through FPN1, and its expression increases in macrophages within an hour after erythrophagocytosis [49,125]. Moreover, erythrophagocytosis-activated macrophages have concomitant high levels of heavy (H-) and light (L-)ferritin content, even several hours after erythrophagocytosis has occurred; this indicates that a substantial proportion of iron can be stored intracellularly within ferritin after erythrophagocytosis in the liver and the spleen [29,126]. An increase of cellular iron upon systemic hemolysis, when iron storage in KCs of the liver is saturated, iron has been found to be transferred into hepatocytes for long-term storage [127].
8. Stress-Induced Erythrophagocytosis and Iron Metabolism
Under steady-state conditions, heme iron is preferentially digested by RPMs in the spleen, whereas, under stress conditions, macrophages in the liver become essential for erythrophagocytosis. For example, during systemic hemolysis, Ly6C+ monocytes migrate to the liver and, thereupon, differentiate into iron-recycling macrophages, but not to KCs [127]. Interestingly, colony stimulating factor 1 (CSF1) and nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor promote the development of these iron-recycling FPN1+Tim4neg macrophages from blood-borne monocytes in the liver [127]. Moreover, iron recycling by RPMs in the spleen is not ameliorated to compensate for the increased iron need. Hence, FPN1+Tim4neg iron-recycling liver handle and recycle heme and iron from senescent and damaged RBCs after massive hemolysis [10].
Macrophages have developed further elaborate mechanisms to remove systemic heme during hemolysis. The body responds immediately against toxic free Hgb and heme by producing the Hgb-scavenging protein haptoglobin and the heme-binding serum protein hemopexin. Splenic RPMs and liver KCs express the receptors CD163 and CD91: these are involved in the clearance of excessive labile heme and, importantly, offer an acute protection mechanism [128,129]. Macrophages possess ferroxidases (such as Cp and HEPH) and proteins promoting ferrireductase activity (such as FtH and Dcytb); these enable the quick oxidation and reduction of iron, and ensure efficient transportation and utilization of iron within a cell [130]. Additionally, as described above, heme-mediated Spi-C induction in monocytes promotes the differentiation of erythrophagocytosing RPMs in the spleen and induces the expression of important heme- and iron-regulating proteins [100]. This facilitates a quick heme and iron processing and inhibits iron-induced toxicity in the cells [21,29,100,101] (Figure 3).
Recently, it was demonstrated that labile heme released after massive hemolysis, following bacterial infection, suppresses the phagocytosing function of macrophages [131]. It is conceivable that the liberation of heme when erythrophagocytosis commences may function as a negative feedback loop and impairs further erythrophagocytosis by macrophages as a protective mechanism to sustain oxygen supply.
9. Bone Marrow Macrophages and Erythroblastic Islands
Erythropoiesis is a dynamic process that is regulated through environmental signals, nutrient availability, and cellular interplay. Most of the transferrin-bound iron is utilized for heme synthesis during definitive erythropoiesis in the BM. In the BM, CD169 (Sialoadhesin or Siglecß1) is expressed on certain macrophages—referred to as nurse macrophages. These form an erythroblastic island, in which the central nurse macrophage is surrounded by erythroid progenitors at different developmental stages and provides a niche for erythropoietic regulation [132,133,134] (Figure 3). Thus, the nursing macrophage supports erythropoiesis at different stages through varying the stimulating and repressing signals to control proliferation versus differentiation of erythroid progenitors, and to sustain survival signals [134,135]. Depending on the developmental stage, different soluble factors, such as BMP4, insulin-like growth factor 1 (ILGF1), IL-3, and granulocyte-macrophage colony-stimulating factor (GM-CSF) are secreted from the central nurse macrophages and induce proliferative and differential signals in erythroid progenitors [136,137]. In addition, negative regulators of erythropoiesis can be released by BM macrophages, including GATA binding protein 1 (GATA1), TGF-β, interferon gamma (IFNγ), and tumor necrosis factor alpha (TNFα) [136,138].
Central nurse macrophages are thought to be involved in phagocytosing the extruded nuclei from erythroblasts during their development. This process is based on recognition through PS exposure, TIM4 and MER proto-oncogene tyrosine kinase (MerTK) [2,139]. Because developing erythroblasts adhere to the central macrophage, it has also been hypothesized that BM macrophages regulate and determine the release of mature reticulocytes into the blood circulation. Prominent adhesion molecules that are involved in cell–cell interaction, signal transduction, and reticulocyte release controlled by macrophages include erythroblast macrophage protein, vascular cell adhesion protein 1 (VCAM1) and a4b1- or a4b5-integrins [136,137,140]. Ablation of macrophages, or one of the adhesion molecules, delays proerythroblast proliferation and differentiation at different developmental stages and to destroy the erythroblastic island [136,137,141]. Moreover, Spi-C-mediated signals are also essential for erythropoiesis, since Spi-C ablation causes a reduction in erythroblastic island numbers and a delay in RBC formation [100]. Interestingly, CD169+ macrophage ablation, which markedly reduces the number of erythroblasts in the bone marrow, only leads to a mild reduction of erythroid cells without overt anemia suggesting that erythropoietic compensatory mechanisms exist during homeostasis [141]. However, the induction of stress-induced anemia (for example, through phenylhydrazin treatment) promoted more severe delays in erythropoietic recovery in CD169+-ablated animals. This indicates that BM macrophages promote fast recovery responses to stress-induced erythropoiesis [132]. On the contrary, pathological erythropoiesis in mouse models of polycythemia vera (PV) can be reversed through the ablation of central macrophages [142].
10. Regulation of Iron Transfer by Central Nurse Macrophages
Transferrin-bound iron is the main iron source for erythropoiesis. Iron-bound transferrin and its receptor are internalized by receptor-mediated endocytosis [143]. TfR expression on erythroblasts increases during erythrocyte development, but is downregulated after the release of reticulocytes [144]. Loss of transferrin impedes erythropoiesis and leads to an accumulation of iron in the storage organs; this indicates the importance of transferrin-bound iron as a crucial source for erythropoiesis [145]. BM nurse macrophages contribute to iron-loading of the erythroblasts, which might be especially pronounced during stress conditions. In the macrophage, iron atoms are released from transferrin in the acidic condition within endosomes (pH 4.5), and transferrin, along with TfRs, is recycled back onto the macrophage surface. Ferric irons (Fe3+) in the endosomes are reduced to ferrous irons (Fe2+) by six-transmembrane epithelial antigen of the prostate 3 (STEAP3)—an endosomal ferrireductase located in endosomes [146]. Ferrous iron (Fe2+) is then transported by means of DMT1 through the endosomal membrane and is released into the cytosol. It is not entirely clear how iron is subsequently translocated through the cellular cytosol and utilized for erythropoiesis. The high expression of FPN1 on the nurse macrophages and the observation that FPN1 deletion leads to an accumulation of iron in BM macrophages and a blockage of erythroblast development suggest that iron is released from macrophages prior to being loaded into the erythroblasts [147,148]. It is also possible that NRAMP1, which participates in iron transport in BM macrophages, is responsible for FPN1-associated iron release during erythropoiesis [124,149]. Interestingly, in the absence of transferrin-bound iron, macrophages utilize other forms of iron, such as secreted ferritin iron and heme iron to support erythropoiesis [150,151]. However, the importance of these alternative pathways in macrophage-dependent iron transfer remains to be determined.
11. Concluding Remarks
Efficient iron metabolism is a matter that decides between life and death and depends on complex parallel, compensatory, and complementary networks to meet the iron demand for erythropoiesis, as well as other cellular functions, but to prevent iron overload, which cause cellular and organismal toxicity. Tissue macrophages take center stage in iron homeostasis and their regulatory mechanisms are increasingly recognized. Except duodenal iron uptake, macrophages are implicated in every step of iron recycling and metabolism. Future studies may identify new functions of macrophages that regulate tissue-specific iron metabolism and contribute to metabolic disorders. One emerging theme from knock-out mice suggests that macrophage-mediated iron metabolism is particularly important during stress conditions. Whereas these mouse models show that under homeostatic conditions, compensatory mechanisms exist that maintain systemic iron metabolism, macrophages may still be the main iron regulators in unaltered wild-type mice. Evidence implicates the importance of the central macrophages in erythropoietic regulation, however, it is not known how macrophages sense erythropoietic need and whether macrophages coordinate erythropoiesis dependent on iron availability and oxygen demand. The sensing of these factors is essential for the proper regulation of RBC maturation. Thus, HIF-regulatory mechanisms may be an attractive candidate, because they can sense both iron and oxygen. The understanding of cellular and systemic regulation of iron homeostasis will help to develop novel therapeutic possibilities in anemic diseases and other conditions, such as malaria, acute blood loss, hematopoietic stem cell transplantation, or chronic kidney disease.
Author Contributions
N.S. and T.W. wrote, reviewed, and edited this manuscript.
Funding
Supported by grants from the Austrian Science Fund (FWF) P27701-B20 and P30857-B28 and the Foundation for Sarcoidosis Research. N.S. is supported by the [DOC] Doctoral Fellowship Programme of the Austrian Academy of Sciences. Figures are illustrated using Servier Medical Art.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACO2 | aconitase 2 |
| ALAS1 | delta-aminolevulinate synthase 1 |
| BACH1 | BTB domain and CNC homolog 1 |
| BM | bone marrow |
| BMP | Bone morphogenetic protein |
| Cp | ceruloplasmin |
| DCT1 | divalent cation transporter 1 |
| DcytB | duodenal cytochrome B |
| DMT1 | divalent metal transporter 1 |
| E3 | E3 ubiquitin ligase |
| Epo | erythroid factor erythropoietin |
| FLVCR1 | Feline leukemia virus group C cellular receptor 1 |
| FPN1 | ferroportin 1 |
| FtMt | Mitochondrial ferritin |
| FtH | heavy chain H-ferritin |
| FtL | light chain L-ferritin |
| GDF15 | growth differentiation factor 15 |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| Hamp | hepcidin antimicrobial peptide |
| HEPH | hephaestin |
| HCP1 | heme-carrier protein 1 |
| HFE | homeostatic iron regulator |
| Hgb | hemoglobin |
| HIF1α/HIF2α | hypoxia-inducible factor 1α/2α |
| HJV | hemojuvelin |
| HMOX1 | heme oxygenase 1 |
| Hp | haptoglobin |
| Hpx | hemopexin |
| HRE | hypoxia-responsive element |
| HRG1 | heme responsive gene 1 protein |
| ILGF1 | insulin-like growth factor 1 |
| IFNγ | interferon gamma |
| IRE | iron-regulatory element |
| IRP1/IRP2 | iron-regulatory protein1/2 |
| ISC | iron-sulfur cluster |
| KC | Kupffer cell |
| LIP | Labile iron pool |
| Mfrn1/2 | Mitoferrin 1/2 |
| MerTK | MER proto-oncogene tyrosine kinase |
| NTBI | non-transferrin-bound iron |
| NRAMP1 | natural resistance-associated macrophage protein 1 |
| PHDs | oxygen prolyl hydroxylases |
| PS | phosphatidylserine |
| PV | polycythemia vera |
| RBC | red blood cell |
| RPM | red pulp macrophages |
| SIRPα | signal-regulatory protein alpha |
| Spi-C | Spi-1/PU.1 related transcription factor |
| SR-AI | scavenger receptor type A member I |
| STAT3 | activator of transcription 3 |
| STEAP3 | six-transmembrane epithelial antigen of the prostate 3 |
| Tf | transferrin |
| TfR1/2 | transferrin receptor 1/2 |
| TNFα | tumor necrosis factor alpha |
| TGF-β | transforming growth factor beta |
| TWSG1 | twisted gastrulation BMP signaling modulator 1 |
| VCAM1 | vascular cell adhesion protein 1 |
| UTR | untranslated region |
References
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Hamza, I. Macrophages and iron metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef] [PubMed]
- Fabiano, A.; Brilli, E.; Mattii, L.; Testai, L.; Moscato, S.; Citi, V.; Tarantino, G.; Zambito, Y. Ex vivo and in vivo study of sucrosomial® iron intestinal absorption and bioavailability. Int. J. Mol. Sci. 2018, 19, 2722. [Google Scholar] [CrossRef] [PubMed]
- Orr, J.S.; Kennedy, A.; Anderson-Baucum, E.K.; Webb, C.D.; Fordahl, S.C.; Erikson, K.M.; Zhang, Y.; Etzerodt, A.; Moestrup, S.K.; Hasty, A.H. Obesity alters adipose tissue macrophage iron content and tissue iron distribution. Diabetes 2014, 63, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Kim, M.S.; Han, S.N. Diet-induced obesity leads to decreased hepatic iron storage in mice. Nutr. Res. 2011, 31, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Festa, M.; Ricciardelli, G.; Mele, G.; Pietropaolo, C.; Ruffo, A.; Colonna, A. Overexpression of H ferritin and up-regulation of iron regulatory protein genes during differentiation of 3T3-L1 pre-adipocytes. J. Biol. Chem. 2000, 275, 36708–36712. [Google Scholar] [CrossRef]
- Fleming, M.D.; Andrews, N.C. Mammalian iron transport: An unexpected link between metal homeostasis and host defense. J. Lab. Clin. Med. 1998, 132, 464–468. [Google Scholar] [CrossRef]
- Nairz, M.; Theurl, I.; Swirski, F.K.; Weiss, G. “Pumping iron”-how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch. 2017, 469, 397–418. [Google Scholar] [CrossRef]
- Gruenheid, S.; Pinner, E.; Desjardins, M.; Gros, P. Natural resistance to infection with intracellular pathogens: The Nramp1 protein is recruited to the membrane of the phagosome. J. Exp. Med. 1997, 185, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.R.; Gros, P. Iron, manganese, and cobalt transport by Nramp1 (Slc11a1) and Nramp2 (Slc11a2) expressed at the plasma membrane. Blood 2003, 102, 1884–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soe-Lin, S.; Sheftel, A.D.; Wasyluk, B.; Ponka, P. Nramp1 equips macrophages for efficient iron recycling. Exp. Hematol. 2008, 36, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Nakai, Y.; Ueda, S.; Kamigaso, S.; Ohta, K.; Hatakeyama, M.; Hayashi, Y.; Otagiri, M.; Yuasa, H. Functional characterization of PCFT/HCP1 as the molecular entity of the carrier-mediated intestinal folate transport system in the rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G660–G668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Schaer, C.A.; Vallelian, F.; Imhof, A.; Schoedon, G.; Schaer, D.J. Heme carrier protein (HCP-1) spatially interacts with the CD163 hemoglobin uptake pathway and is a target of inflammatory macrophage activation. J. Leukoc. Biol. 2008, 83, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef]
- Zhang, J.; Chambers, I.; Yun, S.; Phillips, J.; Krause, M.; Hamza, I. Hrg1 promotes heme-iron recycling during hemolysis in the zebrafish kidney. PLoS Genet. 2018, 14, e1007665. [Google Scholar] [CrossRef]
- Delaby, C.; Rondeau, C.; Pouzet, C.; Willemetz, A.; Pilard, N.; Desjardins, M.; Canonne-Hergaux, F. Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS ONE 2012, 7, e42199. [Google Scholar] [CrossRef]
- Li, J.Y.; Paragas, N.; Ned, R.M.; Qiu, A.; Viltard, M.; Leete, T.; Drexler, I.R.; Chen, X.; Sanna-Cherchi, S.; Mohammed, F.; et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev. Cell 2009, 16, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Seaman, W.E.; Di, X.; Wang, W.; Willingham, M.; Torti, F.M.; Torti, S.V. Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS ONE 2011, 6, e23800. [Google Scholar] [CrossRef] [PubMed]
- Breuer, W.; Shvartsman, M.; Cabantchik, Z.I. Intracellular labile iron. Int. J. Biochem. Cell Biol. 2008, 40, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Rauen, U.; Springer, A.; Weisheit, D.; Petrat, F.; Korth, H.-G.; de Groot, H.; Sustmann, R. Assessment of chelatable mitochondrial iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities. Chembiochem 2007, 8, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Yewdall, S.J.; Harrison, P.M.; Santambrogio, P.; Cozzi, A.; Rovida, E.; Albertini, A.; Arosio, P. Evidence of H- and L-chains have co-operative roles in the iron-uptake mechanism of human ferritin. Biochem. J. 1992, 288 Pt 2, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Delaby, C.; Pilard, N.; Hetet, G.; Driss, F.; Grandchamp, B.; Beaumont, C.; Canonne-Hergaux, F. A physiological model to study iron recycling in macrophages. Exp. Cell Res. 2005, 310, 43–53. [Google Scholar] [CrossRef]
- Ferreira, C.; Santambrogio, P.; Martin, M.E.; Andrieu, V.; Feldmann, G.; Hénin, D.; Beaumont, C. H ferritin knockout mice: A model of hyperferritinemia in the absence of iron overload. Blood 2001, 98, 525–532. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Regulation of iron metabolism by hepcidin. Annu. Rev. Nutr. 2006, 26, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.D.; Zhang, A.-S.; Brown, C.; Shirihai, O.S.; Ponka, P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 2007, 110, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Montosi, G.; Donovan, A.; Totaro, A.; Garuti, C.; Pignatti, E.; Cassanelli, S.; Trenor, C.C.; Gasparini, P.; Andrews, N.C.; Pietrangelo, A. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J. Clin. Investig. 2001, 108, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Njajou, O.T.; Vaessen, N.; Joosse, M.; Berghuis, B.; van Dongen, J.W.; Breuning, M.H.; Snijders, P.J.; Rutten, W.P.; Sandkuijl, L.A.; Oostra, B.A.; et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat. Genet. 2001, 28, 213–214. [Google Scholar] [CrossRef]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Dini, L.; Carbonaro, M.; Musci, G.; Calabrese, L. The interaction of ceruloplasmin with Kupffer cells. Eur. J. Cell Biol. 1990, 52, 207–212. [Google Scholar]
- De Domenico, I.; Ward, D.M.; di Patti, M.C.B.; Jeong, S.Y.; David, S.; Musci, G.; Kaplan, J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007, 26, 2823–2831. [Google Scholar] [CrossRef] [Green Version]
- Harris, Z.L.; Durley, A.P.; Man, T.K.; Gitlin, J.D. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. USA 1999, 96, 10812–10817. [Google Scholar] [CrossRef] [Green Version]
- Harris, Z.L.; Takahashi, Y.; Miyajima, H.; Serizawa, M.; MacGillivray, R.T.; Gitlin, J.D. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. USA 1995, 92, 2539–2543. [Google Scholar] [CrossRef] [PubMed]
- Kennard, M.L.; Richardson, D.R.; Gabathuler, R.; Ponka, P.; Jefferies, W.A. A novel iron uptake mechanism mediated by GPI-anchored human p97. EMBO J. 1995, 14, 4178–4186. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Frazer, D.M.; Wilkins, S.J.; Becker, E.M.; Murphy, T.L.; Vulpe, C.D.; McKie, A.T.; Anderson, G.J. A rapid decrease in the expression of DMT1 and Dcytb but not Ireg1 or hephaestin explains the mucosal block phenomenon of iron absorption. Gut 2003, 52, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupic, F.; Fruchon, S.; Bensaid, M.; Loreal, O.; Brissot, P.; Borot, N.; Roth, M.P.; Coppin, H. Duodenal mRNA expression of iron related genes in response to iron loading and iron deficiency in four strains of mice. Gut 2002, 51, 648–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.S.; Sheftel, A.D.; Ponka, P. The anemia of “haemoglobin-deficit” (hbd/hbd) mice is caused by a defect in transferrin cycling. Exp. Hematol. 2006, 34, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, J.N.; Conrad, M.E.; Moore, E.G.; Latour, L.F. Iron absorption and cellular transport: The mobilferrin/paraferritin paradigm. Semin. Hematol. 1998, 35, 13–26. [Google Scholar] [PubMed]
- Zhang, D.-L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- Yeh, K.-Y.; Yeh, M.; Glass, J. Interactions between ferroportin and hephaestin in rat enterocytes are reduced after iron ingestion. Gastroenterology 2011, 141, 292–299.e1. [Google Scholar] [CrossRef]
- Yeh, K.-Y.; Yeh, M.; Mims, L.; Glass, J. Iron feeding induces ferroportin 1 and hephaestin migration and interaction in rat duodenal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G55–G65. [Google Scholar] [CrossRef] [PubMed]
- Cherukuri, S.; Potla, R.; Sarkar, J.; Nurko, S.; Harris, Z.L.; Fox, P.L. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab. 2005, 2, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, T.A.M.; Mauk, A.G.; MacGillivray, R.T.A. Recombinant expression and functional characterization of human hephaestin: A multicopper oxidase with ferroxidase activity. Biochemistry 2005, 44, 14725–14731. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Preza, G.C.; Jung, C.-L.; Kaplan, J.; Waring, A.J.; Ganz, T. The N-terminus of hepcidin is essential for its interaction with ferroportin: Structure-function study. Blood 2006, 107, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Kulaksiz, H.; Gehrke, S.G.; Janetzko, A.; Rost, D.; Bruckner, T.; Kallinowski, B.; Stremmel, W. Pro-hepcidin: Expression and cell specific localisation in the liver and its regulation in hereditary haemochromatosis, chronic renal insufficiency, and renal anaemia. Gut 2004, 53, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.-J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Valore, E.V.; Nemeth, E.; Goodnough, J.B.; Gabayan, V.; Ganz, T. Iron transferrin regulates hepcidin synthesis in primary hepatocyte culture through hemojuvelin and BMP2/4. Blood 2007, 110, 2182–2189. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.; Bennoun, M.; Porteu, A.; Mativet, S.; Beaumont, C.; Grandchamp, B.; Sirito, M.; Sawadogo, M.; Kahn, A.; Vaulont, S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. USA 2002, 99, 4596–4601. [Google Scholar] [CrossRef] [Green Version]
- Altamura, S.; Kessler, R.; Gröne, H.-J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 2014, 20, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Roetto, A.; Papanikolaou, G.; Politou, M.; Alberti, F.; Girelli, D.; Christakis, J.; Loukopoulos, D.; Camaschella, C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 2003, 33, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef]
- Fleming, R.E.; Sly, W.S. Hepcidin: A putative iron-regulatory hormone relevant to hereditary hemochromatosis and the anemia of chronic disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8160–8162. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.B.; Chen, J.; Murchison, N.; Green, F.A.; Enns, C.A. Transferrin receptor 2: Evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol. Biol. Cell 2007, 18, 743–754. [Google Scholar] [CrossRef]
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008, 7, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Viatte, L.; Lou, D.-Q.; Bennoun, M.; Beaumont, C.; Kahn, A.; Andrews, N.C.; Vaulont, S. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat. Genet. 2003, 34, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, J.; Kramer, M.; Tsukamoto, H.; Zhang, A.-S.; Enns, C.A. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009, 9, 217–227. [Google Scholar] [CrossRef]
- Ajioka, R.S.; Levy, J.E.; Andrews, N.C.; Kushner, J.P. Regulation of iron absorption in Hfe mutant mice. Blood 2002, 100, 1465–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.F.; Summerville, L.; Crampton, E.M.; Frazer, D.M.; Anderson, G.J.; Subramaniam, V.N. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 2009, 50, 1992–2000. [Google Scholar] [CrossRef]
- Ramey, G.; Deschemin, J.-C.; Vaulont, S. Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 2009, 94, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H.; Fleming, R.E.; Gui, D.; Moon, S.Y.; Saitoh, T.; O’Kelly, J.; Umehara, Y.; Wano, Y.; Said, J.W.; Koeffler, H.P. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood 2005, 105, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Armitage, A.E.; Eddowes, L.A.; Gileadi, U.; Cole, S.; Spottiswoode, N.; Selvakumar, T.A.; Ho, L.-P.; Townsend, A.R.M.; Drakesmith, H. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011, 118, 4129–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Zinkernagel, A.S.; Datta, V.; Lauth, X.; Johnson, R.S.; Nizet, V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurl, I.; Theurl, M.; Seifert, M.; Mair, S.; Nairz, M.; Rumpold, H.; Zoller, H.; Bellmann-Weiler, R.; Niederegger, H.; Talasz, H.; et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 2008, 111, 2392–2399. [Google Scholar] [CrossRef]
- Soares, M.P.; Weiss, G. The Iron age of host-microbe interactions. EMBO Rep. 2015, 16, 1482–1500. [Google Scholar] [CrossRef] [Green Version]
- Pasricha, S.-R.; McHugh, K.; Drakesmith, H. Regulation of hepcidin by erythropoiesis: The story so far. Annu. Rev. Nutr. 2016, 36, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.P.; Ribeiro, S.; Pontes, H.; Thowfeequ, S.; Tosh, D.; Carvalho, F.; Porto, G. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPalpha. Blood 2008, 111, 5727–5733. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.-H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.C.; Wang, R.-H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.-J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C. Iron regulatory elements (IREs): A family of mRNA non-coding sequences. Biochem. J. 1994, 304 Pt 1, 1–11. [Google Scholar] [CrossRef]
- Hentze, M.W.; Caughman, S.W.; Rouault, T.A.; Barriocanal, J.G.; Dancis, A.; Harford, J.B.; Klausner, R.D. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 1987, 238, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.L.; Hentze, M.W.; Koeller, D.M.; Caughman, S.W.; Rouault, T.A.; Klausner, R.D.; Harford, J.B. Iron-responsive elements: Regulatory RNA sequences that control mRNA levels and translation. Science 1988, 240, 924–928. [Google Scholar] [CrossRef]
- Dandekar, T.; Hentze, M.W. Finding the hairpin in the haystack: Searching for RNA motifs. Trends Genet. 1995, 11, 45–50. [Google Scholar] [CrossRef]
- Evstatiev, R.; Gasche, C. Iron sensing and signalling. Gut 2012, 61, 933–952. [Google Scholar] [CrossRef]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fillebeen, C.; Chen, G.; Biederbick, A.; Lill, R.; Pantopoulos, K. Iron-dependent degradation of apo-IRP1 by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 2007, 27, 2423–2430. [Google Scholar] [CrossRef]
- Wang, J.; Chen, G.; Lee, J.; Pantopoulos, K. Iron-dependent degradation of IRP2 requires its C-terminal region and IRP structural integrity. BMC Mol. Biol. 2008, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Allerson, C.R.; Polycarpou-Schwarz, M.; Rofts, A.; Rogers, J.T.; Kishi, F.; Hentze, M.W.; Rouault, T.A.; Andrews, N.C.; Hediger, M.A. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001, 509, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Galy, B.; Ferring, D.; Minana, B.; Bell, O.; Janser, H.G.; Muckenthaler, M.; Schümann, K.; Hentze, M.W. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 2005, 106, 2580–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Iwai, K.; LaVaute, T.; Brazzolotto, X.; Berger, U.V.; Land, W.; Ollivierre-Wilson, H.; Grinberg, A.; Love, P.; et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004, 23, 386–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.A.; Nizzi, C.P.; Chang, Y.-I.; Deck, K.M.; Schmidt, P.J.; Galy, B.; Damnernsawad, A.; Broman, A.T.; Kendziorski, C.; Hentze, M.W.; et al. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013, 17, 282–290. [Google Scholar] [CrossRef]
- Cooperman, S.S.; Meyron-Holtz, E.G.; Olivierre-Wilson, H.; Ghosh, M.C.; McConnell, J.P.; Rouault, T.A. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 2005, 106, 1084–1091. [Google Scholar] [CrossRef]
- Ferring-Appel, D.; Hentze, M.W.; Galy, B. Cell-autonomous and systemic context-dependent functions of iron regulatory protein 2 in mammalian iron metabolism. Blood 2009, 113, 679–687. [Google Scholar] [CrossRef]
- Haldar, M.; Kohyama, M.; So, A.Y.-L.; Kc, W.; Wu, X.; Briseño, C.G.; Satpathy, A.T.; Kretzer, N.M.; Arase, H.; Rajasekaran, N.S.; et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 2014, 156, 1223–1234. [Google Scholar] [CrossRef]
- Kohyama, M.; Ise, W.; Edelson, B.T.; Wilker, P.R.; Hildner, K.; Mejia, C.; Frazier, W.A.; Murphy, T.L.; Murphy, K.M. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 2009, 457, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Kurotaki, D.; Uede, T.; Tamura, T. Functions and development of red pulp macrophages. Microbiol. Immunol. 2015, 59, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef] [PubMed]
- Warnatz, H.-J.; Schmidt, D.; Manke, T.; Piccini, I.; Sultan, M.; Borodina, T.; Balzereit, D.; Wruck, W.; Soldatov, A.; Vingron, M.; et al. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J. Biol. Chem. 2011, 286, 23521–23532. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Schimanski, L.M.; Ormerod, E.; Merryweather-Clarke, A.T.; Viprakasit, V.; Edwards, J.P.; Sweetland, E.; Bastin, J.M.; Cowley, D.; Chinthammitr, Y.; et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood 2005, 106, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flashman, E.; Davies, S.L.; Yeoh, K.K.; Schofield, C.J. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 2010, 427, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappalardi, M.B.; McNulty, D.E.; Martin, J.D.; Fisher, K.E.; Jiang, Y.; Burns, M.C.; Zhao, H.; Ho, T.; Sweitzer, S.; Schwartz, B.; et al. Biochemical characterization of human HIF hydroxylases using HIF protein substrates that contain all three hydroxylation sites. Biochem. J. 2011, 436, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, N.; Hentze, M.W. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: Implications for regulation and cellular function. Proc. Natl. Acad. Sci. USA 2002, 99, 12345–12350. [Google Scholar] [CrossRef] [Green Version]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.-Y.; Hughes, R.; Murdoch, C.; Coffelt, S.B.; Biswas, S.K.; Harris, A.L.; Johnson, R.S.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Fiorito, V.; Marro, S.; Silengo, L.; Altruda, F.; Tolosano, E. Cell-specific regulation of Ferroportin transcription following experimentally-induced acute anemia in mice. Blood Cells Mol. Dis. 2013, 50, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrogiannaki, M.; Matak, P.; Mathieu, J.R.R.; Delga, S.; Mayeux, P.; Vaulont, S.; Peyssonnaux, C. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica 2012, 97, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Nathan, D.G.; Ginsburg, D.; Look, A.T.; Fisher, D.E.; Lux, S. Nathan and Oski’s Hematology of Infancy and Childhood E-Book; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- de Back, D.Z.; Kostova, E.B.; van Kraaij, M.; van den Berg, T.K.; van Bruggen, R. Of macrophages and red blood cells; a complex love story. Front. Physiol. 2014, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Ulyanova, T.; Padilla, S.M.; Papayannopoulou, T. Stage specific functional roles of integrins in erythropoiesis. Exp. Hematol. 2014, 2, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Bratosin, D.; Mazurier, J.; Tissier, J.P.; Estaquier, J.; Huart, J.J.; Ameisen, J.C.; Aminoff, D.; Montreuil, J. Cellular and molecular mechanisms of senescent erythrocyte phagocytosis by macrophages. A review. Biochimie 1998, 80, 173–195. [Google Scholar] [CrossRef]
- Marro, S.; Chiabrando, D.; Messana, E.; Stolte, J.; Turco, E.; Tolosano, E.; Muckenthaler, M.U. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position −7007 of the FPN1 promoter. Haematologica 2010, 95, 1261–1268. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Beaumont, C.; Delaby, C. Recycling iron in normal and pathological states. Semin. Hematol. 2009, 46, 328–338. [Google Scholar] [CrossRef]
- Gottlieb, Y.; Truman, M.; Cohen, L.A.; Leichtmann-Bardoogo, Y.; Meyron-Holtz, E.G. Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica 2012, 97, 1489–1493. [Google Scholar] [CrossRef]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [PubMed]
- Kovtunovych, G.; Ghosh, M.C.; Ollivierre, W.; Weitzel, R.P.; Eckhaus, M.A.; Tisdale, J.F.; Yachie, A.; Rouault, T.A. Wild-type macrophages reverse disease in heme oxygenase 1-deficient mice. Blood 2014, 124, 1522–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soe-Lin, S.; Apte, S.S.; Andriopoulos, B.; Andrews, M.C.; Schranzhofer, M.; Kahawita, T.; Garcia-Santos, D.; Ponka, P. Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5960–5965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, F.; An, P.; Guo, X.; Shen, Y.; Tao, Y.; Wu, Q.; Zhang, Y.; Yu, Y.; Ning, B.; et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011, 118, 1912–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, L.A.; Gutierrez, L.; Weiss, A.; Leichtmann-Bardoogo, Y.; Zhang, D.; Crooks, D.R.; Sougrat, R.; Morgenstern, A.; Galy, B.; Hentze, M.W.; et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010, 116, 1574–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.S.; Holderried, T.A.W.; Seifert, M.; et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabriek, B.O.; van Bruggen, R.; Deng, D.M.; Ligtenberg, A.J.M.; Nazmi, K.; Schornagel, K.; Vloet, R.P.M.; Dijkstra, C.D.; van den Berg, T.K. The macrophage scavenger receptor CD163 functions as an innate immune sensor for bacteria. Blood 2009, 113, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Graversen, J.H.; Madsen, M.; Moestrup, S.K. CD163: A signal receptor scavenging haptoglobin-hemoglobin complexes from plasma. Int. J. Biochem. Cell Biol. 2002, 34, 309–314. [Google Scholar] [CrossRef]
- Eid, R.; Zhou, D.R.; Arab, N.T.T.; Boucher, E.; Young, P.G.; Mandato, C.A.; Greenwood, M.T. Heterologous expression of anti-apoptotic human 14-3-3β/α enhances iron-mediated programmed cell death in yeast. PLoS ONE 2017, 12, e0184151. [Google Scholar] [CrossRef]
- Martins, R.; Maier, J.; Gorki, A.-D.; Huber, K.V.M.; Sharif, O.; Starkl, P.; Saluzzo, S.; Quattrone, F.; Gawish, R.; Lakovits, K.; et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat. Immunol. 2016, 17, 1361–1372. [Google Scholar] [CrossRef]
- Chow, A.; Huggins, M.; Ahmed, J.; Hashimoto, D.; Lucas, D.; Kunisaki, Y.; Pinho, S.; Leboeuf, M.; Noizat, C.; van Rooijen, N.; et al. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 2013, 19, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Lucas, D.; Hidalgo, A.; Méndez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessis, M. Erythroblastic island, functional unity of bone marrow. Rev. Hematol. 1958, 13, 8–11. [Google Scholar] [PubMed]
- Mohandas, N.; Prenant, M. Three-dimensional model of bone marrow. Blood 1978, 51, 633–643. [Google Scholar] [PubMed]
- Klei, T.R.L.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the cradle to the grave: The role of macrophages in erythropoiesis and erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef]
- Sadahira, Y.; Yoshino, T.; Monobe, Y. Very late activation antigen 4-vascular cell adhesion molecule 1 interaction is involved in the formation of erythroblastic islands. J. Exp. Med. 1995, 181, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zermati, Y.; Fichelson, S.; Valensi, F.; Freyssinier, J.M.; Rouyer-Fessard, P.; Cramer, E.; Guichard, J.; Varet, B.; Hermine, O. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp. Hematol. 2000, 28, 885–894. [Google Scholar] [CrossRef]
- Toda, S.; Segawa, K.; Nagata, S. MerTK-mediated engulfment of pyrenocytes by central macrophages in erythroblastic islands. Blood 2014. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Iglesias, A.; Potocnik, A.J.; Hartmann, U.; Fässler, R. Impaired migration but not differentiation of haematopoietic stem cells in the absence of β1 integrins. Nature 1996, 380, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Heideveld, E.; van den Akker, E. Digesting the role of bone marrow macrophages on hematopoiesis. Immunobiology 2017, 222, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.; Casu, C.; Gardenghi, S.; Breda, L.; Crielaard, B.J.; Guy, E.; Marongiu, M.F.; Gupta, R.; Levine, R.L.; Abdel-Wahab, O.; et al. Macrophages support pathological erythropoiesis in polycythemia vera and β-thalassemia. Nat. Med. 2013, 19, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolnek, T.; Hamza, I. Macrophages and iron trafficking at the birth and death of red cells. Blood 2015, 125, 2893–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacchini, L.; Gammella, E.; De Ponti, C.; Recalcati, S.; Cairo, G. Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J. Biol. Chem. 2008, 283, 20674–20686. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N.C. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Lambe, T.; Simpson, R.J.; Dawson, S.; Bouriez-Jones, T.; Crockford, T.L.; Lepherd, M.; Latunde-Dada, G.O.; Robinson, H.; Raja, K.B.; Campagna, D.R.; et al. Identification of a Steap3 endosomal targeting motif essential for normal iron metabolism. Blood 2009, 113, 1805–1808. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D.; Oukka, M.; Koss, L.M.; Aydemir, F.; Wessling-Resnick, M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. USA 2005, 102, 1324–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Song, Y.; Zhang, Z.; Li, D.; Zhu, H.; Liang, R.; Gu, Y.; Pang, Y.; Qi, J.; Wu, H.; et al. Distinct role of heme oxygenase-1 in early- and late-stage intracerebral hemorrhage in 12-month-old mice. J. Cereb. Blood Flow Metab. 2016, 37, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaer, C.A.; Deuel, J.W.; Schildknecht, D.; Mahmoudi, L.; Garcia-Rubio, I.; Owczarek, C.; Schauer, S.; Kissner, R.; Banerjee, U.; Palmer, A.F.; et al. Haptoglobin Preserves Vascular Nitric Oxide Signaling during Hemolysis. Am. J. Respir. Crit. Care Med. 2016, 193, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Leimberg, M.J.; Prus, E.; Konijn, A.M.; Fibach, E. Macrophages function as a ferritin iron source for cultured human erythroid precursors. J. Cell Biochem. 2008, 103, 1211–1218. [Google Scholar] [CrossRef]
- Leimberg, J.M.; Konijn, A.M.; Fibach, E. Developing human erythroid cells grown in transferrin-free medium utilize iron originating from extracellular ferritin. Am. J. Hematol. 2003, 73, 211–212. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Iron metabolism by macrophages. General steps of iron uptake, acquisition, storage and release in the macrophage: In addition to iron extraction from red blood cells by erythrophagocytosis, macrophages express a variety of receptors to acquire iron from different sources. Transferrin-bound iron can be taken up by the transferrin receptor (TfR or CD71) and the complex is internalized by clathrin coated endocytosis followed by iron release at a low endosomal pH. Empty apo-transferrin and transferrin receptor complex are recycled again (grey arrows). Non-transferrin iron can be acquired directly via divalent metal transporter 1 (DMT-1) that is associated with duodenal cytochrome B (DcytB) (blue arrow). CD91 (LRP1) receptors scavenge hemopexin-bound heme (Hpx-Heme) iron, whereas haptoglobin- hemoglobin (Hp-Hgb) complex is taken up through CD163 receptor (red arrows). Iron transport across the endosomal membrane into the cytosol is accomplished through DMT1 after it is reduced by the endosomal reductase six-transmembrane epithelial antigen of the prostate 3 (STEAP3). The resulting intracellular free iron joins the labile iron pool (LIP), which is then either stored in ferritin or utilized in cellular processes for example in mitochondrial iron metabolism as ISC [Fe-S] after being transported through Mitoferrin 1/2 (Mfrn1/2). Iron can be exported through ferroportin 1 (FPN1) out of the cell, supported by the ferroxidase ceruloplasmin (Cp). Iron is then loaded to transferrin in order to be carried to the target cells.
Figure 1.
Iron metabolism by macrophages. General steps of iron uptake, acquisition, storage and release in the macrophage: In addition to iron extraction from red blood cells by erythrophagocytosis, macrophages express a variety of receptors to acquire iron from different sources. Transferrin-bound iron can be taken up by the transferrin receptor (TfR or CD71) and the complex is internalized by clathrin coated endocytosis followed by iron release at a low endosomal pH. Empty apo-transferrin and transferrin receptor complex are recycled again (grey arrows). Non-transferrin iron can be acquired directly via divalent metal transporter 1 (DMT-1) that is associated with duodenal cytochrome B (DcytB) (blue arrow). CD91 (LRP1) receptors scavenge hemopexin-bound heme (Hpx-Heme) iron, whereas haptoglobin- hemoglobin (Hp-Hgb) complex is taken up through CD163 receptor (red arrows). Iron transport across the endosomal membrane into the cytosol is accomplished through DMT1 after it is reduced by the endosomal reductase six-transmembrane epithelial antigen of the prostate 3 (STEAP3). The resulting intracellular free iron joins the labile iron pool (LIP), which is then either stored in ferritin or utilized in cellular processes for example in mitochondrial iron metabolism as ISC [Fe-S] after being transported through Mitoferrin 1/2 (Mfrn1/2). Iron can be exported through ferroportin 1 (FPN1) out of the cell, supported by the ferroxidase ceruloplasmin (Cp). Iron is then loaded to transferrin in order to be carried to the target cells.

Figure 2.
Main regulatory mechanisms of iron. Regulation of iron metabolism through hepcidin, the IRP/IRE, Spi-C/Bach1, and HIF/HRE. For details see text.
Figure 2.
Main regulatory mechanisms of iron. Regulation of iron metabolism through hepcidin, the IRP/IRE, Spi-C/Bach1, and HIF/HRE. For details see text.

Figure 3.
Macrophages in erythrophagocytosis and erythropoiesis. The majority of iron in a macrophage is obtained during erythrophagocytosis. Splenic red pulp macrophages (RPMs) and liver Kupffer cells (KCs) is the main source of iron in the body. Senescent red blood cells (RBCs) are recognized by macrophages and digested in the lysosomes. Heme is extracted in the phagolysosomes and transferred to the cytosol by heme transporters heme-carrier protein 1 (HCP1) and heme responsive gene 1 protein (HRG1). Heme degradation by heme oxygenase 1 (HMOX1) in the cytosol provides ferrous iron that enters the intracellular labile iron pool. Iron is either stored in ferritin or exported by ferroportin 1 (FPN1) and ceruloplasmin (Cp). The transcription factor (Spi-C) protects iron processing RPMs in the spleen and BM nurse macrophages. Central nurse macrophages in the bone marrow (BM) promote erythropoiesis in the erythroblastic island niche. These macrophages ubiquitously express transferrin receptors and thus, probably mainly utilize transferrin (Tf)-bound iron. Nurse macrophages are important for the development of erythroblasts. This is mediated by adhesion molecules that control proliferation and differentiation of erythroblasts, enable erythroblast nuclei ingestion, and control the subsequent release of reticulocytes into the blood stream. Central macrophages are supposed to be involved in supplying maturing erythrocytes with iron to trigger heme synthesis.
Figure 3.
Macrophages in erythrophagocytosis and erythropoiesis. The majority of iron in a macrophage is obtained during erythrophagocytosis. Splenic red pulp macrophages (RPMs) and liver Kupffer cells (KCs) is the main source of iron in the body. Senescent red blood cells (RBCs) are recognized by macrophages and digested in the lysosomes. Heme is extracted in the phagolysosomes and transferred to the cytosol by heme transporters heme-carrier protein 1 (HCP1) and heme responsive gene 1 protein (HRG1). Heme degradation by heme oxygenase 1 (HMOX1) in the cytosol provides ferrous iron that enters the intracellular labile iron pool. Iron is either stored in ferritin or exported by ferroportin 1 (FPN1) and ceruloplasmin (Cp). The transcription factor (Spi-C) protects iron processing RPMs in the spleen and BM nurse macrophages. Central nurse macrophages in the bone marrow (BM) promote erythropoiesis in the erythroblastic island niche. These macrophages ubiquitously express transferrin receptors and thus, probably mainly utilize transferrin (Tf)-bound iron. Nurse macrophages are important for the development of erythroblasts. This is mediated by adhesion molecules that control proliferation and differentiation of erythroblasts, enable erythroblast nuclei ingestion, and control the subsequent release of reticulocytes into the blood stream. Central macrophages are supposed to be involved in supplying maturing erythrocytes with iron to trigger heme synthesis.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. https://0-doi-org.brum.beds.ac.uk/10.3390/ph11040137
AMA Style
Sukhbaatar N, Weichhart T. Iron Regulation: Macrophages in Control. Pharmaceuticals. 2018; 11(4):137. https://0-doi-org.brum.beds.ac.uk/10.3390/ph11040137
Chicago/Turabian StyleSukhbaatar, Nyamdelger, and Thomas Weichhart. 2018. "Iron Regulation: Macrophages in Control" Pharmaceuticals 11, no. 4: 137. https://0-doi-org.brum.beds.ac.uk/10.3390/ph11040137
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.